
Abstract
Aberrant regulation of the Wnt/β-catenin signaling pathway is one of the major causes of colorectal cancer (CRC). In this study, we examined the effect of polymethoxyflavones present in citrus peels on Wnt/β-catenin signaling in the HCT116 CRC cell line. We found that 5,7,3′,4′-tetra-methoxyflavone (TMF) and 7,8,3′,4′-TMF inhibited the expression of target genes of Wnt/β-catenin signaling and the transcriptional activities of β-catenin/Tcf and suppressed the motility of HCT116 cells. Because the binding of β-catenin to Tcf-4 was disrupted by 5,7,3′,4′-TMF and 7,8,3′,4′- TMF, we suggest that they are inhibitors of the Wnt/β-catenin signaling and may have potential applications in CRC prevention.
Introduction
Abnormalities in the Wnt/β-catenin pathway, including the activating mutations on β-catenin and inactivation of adenomatous polyposis coli (APC) genes, have been reported in human colorectal cancers (CRC) (Citation1–3). The inhibition of β-catenin ubiquitin-proteasome degradation, caused by mutations in APC or β-catenin, leads to the accumulation of β-catenin (Citation4). Stabilized β-catenin is translocated into the nucleus, where it binds to T cell factor (TCF)/lymphoid enhancer-binding factor (LEF) transcription factors, leading to the regulation of the transcription of Wnt target genes, including cyclin D1, c-Myc, Axin2, and vimentin (Citation5–7). Activation of Wnt/β-catenin signaling is one of the earliest events during the development of colon cancer and is required for tumor progression and metastasis. (Citation8–11). Therefore, the targeted inhibition of Wnt/β-catenin signaling is an effective approach for the prevention of CRC.
Epidemiological evidence suggests that a plant-based diet rich in flavonoids has anticancer effects (Citation12–14). In various cancer cell lines, flavonoids can modulate the Wnt/β-catenin signaling pathway (Citation15). However, little is known about the effects of polymethoxyflavone on Wnt/β-catenin pathway-dependent CRC. Polymethoxyflavones are compounds found in citrus peels, black ginger, etc. and have been reported to possess various physiological activities, such as anti-carcinogenic and anti-inflammatory properties (Citation16–18). To explore how food ingredients directly affect cells, we utilized the epithelial-like CRC cell line HCT116. HCT116 cells have a mutation in a CTNNB1 gene that encodes β-catenin, thereby enhancing the Wnt/β-catenin pathway (Citation19). In this study, we identified polymethoxyflavones that inhibit the Wnt/β-catenin pathway and evaluated their effect on the motility of HCT116 cells. Our findings make a significant contribution to the potential of TMF in the prevention of CRC.
Materials and Methods
Reagents
5,7,3′,4′-Tetra-methoxyflavone (TMF) (Funakoshi, Tokyo, Japan), 7,8,3′,4′-TMF (Extrasynthese, Genay, France), and dimethyl sulfoxide (DMSO) (Fujifilm Wako Pure Chemical Corporation, Osaka, Japan) were purchased commercially. Each TMF sample was dissolved in 100% DMSO. Control cells were treated with 0.4% DMSO. The following antibodies were purchased from commercial sources: anti-c-Myc (ab32072, Abcam, Cambridge, MA), anti-vimentin (LS-B1870, LSBio, Seattle, WA, USA), anti-Axin2 (Abcam ab109307), anti-β-catenin (NBP1-54467, Novus Biologicals, Littleton, CO, USA), anti-Tcf-4 (6H5-3, Merck, Darmstadt, Germany), anti-ERK1/ERK2 (GTX134462, GeneTex, Irvine, CA, USA), anti-phospho-ERK1/ERK2 (Thr185, Tyr187) (Thermo Fisher Scientific, MA, USA), anti-AKT (GTX121937, GeneTex, Irvine, CA, USA), anti-phospho-AKT (S473) (Abgent, San Diego, CA, USA), anti-β-actin (2F3, Wako, Pure Chemical Corporation, Osaka, Japan), anti-GAPDH (14C10, Cell Signaling Technology, Danvers, MA, USA), and FITC-conjugated goat anti-mouse IgG (Cosmo Bio, Tokyo, Japan).
Cell Culture
Human colorectal cancer cell line HCT116 was provided by RIKEN Bio-Resource Research Center through the National Bio-Resource Project of the NEXT (Japan) and cultured in RPMI-1640 medium (Wako) supplemented with 10% fetal bovine serum (Cytiva, Marlborough MA, USA) and 1% penicillin-streptomycin solution (Wako).
RT-PCR
HCT116 cells were treated with 30 μM 5,7,3′,4′-TMF (0.4% final DMSO concentration) or 7,8,3′,4′-TMF for 72 h. Total RNA was purified using an RNeasy Minikit (QIAGEN, Valencia, CA, USA) according to the manufacturer’s protocols and converted to cDNA using Moloney murine leukemia virus reverse transcriptase (Invitrogen, Carlsbad, CA, USA). The oligonucleotides used for the amplification of each cDNA were as follows: c-Myc, 5′-GCAGCCCCGAGCCCCTGGTGCTCCATGA-3′ and 5′-GAGACGTGGCACCTCTTGAGGACCAGTG-3′; vimentin, 5′-GAAATTGCAGGAGGAGATGC-3′ and 5′-GCCTGCAGCTCCTGGATTTCCTCTTCGTG-3′; Axin2, 5′-TGGGGGACTCGGGAGCCTAAAGGTCGTG-3′ and 5′-CATCTCCTTGGGCAGGCGGTGGGTTCTC-3′; β-actin, 5′-TCCTCCCTGGAGAAGAGCTAC-3′ and 5′-TCCTGCTTGCTGATCCACAT-3′.
Western Blotting, Immunoprecipitation, and Immunocytochemistry
HCT116 cells were treated with 30 μM 5,7,3′,4′-TMF or 7,8,3′,4′-TMF for 72 h and lysed in ice-cold Triton X-100 lysis buffer, which comprised 0.5% Triton X-100, 10 mM HEPES (pH 7.9), 50 mM NaCl, 100 mM EDTA, and 0.5 M sucrose, supplemented with a protease inhibitor cocktail Set I (Wako), for 30 min on ice. Lysates were centrifuged at 12,000 × g for 30 min at 4 °C, separated using SDS-polyacrylamide gels, and electroblotted.
For immunoprecipitation, HCT116 cells were treated with 30 μM 5,7,3′,4′-TMF or 7,8,3′,4′-TMF for 72 h and incubated in ice-cold Triton X-100 lysis buffer with protease inhibitor cocktail. Cell lysates were then incubated with anti-Tcf-4 antibodies for 1 h. The immune complexes were incubated with Dynabeads Protein A (Invitrogen) for 1 h, rinsed three times with ice-cold Triton X-100 lysis buffer, and boiled with Laemmli SDS-polyacrylamide gel electrophoresis sample buffer for 5 min. Total cell extracts and immunoprecipitates were subjected to western blotting.
For immunocytochemistry, HCT116 cells grown on poly L-lysine-coated cover glass (Matsunami Glass Ind. Ltd, Osaka, Japan) were treated with 30 μM 5,7,3′,4′-TMF or 7,8,3′,4′-TMF for 72 h. The cells were fixed in PBS containing 4% paraformaldehyde for 15 min and permeabilized with PBS containing 0.5% Triton X-100 a for 20 min. They were incubated for 1 h with 1% bovine serum albumin containing anti-β-catenin antibodies and visualized using fluorescein-conjugated goat anti-mouse IgG under fluorescence microscope (Axio Imager A1; Carl Zeiss Co. Ltd., Oberkochen, Germany).
Cell Proliferation Assay
HCT116 cells were plated onto 96-well plates (5 × 103 cells/well) and treated with 15-60 μM 5,7,3′,4′-TMF or 7,8,3′,4′-TMF for 72 h. Cell proliferation was assessed using the MTT Cell Proliferation and Cytotoxicity Assay Kit (Boster Immunoleader, Fremont, CA, USA) and Cell Counting Kit-8 (CCK-8) (Dojindo, Kumamoto, Japan) according to the manufacturer’s protocol. CCK-8 assay is based on the ability of dehydrogenases in viable cells to convert water-soluble tetrazolium salt to a water-soluble formazan dye. The amount of the formazan dye is directly proportional to the number of viable cells. Absorbance was measured at 570 (in the MTT assay) and 450 (in the CCK-8 assay) nm respectively using an Infinite™ M200PRO plate reader (Tecan, Grödig, Austria).
Wound-Healing Assay
After 72 h of cell treatment with 30 μM 5,7,3′,4′-TMF or 7,8,3′,4′-TMF, the cell monolayer was scratched using a sterile pipette. The wounded monolayers were washed three times with PBS and incubated for 48 h in RPMI-1640 culture medium with 30 μM 5,7,3′,4′-TMF or 7,8,3′,4′-TMF. Images were obtained using a microscope (CKX41; Olympus). Trypan blue dye exclusion assay was carried out to determine the amount of viable cells and total cell counts. At the end of the wound-healing assay, an equal volume of 0.5% trypan blue stain was added to the collected cell suspension and live cells were counted using a hemocytometer.
Cell Invasion Assay
To investigate the effect of TMFs on cell invasion, a CytoSelectTM 24-Well Cell Invasion Assay Kit (basement membrane colorimetric format) was used according to the manufacturer’s recommendations (Cell Biolabs Inc. San Diego, CA, USA). The basement membrane layer of the upper insert chambers was rehydrated with 300 μl of serum-free RPMI-1640. Sixty hours after treatment with 30 μM TMF or no TMF, cells were harvested, and 1.6 × 105 cells were seeded into the upper insert in serum-free RPMI-1640 containing 30 μM TMF or no TMF. Bottom chambers were filled with RPMI-1640 supplemented with 10% FBS containing 30 μM TMF or no TMF. After 48 h of incubation, cell invasion was assessed by a cell-staining solution (Cell Biolabs Inc.). Following the manufacturer’s protocol, stained inserts were placed in the extraction solution, and absorbance at 560 nm was determined using an Infinite™ M200PRO plate reader.
Luciferase Assay
TOPFLASH contained a luciferase reporter gene controlled by a minimum promoter containing multiple TCF binding sites to assess Wnt/β-catenin pathway activity. As a negative control, FOPFLASH was used, which contained a mutant instead of normal TCF-binding sites. HCT116 cells were plated onto 96-well plates and transfected with the TOPFLASH or FOPFLASH reporter plasmid carrying the firefly luciferase gene (Merck) and the reference plasmid pRL-SV40 carrying the Renilla luciferase gene (Promega, Madison, WI, USA). Twenty-four hours after transfection, cells were treated with 30 μM 5,7,3′,4′-TMF or 30 μM 7,8,3′,4′-TMF for 72 h. The cells were lysed in a passive lysis buffer (Promega). Firefly luciferase and Renilla luciferase activities were measured using the Dual-Luciferase Reporter Assay System (Promega) and an Infinite™ M200PRO plate reader.
Results
Screening of Polymethoxyflavones with Inhibitory Effect on Wnt/β-Catenin Signaling
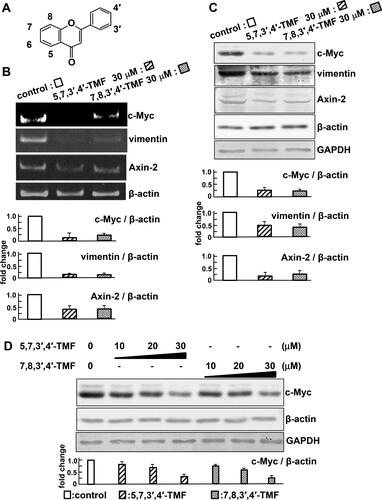
Polymethoxyflavones have a C6-C3-C6 skeleton and undergo various reactions, such as methylation (). We evaluated compounds that suppress the expression level of c-Myc, a target gene of Wnt/β-catenin signaling. Treatments with 5,7,4′-tri-methoxyflavone, tangeretin (5,6,7,8,4′-penta-methoxyflavone), and nobiletine (5,6,7,8,3′,4′-hexa-methoxyflavone) did not affect the level of c-Myc mRNA in HCT116 cells (). By contrast, the level of c-Myc mRNA was inhibited by treatment with 5,7,3′,4′-TMF or 7,8,3′,4′-TMF compared with the control (). Moreover, TMFs suppressed the transcription of Axin2 and vimentin, target genes of Wnt/β-catenin signaling pathway (). Consistent with the RT-PCR results, TMFs downregulated the protein levels of c-Myc, Axin2, and vimentin (). At concentrations below 30 μM, there was no change in the expression level of the internal controls, β-actin and GAPDH. On the contrary, the protein level of c-Myc was most suppressed in the treatment of TMFs at a concentration of 30 μM. Therefore, the following experiments were performed at 30 μM ().
Figure 1. 5,7,3′,4′-TMF or 7,8,3′,4′-TMF inhibits the target genes of Wnt/β-catenin signaling. (A) C6-C3-C6 skeleton of polymethoxyflavones. (B) mRNA levels of Wnt/β-catenin target genes, c-Myc, vimentin, and Axin2, were detected by RT-PCR. (C) Protein levels of c-Myc, vimentin, and Axin2. (D) Concentration-dependent effects of TMFs on protein levels of c-Myc and the internal standard, β-actin. Cell lysates were subjected to western blotting using the indicated antibodies. Results are normalized to β-actin mRNA or protein expression and presented as fold-change relative to the control. The results are representative of five independent experiments. These results are represented as the mean ± SE and are presented in a bar graph below the image. Statistical analyses of the images using the ImageJ software indicate that TMFs suppress the transcription and expression of c-Myc, vimentin, and Axin2.
5,7,3′,4′-TMF or 7,8,3′,4′-TMF inhibits the motility of HCT116 cells
To determine the effect of 5,7,3′,4′-TMF and 7,8,3′,4′-TMF on the proliferation or cytotoxicity in HCT116 cells, we investigated cell viability. In the MTT assay, a reduction in the proportion of viable cells was observed at 45 and 60 μM TMFs (). Furthermore, when the cell viability at 30 μM TMFs was confirmed using the CCK-8 assay, no significant change was observed after treatment with TMFs for 72 h. (). Next, we examined the effect of TMFs on extracellular signal-regulated kinase (ERK1/2) and the AKT pathway because the HCT116 cell line contains a heterozygous KRAS mutation. As shown in , TMFs were not involved in their activation. These findings suggest that TMFs are not associated with HCT116 cell proliferation at concentrations below 30 μM.
Figure 2. 5,7,3′,4′-TMF or 7,8,3′,4′-TMF inhibits the motility of HCT116 cells. (A) Effects of TMFs on cell viability and proliferation of HCT116. Left panel: MTT assay. Cells were treated with TMFs at the indicated concentrations and incubated for 72 h. Right panel: CCK-8 assay. Cells were treated with 30 μM TMF and incubated for 72 h. The amount of the formazan dye generated by dehydrogenase was assayed and presented as fold-change relative to the control or without dimethyl sulfoxide (DMSO). Data are presented as the mean ± SE. MTT and CCK-8 assays were performed in sextuplicate culture wells and repeated three times each. (B) Effects of TMFs on cell migration: control (a, b, and c), 5,7,3′,4′-TMF (d, e, and f), 7,8,3′,4′-TMF (g, h, and i) at 0 (a, d, and g), 24 (b, e, and h), and 48 h (c, f, and i) after wounding. Indicated lines show the scratch width. Representative images from the results of three independent wound healing assays (j). The wound area was measured and analyzed using the ImageJ software. Data are presented as the mean ± SE. At 48 h after wounding, the percentage of viable cells (k) and the total cell counts (l) were determined using a trypan blue dye exclusion assay. Experiments were performed in triplicate and repeated three times. (C) Effects of TMFs on cell invasion. Left: Invaded cells were visualized by cell staining. Right: Invasive cells were quantified at an optical density (OD) of 560 nm after extraction. Data represent the mean ± SE of three independent experiments. Statistical analyses indicated that TMFs were not associated with proliferation at concentrations below 30 μM and that they inhibit the motility of HCT116 cells.
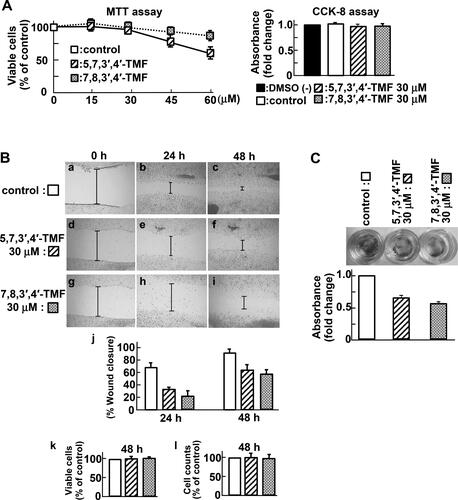
Next, we focused on reducing vimentin expression upon TMF stimulation. Vimentin, an intermediate filament protein, is correlated with the upregulation of cell motility and is a marker of epithelial-to-mesenchymal transition (EMT). Thus, we examined the effect of TMFs on the motility of HCT116 cells. Wound-healing assay revealed that TMFs decreased the migration distance of HCT116 cells (), suggesting an inhibitory effect. We further confirmed that there was no difference in the percentage of viable cells () or in the total cell counts () at the end of the wound-healing assay. An In Vitro invasion assay using Matrigel as a basement membrane barrier indicated that TMF-treated cells had markedly weaker invasion capacity than the control cells (). On the contrary, nobiletine- treatment did not reduce invasion (data not shown). Taken together, these data indicate that 5,7,3′,4′- TMF and 7,8,3′,4′-TMF inhibited the motility of HCT116 cells.
5,7,3′,4′-TMF or 7,8,3′,4′-TMF inhibits TCF-dependent transcription of HCT116 cells
To clarify the mechanism underlying the inhibitory effect of TMFs on the Wnt/β-catenin pathway, we examined the localization of β-catenin using immunocytochemistry. In the control group, β-catenin accumulation was observed in the cytoplasm and nucleus. Compared with the control, the TMF-treated group showed no difference in the expression level and localization of β-catenin ().
Figure 3. 5,7,3′,4′-TMF or 7,8,3′,4′-TMF inhibits TCF-dependent transcription of HCT116 cells. (A) Effects of TMFs on the activation of TCF-4. HCT116 cells transfected with pRL-SV40 reference plasmid (■ and □), TOPFLASH-Luc (■), or FOPFLASH-Luc (□) reporter plasmid were treated with DMSO (control), 30 μM 5,7,3′,4′-TMF, and 30 μM 7,8,3′,4′-TMF for 72 h. Results were measured as the ratio of firefly luciferase activity to Renilla luciferase activity and expressed as the fold-change relative to the control. Data represent the mean ± SE of three independent experiments. (B) Effects of TMFs on the formation of the TCF-4 and β-catenin complex. HCT116 cells were treated with DMSO, 30 μM 5,7,3′,4′-TMF, or 30 μM 7,8,3′,4′-TMF for 72 h. The lysates were probed with the indicated antibodies (lanes 1, 2, and 3) and immunoprecipitated with anti- TCF-4 antibodies (lanes 4, 5, and 6). IP, immunoprecipitation. Experiments were repeated three times. Protein levels of β-catenin and Tcf-4 are normalized to β-actin protein expression and presented as fold-change relative to the control. Data are presented as the mean ± SE. Results are representative of five independent experiments. Statistical analyses indicated that TMFs do not affect the expression levels of TCF-4 and β-catenin but interfere with the TCF-4 and β-catenin complex formation.
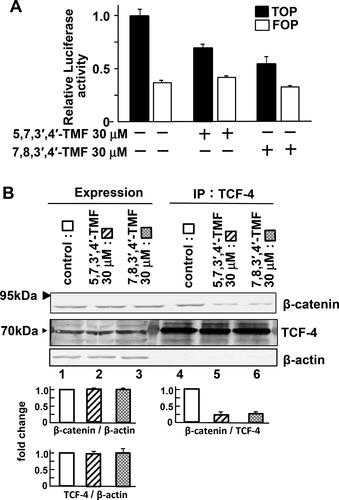
TOPFLASH and FOPFLASH constructs are widely used to evaluate TCF-dependent signaling (Citation20, Citation21). As shown in , there was no difference in FOPFLASH activity; however, TMFs effectively suppressed the TCF-dependent transcription (TOPFLASH) in the dual luciferase reporter assay. These findings suggest that TMFs are not associated with the proliferation of HCT116 cells at concentrations below 30 μM, indicating that TMFs may inhibit the binding between TCF and β-catenin or TCF and promoter.
To determine whether TMFs affect the formation of a complex between TCF-4 and β-catenin, immunoprecipitation experiments were performed. The TMF-treated cells and the control showed no difference in the expression levels of TCF-4 and β-catenin. Following TMF treatment and immunoprecipitation with anti-TCF-4 antibodies, we observed that the formation of the TCF-4 and β-catenin complex was reduced (). These findings suggest that TMFs interfere with the complex formation between TCF-4 and β-catenin.
Discussion
The multi-stage theory of carcinogenesis in CRC has been clarified. In the first stage, Wnt/β-catenin signaling is activated by β-catenin or APC gene mutation; in the second stage, KRAS is activated; in the third stage, the tumor suppressor gene p-53 is inactivated, and other factors contribute to malignancy (Citation22). In this study, we analyzed HCT116 cell lines with genetic mutations in β-catenin and KRAS(G12D). Mutations in KRAS cause the activation of downstream AKT and ERK and are involved in cell survival signal. Therefore, HCT116 cell line is an advanced cancer cell in which the Ras-dependent survival signaling is activated in addition to the activation of Wnt/β-catenin signaling. We found that TMFs suppressed the Wnt target gene involved in cell proliferation but did not affect cell survival and the activation of AKT and ERK. These data suggest that the limited concentration of TMFs cannot suppress the cell survival signal by KRAS activation. However, TMFs suppressed EMT-related vimentin expression and cell motility, which may inhibit cancer metastasis. In a CRC mouse model with APC gene mutation, nuclear accumulation of β-catenin is observed at the site of intestinal polyps but not in the normal epithelium. It has been reported that fibroblast-secreted factors partially activate the nuclear accumulation of β-catenin (Citation23). Since such a microenvironment is the first step in adenoma formation, early suppression of Wnt/β-catenin signaling leads to inhibition of adenoma formation. This may prevent progression to the next step, that is, the transition to adenocarcinoma. TMFs did not affect the survival; however, this chemopreventive approach may inhibit metastasis or adenoma formation by specifically inhibiting Wnt/β-catenin signaling.
Quercetin is a polyphenolic flavonoid widely distributed in plants and has been reported to suppress the binding of β-catenin to Tcf-4 in the colon cancer cell line SW480 (Citation24, Citation25). Our experimental data are consistent with previous findings as the TMFs we studied have a C6-C3-C6 skeleton similar to quercetin. Because flavonoids with a methoxy group can easily pass through the cell membrane, TMFs may have a more effective suppression effect on the binding of β-catenin to Tcf-4 than quercetin, which has a hydroxyl group.
In general, compounds with fewer methoxy groups exert stronger antioxidant effects. However, in this study, we found no correlation between the number of methoxy groups and the inhibitory effect on Wnt target gene expression. Because flavones having four methoxy groups have an inhibitory effect on the expression of Wnt target genes, the structure of the compound may be involved in the inhibition. As there was no change in the protein level and localization of β-catenin, we propose that TMF inhibited the formation of the complex between β-catenin and the transcription factor Tcf/Lef in the nucleus or that between Tcf/Lef and the promoter. In this study, the analysis used cultured cells, which is different from the environment In Vivo. In addition, because β-catenin is accumulated by APC mutations in many CRCs, it is necessary to verify whether TMF can inhibit the APC mutation-dependent activation of Wnt/β-catenin signaling. To validate this hypothesis, we are currently conducting In Vivo studies using a CRC mouse model. Control of the Wnt/β-catenin pathway by food components such as flavonoids has the potential to contribute to innovative treatment strategies for CRC.
Conclusion
In conclusion, we found that 5,7,3′,4′-TMF and 7,8,3′,4′-TMF suppressed the expression of target genes of Wnt/β-catenin signaling, inhibited cell motility, and interfered with complex formation between TCF-4 and β-catenin. TMFs have potential applications in CRC prevention.
Supplemental Material
Download Zip (306.2 KB)Disclosure statement
The authors report no conflict of interest.
Additional information
Funding
References
- Novellasdemunt L, Antas P, Li VS. Targeting Wnt signaling in colorectal cancer: a review in the theme: cell signaling: proteins, pathways and mechanisms. Am J Physiol Cell Physiol 2015;309(8):C511–C521. doi:10.1152/ajpcell.00117.2015
- Zhang L, Shay JW. Multiple roles of APC and its therapeutic implications in colorectal cancer. J Natl Cancer Inst 2017;109:djw332. doi:10.1093/jnci/djw332
- Yang X, Zhong J, Zhang Q, Feng L, Zheng Z, Zhang J, Lu S. Advances and insights of APC-Asef inhibitors for metastatic colorectal cancer therapy. Front Mol Biosci 2021;8:662579. doi:10.3389/fmolb.2021.662579
- Zhang Y, Wang X. Targeting the Wnt/β-catenin signaling pathway in cancer. J Hematol Oncol 2020;13(1):165. doi:10.1186/s13045-020-00990-3
- Lecarpentier Y, Schussler O, Hébert JL, Vallée A. Multiple targets of the canonical Wnt/β-catenin signaling in cancers. Front Oncol 2019;9:1248. doi:10.3389/fonc.2019.01248
- Lazarova DL, Bordonaro M. Vimentin, colon cancer progression and resistance to butyrate and other HDACis. J Cell Mol Med 2016;20(6):989–93. doi:10.1111/jcmm.12850
- Strouhalova K, Přechová M, Gandalovičová A, Brábek J, Gregor M, Rosel D. Vimentin intermediate filaments as potential target for cancer treatment. Cancers 2020;12(1):184. doi:10.3390/cancers12010184
- Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature 2005;434(7035):843–50. doi:10.1038/nature03319
- Humphries A, Wright NA. Colonic crypt organization and tumorigenesis. Nat Rev Cancer 2008;8(6):415–24. doi:10.1038/nrc2392
- Scholer-Dahirel A, Schlabach MR, Loo A, Bagdasarian L, Meyer R, Guo R, Woolfenden S, Yu KK, Markovits J, Killary K, et al. Maintenance of adenomatous polyposis coli (APC)-mutant colorectal cancer is dependent on Wnt/beta-catenin signaling . Proc Natl Acad Sci U S A 2011;108(41):17135–40. doi:10.1073/pnas.1104182108
- Cheriyamundath S, Ben ZA. Wnt/β-catenin target genes in colon cancer metastasis: the special case of L1CAM. Cancers 2020;12(11):3444. doi:10.3390/cancers12113444
- Kundu JK, Surh YJ. Breaking the relay in deregulated cellular signal transduction as a rationale for chemoprevention with anti-inflammatory phytochemicals. Mutat Res 2005;591(1-2):123–46. doi:10.1016/j.mrfmmm.2005.04.019
- Khan N, Afaq F, Mukhtar H. Cancer chemoprevention through dietary antioxidants: progress and promise. Antioxid Redox Signal 2008;10(3):475–510. doi:10.1089/ars.2007.1740
- Rodríguez-García C, Sánchez-Quesada C, Gaforio JJ. Dietary flavonoids as cancer chemopreventive agents: an updated review of human studies. Antioxidants 2019;8(5):137. doi:10.3390/antiox8050137
- Amado NG, Fonseca BF, Cerqueira DM, Neto VM, Abreu JG. Flavonoids: potential Wnt/beta-catenin signaling modulators in cancer. Life Sci 2011;89(15-16):545–54. doi:10.1016/j.lfs.2011.05.003
- Kawabata K, Murakami A, Ohigashi H. Nobiletin, a citrus flavonoid, down-regulates matrix metalloproteinase-7 (matrilysin) expression in HT-29 human colorectal cancer cells. Biosci Biotechnol Biochem 2005;69(2):307–14. doi:10.1271/bbb.69.307
- Lai C-S, Li S, Chai C-Y, Lo C-Y, Dushenkov S, Ho C-T, Pan M-H, Wang Y-J. Anti-inflammatory and antitumor promotional effects of a novel urinary metabolite, 3’,4’-didemethylnobiletin, derived from nobiletin. Carcinogenesis 2008;29(12):2415–24. doi:10.1093/carcin/bgn222
- Li N, Zhang Z, Jiang G, Sun H, Yu D. Nobiletin sensitizes colorectal cancer cells to oxaliplatin by PI3K/Akt/MTOR pathway. Front Biosci (Landmark Ed) 2019;24(2):303–12. doi:10.2741/4719
- Arnold A, Tronser M, Sers C, Ahadova A, Endris V, Mamlouk S, Horst D, Möbs M, Bischoff P, Kloor M, et al. The majority of β-catenin mutations in colorectal cancer is homozygous. BMC Cancer 2020;20(1):1038. doi:10.1186/s12885-020-07537-2
- Hino S-I, Tanji C, Nakayama K-I, Kikuchi A. Phosphorylation of beta-catenin by cyclic AMP-dependent protein kinase stabilizes beta-catenin through inhibition of its ubiquitination . Mol Cell Biol 2005;25(20):9063–72. doi:10.1128/MCB.25.20.9063-9072.2005
- Lu B, Green BA, Farr JM, Lopes FC, Van Raay TJ. Wnt drug discovery: weaving through the screens, patents and clinical trials. Cancers 2016;8(9):82. doi:10.3390/cancers8090082
- Yamada Y, Mori H. Multistep carcinogenesis of the colon in Apc(Min/+) mouse. Cancer Sci 2007;98(1):6–10. doi:10.1111/j.1349-7006.2006.00348.x
- Vermeulen L, De Sousa E Melo F, van der Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M, Merz C, Rodermond H, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol 2010;12(5):468–76. doi:10.1038/ncb2048
- Park CH, Chang JY, Hahm ER, Park S, Kim H-K, Yang CH. Quercetin, a potent inhibitor against beta-catenin/Tcf signaling in SW480 colon cancer cells. Biochem Biophys Res Commun 2005;328(1):227–34. doi:10.1016/j.bbrc.2004.12.151
- Shan BE, Wang MX, Li RQ. Quercetin inhibit human SW480 colon cancer growth in association with inhibition of cyclin D1 and survivin expression through Wnt/beta-catenin signaling pathway . Cancer Invest 2009;27(6):604–12. doi:10.1080/07357900802337191