A Czech warmblood filly was presented for the first time at the clinic at the age of five months. Following delivery, she was weak and needed assistance to stand up for the first few days but she was able to suck without any problems. After a few days, movement and the ability to stand up improved according to the owner. At the age of one month, the movement of the foal was deemed abnormal. Behaviour and appetite were regarded normal although the foal was thin. The abnormal movement did not change during exercise or over a long-term period.
The filly's body condition score was 4/9 at admission. The M. gluteus medius, M. quadriceps femoris, M. semitendinosus and M. biceps femoris were hypertrophied, whereas M. gluteus superficialis was atrophied. The muscles of the neck and trunk were poorly developed. During the first few minutes of movement, the filly demonstrated toe-touching which later disappeared when she was then able to fully tread on the foot. Movement was stiff with a prolongation of the swing phase (visible mainly in trot). Weakness was not visible. In gallop, the foal showed bunny-hopping (Video 11). Spontaneous muscle dimpling was noticed but percussion of the rear muscles did not elicit muscle contraction. Needle EMG was not performed because of the unavailability of this examination at that time. Clinical examination did not reveal any other abnormalities. Blood analysis did not reveal abnormalities, except for slightly elevated muscle enzyme activities (CK 491 IU/L [reference range 204–263 IU/L]; AST 447 IU/L [reference range 329–337 IU/L]; LDH 1071 IU/L [reference range 569–917 IU/L]).
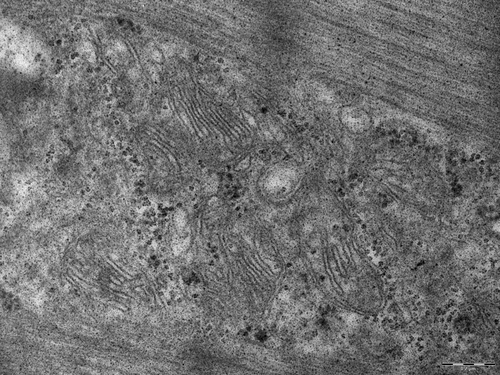
A needle biopsy taken from the gluteus medius muscle (according to Dubowitz and Sewry Citation2007) revealed a pattern of muscle dystrophy. In addition to the general myopathic pattern (variation in fibre size, 10% inner nuclei, splitting of the hypertrophic fibres, regenerating fibres, endo-perimysial fibrosis) and fibre-type grouping, the most typical and characteristic finding was the presence of cytoplasmic inclusions mainly present in type 1 fibres. These were identified in routinely stained sections (H&E, Gomori) as irregularly shaped, weakly stained amorphous areas (). Periodic acid-Schiff (PAS) staining showed the presence of dispersed (amylase-sensitive) glycogen granules mostly at the periphery of the inclusions. The inclusions were unstained or only very faintly stained, but sharply demarcated by the reaction to mitochondrial dehydrogenases. The reaction to desmin (Dako, Glostrup, Denmark) and myotilin (Novocastra Reagents, Leica Biosystems Ltd, Newcastle upon Tyne, UK) showed fluctuating reaction intensity inside the inclusions but neither accumulation nor depletion of the filaments. The reaction of the inclusions to myofibrillar ATPase (pH 9.4) was preserved but usually weaker. Electron microscopic examination (according to Dubowitz and Sewry Citation2007) of the inclusions revealed an absence or disorganization of the myofibrils: some inclusions at the periphery of the myofibres contained homogenous granulofilamentous material, sporadically apoptotic nuclei or apoptotic bodies. The myofibrils showed sporadically streamed Z-lines. In addition to the inclusions, there were other spaces with altered myofibrillar texture intensively stained by Gomori trichrome, PAS and SDH. The spaces contained myofilaments, fascicles or fragments of the myofibrils, dispersed glycogen granules, vesicles, myelin figures, irregularly scattered mitochondria and membranes resembling endoplasmic reticulum or Golgi system ().
Figure 1. Gomori trichrome. Hyaline inclusions inside the myofibres (asterisks). Intensively stained spaces at the periphery of myofibres (arrows) (Bar 100 µm, 1st biopsy).
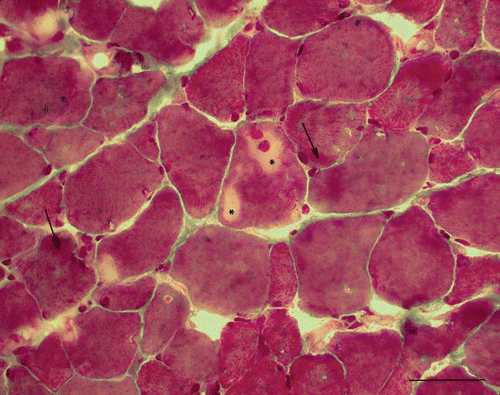
Figure 2. Membranes of sarcoplasmic reticulum and Golgi system in the spaces (1st biopsy).
At the age of two years, movement had improved according to her new owner and hind leg stiffness was visible mainly at the start of movement. Body condition was normal (5/9) and the mare still had hypertrophy and dimpling of the rear muscles.
At the age of three years, M. semitendinosus, M. biceps femoris, M. quadriceps femoris and M. gluteus medius were hypertrophied and M. gluteus superficialis was atrophied similar to foal age. The girdle of the thoracic limb and neck were normally developed, but muscles of the back were poorly developed. Movement of the horse was stiff with prolongation of the swing phase most visible in the trot (Video 2). Clinical examination did not reveal any other abnormalities, whereas muscle enzyme activities were slightly increased (CK 567 IU/L and AST 515 IU/L). Needle EMG examination (Dantec Dynamics, Bristol, UK) showed a mild myogenic pattern with early recruitment of small motor unit action potential (MUPs) in M. gluteus medius. Myotonic discharges were prominent in this muscle as well as myotonic discharges and non-specific complex repetitive discharges. Needle EMG of M. splenius revealed no myotonic discharges with results of MUP analysis similar to control horses (). The uncooperative behaviour of the sedated patient in the stock did not allow for complete MUP analysis. Based on the clinical and associated EMG findings a myotonic disorder with features of myotonic dystrophy or congenital myotonia was considered likely. Histopathological findings in another muscle biopsy from gluteus medius muscle revealed a pattern that was quite different in comparison to the first biopsy. In addition to fibre size variability with atrophic and extremely hypertrophic fibres (up to 200 µm), there were numerous central nuclei (80–95%) (), and sarcoplasmic masses located under the sarcolemma or inside the myofibre () and ring fibres. The masses were strongly positive to PAS, NADH-TR, SDH and non-specific esterase. Splitting of the hypertrophic fibres with inner nuclei was very prevalent. There were no inclusions or spaces present compared to the first biopsy. Electron microscopic examination revealed spaces with disorganized and split myofibrils, fascicles of myofilaments and scattered mitochondria, which corresponded to the sarcoplasmic masses, glycogen granules and rod-like structures. On the other hand, there was no fine granular and filamentous material characteristic of the previous biopsy. Histopathologic findings in the second biopsy were regarded as similar to that in human patients with myotonic dystrophy. Fluorescence in situ hybridization on frozen sections was performed as described by Lukas et al. (Citation2012) to the CUG and CCUG expansion repeats in the corresponding genes encoding both types of myotonic dystrophy with negative results.
Figure 3. H&E. Variation of myofibre size, splitting of the hypertrophic fibres (up to 200 µm) with numerous inner nuclei and endomysial fibrosis (Bar 50 µm, 2nd biopsy).
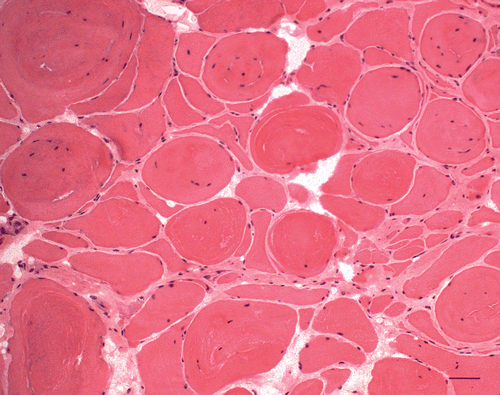
Figure 4. SDH. Sarcoplasmic masses (arrow) (Bar 130 µm, 2nd biopsy).
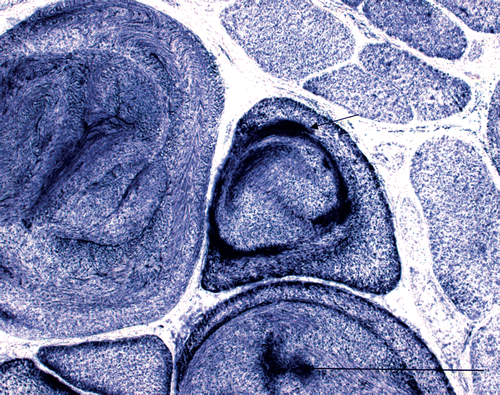
Table 1. Results of the MUP analysis in the index case (M. splenius used).
At the age of four years, she delivered a healthy foal (not examined clinically) and is ridden regularly for pleasure.
The mother of the presented case (Czech Warmblood) had successfully competed in high level show-jumping competitions in the past. She was clinically examined at the age of 17 years without abnormal findings (including symmetrically developed muscles). Needle EMG (M. splenius and M. subclavius) did not reveal any myotonic discharges. The result of MUP analysis was similar to control horses (). The dam was found dead in the stable 8 months later without necropsy being performed.
Table 2. Results of the MUP analysis in the dam (M. splenius and M. subclavius used).
The filly currently described was the dam's first foal, born after a normal pregnancy. The dam's second foal (also a filly) was born weak and unable to stand up after delivery without visible limb deformities. The second filly had a good appetite and did not show signs of dysphagia. However, she died from an unknown cause at the age of 10 days. Neither medical examination nor necropsy of the cadaver was performed. The sire of the second foal was different from the first one.
Muscle dystrophy accompanied by myotonia was described in Quarter Horses (Jamison et al. Citation1987; Reed et al. Citation1988; Hegreberg and Reed Citation1990), Thoroughbreds (Andrews et al. Citation1986; Shirakawa et al. Citation1989), Standardbreds (Sarli et al. Citation1994) and Anglo-Arab Sardinian horse (Montagna et al. Citation2001). Predominant clinical signs in all cases were gait stiffness, muscle hypertrophy or atrophy, weakness and generalized myotonia. The basic electromyographic feature was the presence of myotonic discharges (Reed et al. Citation1988; Hegreberg and Reed Citation1990; Montagna et al. Citation2001). Clinical signs were visible from the first month of life (Reed et al. Citation1988). Testicular atrophy, early cataract and glucose intolerance were also noticed in some cases (Reed et al. Citation1988; Montagna et al. Citation2001).
Histopathological findings in described equine cases included internalized nuclei, fibre size variation, sarcoplasmic masses, ringed fibres, moth-eaten fibres, fibre-type grouping, split fibres intermixed with hypertrophic fibres, and adipose and connective tissue infiltration. Sarcoplasmic masses, which can occur in myotonic dystrophy, are characterized as sharply defined peripheral areas which do not stain to ATPase reaction but with a strong positive reaction to oxidative enzymes and phosphorylase and PAS positivity (Jamison et al. Citation1987; Reed et al. Citation1988; Hegreberg and Reed Citation1990; Montagna et al. Citation2001). The ultrastructure was characterized by distortion of the Z band, sarcolemmal loss with glycogen accumulation and dilation of the sarcotubular system (Montagna et al. Citation2001).
A muscle biopsy is only one piece of the jigsaw and has to be considered together with family history, the clinical history and presentation, and the results of other investigations. In some instances, the changes may be striking and unequivocal. In others, the changes may be more subtle and a systemic approach is required in the evaluation and interpretation. Once the pathology is defined, correlation with clinical features is essential and the pathology must be interpreted in the light of this (Dubowitz and Sewry Citation2007).
The histopathological features of the first biopsy, namely the reaction to myosin-ATPase (pH 9.6), H&E, oxidative enzymes or Gomori trichrome stain and the ultrastructure of the inclusions most closely corresponded to human hyaline body disease (myosin storage myopathy) or myofibrillar myopathy. Preapoptotic and apoptotic nuclei, dislocated membranous organelles and glycogen accumulations in inclusions are described in myofibrillar myopathies similar to inclusions devoid of or diminished of oxidative enzyme activity but with oxidative enzyme activity accentuated around the larger inclusions. Some features of the other spaces, e.g. endoplasmic or Golgi membranes, bundles of myofilaments, fascicles or fragments of the myofibrils, mitochondria and streamed Z-lines are common in different myofibrillar myopathies (Selcen Citation2011). Subsarcolemmal accumulation of hyaline material in type 1 fibres and inclusions unstained in NADH-TR are the most important histopathological feature of myosin storage myopathy (Oldfords Citation2007). Both, myofibrillar myopathy or myosin storage myopathy, as far as we know, have not been described in the horse.
The histopathological pattern of the second biopsy corresponds unequivocally to that in human myotonic dystrophy (Adams Citation1975; Carpenter and Karpati Citation2001; Vattemi et al. Citation2005). It is characterized by fibre size variation, increased number of inner nuclei (80–95%), ring fibres and sarcoplasmic masses that lack properly organized myofibrils and contain mixture of poorly organized myofilaments with mitochondrial sarcotubular elements and glycogen (Carpenter and Karpati Citation2001).
A marked fibre size variation and numerous internal nuclei found in our case were a consistent finding in all described cases of equine myotonic dystrophy (Andrews et al. Citation1986; Jamison et al. Citation1987; Reed et al. Citation1988; Shirakawa et al. Citation1989; Hegreberg and Reed Citation1990; Sarli et al. Citation1994; Montagna et al. Citation2001). Sarcoplasmic masses that were found in our horse were detected in some of them (Reed et al. Citation1988; Hegreberg and Reed Citation1990; Montagna et al. Citation2001). However, it should be realized that sarcoplasmic masses were also described in normal equine skeletal muscles by Aleman et al. (Citation2005).
In all described equine cases of myotonic dystrophy as well as in our case, the clinical signs were noticed for the first time in newborn or suckling foals (Jamison et al. Citation1987; Reed et al. Citation1988; Shirakawa et al. Citation1989; Sarli et al. Citation1994; Montagna et al. Citation2001). In all of them the most affected were hind leg muscles (superficial gluteal, semimembranosus and semitendinosus) and muscles of the back. Front leg muscles were affected only in some cases (Jamison et al. Citation1987; Reed et al. Citation1988; Montagna et al. Citation2001). In the mare described in this report the hind legs were predominantly affected. Front legs were less muscled but it is difficult to distinguish if this was due to lower physical activity (not in training yet; less voluntary activity due to hind limb myotonia) or in accord with myopathy.
Myotonic dystrophy is a slowly progressive disorder in humans. Three of the described equine cases showed progression of clinical signs (Reed et al. Citation1988). In one case, clinical signs did not progress over a period of five years (Montagna et al. Citation2001) similarly as our case which has not been clinically progressive over a period of four years.
Systemic extramuscular involvement (cardiomyopathy, mental retardation, cataracts, ptosis, insulin resistance, hypogonadism, testicular atrophy and neuropathy) is common in myotonic dystrophy in man (Machuca-Tzili et al. Citation2005). Atrophy of the testicles, lenticular opacity and retinal dysplasia was described in some of the equine cases (Reed et al. Citation1988; Montagna et al. Citation2001). In our patient, no systemic extramuscular involvement was detected.
Myotonia was clinically visible in our case as muscle dimpling on the hind legs and as a prolongation of the swing phase most clearly visible in trot. In gallop, the filly showed bunny hopping which was also observed in other equine cases (Reed et al. Citation1988; Hegreberg and Reed Citation1990). Nevertheless, bunny hopping is not pathognomonic for myotonic dystrophy in horses and can be seen in various neurologic or orthopaedic disorders. Also, myotonia is not pathognomonic for myotonic dystrophy either but it can be observed in horses in myotonia congenita (Wijnberg et al. Citation2012) and HYPP (Rudolph et al. Citation1992). In man it has been reported in myofibrillar myopathies (Selcen et al. Citation2004).
In man, weakness is a common clinical sign in most congenital myopathies including myotonic dystrophy (Machuca-Tzili et al. Citation2005), myofibrillar myopathy (Selcen Citation2011) or myosin storage myopathy (Oldfords Citation2007). Some of the described equine cases of myotonic dystrophy suffered from weakness (Reed et al. Citation1988; Hegreberg and Reed Citation1990). In cases described by Jamison et al. (Citation1987) and Montagna et al. (Citation2001) stiffness predominated without weakness similar as in the current case.
CK activity may be mildly elevated in myotonic dystrophy in man. Also, in described suspected equine cases elevation of CK activity was recorded (Jamison et al. Citation1987; Reed et al. Citation1988; Montagna et al. Citation2001) similar to the current case. Elevation of CK is a non-specific feature of myopathies and CK activity within the reference range does not exclude muscle pathology. CK may be normal or mildly elevated in myosin storage myopathy (Goebel and Laing Citation2009). In myofibrillar myopathy, CK activity usually is slightly elevated but may be high as well (Oldfords Citation2007).
The changing or variable histopathological picture seen in the current case has not been described in equine myopathies yet, but it is known in some human neuromuscular diseases especially congenital myopathies (Donner et al. Citation1975; Doriguzzi et al. Citation1999; Jungbluth et al. Citation2008; Romero et al. Citation2010). There are two reports about atypical histopathological findings in congenital or infantile myotonic dystrophy presenting several cases of genetically confirmed myotonic dystrophy as myositis or congenital fibre type disproportion (Lukas et al. Citation2007; Tominaga et al. Citation2010).
The comparison of both histopathological features in the subsequent biopsies suggests irregular and excessive growth of some myofibres with proliferation of myofibrils and muscle hypercontraction followed by splitting of the sarcoplasm in the largest ones and nuclear internalization (which is a process reverse to that occurring in developing muscle fibre) among the biopsies. The presence of inclusions or sarcoplasmic masses may be a consequence of this process.
To the best of our knowledge, only seven other cases of myotonic dystrophy have been described in horses yet. None of these cases described so far, including our case, had a positive family history of any muscle disorder. As a consequence, in horses this condition is more probably caused by de novo mutations, unlike the autosomal dominant inheritance in man.
The relation between human and equine myotonic dystrophy is not clear enough, since the genetic background of the equine disorder has not yet been established. In this report, we tested but did not find – and practically excluded – expanded repetitions CUG or CCUG, typical of human myotonic dystrophy type 1 and type 2, respectively (Brook et al. Citation1992; Fu et al. Citation1992; Liquori et al. Citation2001).
We conclude that the described case displayed myotonic dystrophy. The development of histopathological findings between the first biopsy at the age of five months and the second biopsy performed in adulthood reflects, in our opinion, progression of the disease during this period. Subsequent muscle biopsies are rarely carried out on humans with myotonic disorders. Thus, our case may be of relevance not only in horses, but in human patients as well.
Long-term follow-up of this case indicate possible riding and breeding use of some affected horses and their comfortable life.
tveq_a_749548_sup_30204532.zip
Download Zip (44.2 MB)Acknowledgement
Supported by the Ministry of Education, Youth and Sports of the Czech Republic (Grant No. MSM6215712403).
Notes
1. All supplementary videos can be found online at: http://dx.doi.org/10.1080/01652176.2012.749548
References
- Adams , RD . 1975 . Diseases of muscle , 3rd , Hagerstown (MD) : Harper & Row .
- Aleman , M , LeConteur , RA , Nieto , JE , Williams , DC , Stanke , NJ and Shelton , GD . 2005 . Sarcoplasmic masses in equine skeletal muscle . Neuromuscul Disord , 15 : 147 – 153 .
- Andrews , FM , Spurgeon , TL and Reed , SM . 1986 . Histochemical changes in skeletal muscles of four male horses with neuromuscular disease . Am J Vet Res , 47 : 2078 – 2083 .
- Brook , JD , McCurah , ME , Harley , HG , Buckler , AJ , Church , D , Aburatani , H , Hunter , K , Stanton , VP , Thirion , JP , Hudson , T , Sohn , R , Zemelman , B , Snell , RG , Rundle , SA , Crow , S , Davies , J , Shelbourne , P , Buxton , J , Jones , C , Juvonen , V , Johnson , K , Harper , PS , Shaw , DJ and Housman , DE . 1992 . Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member . Cell , 68 : 799 – 808 .
- Carpenter , S and Karpati , G . 2001 . Pathology of skeletal muscle , 2nd , New York (NY) : Oxford University Press .
- Donner , M , Rapola , J and Somer , H . 1975 . Congenital muscular dystrophy: a clinicopathological and follow-up study of 15 patients . Neuropädiatrie , 6 : 239 – 258 .
- Doriguzzi , C , Palmuzzi , L , Mongini , T , Chiado-Piat , L , Saggiorato , C , Ugo , I and Hoffman , EP . 1999 . Variable histological expression of dystrophinopathy in two females . Acta Neuropathol , 97 : 657 – 660 .
- Dubowitz , V and Sewry , CA . 2007 . Muscle biopsy. A practical approach , 3rd , Philadelphia (PA) : Elsevier Saunders .
- Fu , YH , Pizzuti , A , Fenwick Jr , RG , King , J , Rajnarayan , S , Dunne , PW , Dubel , J , Nasser , GA , Ashizawa , T , Dejong , P , Wieringa , B , Korneluk , R , Perryman , MB , Epstein , HF and Caskey , CT . 1992 . An unstable triplet repeat in a gene related to myotonic dystrophy . Science , 255 : 1256 – 1258 .
- Goebel , HH and Laing , NG . 2009 . Actinopathies and myosinopathies . Brain Pathol , 19 : 516 – 522 .
- Hegreberg , GA and Reed , SM . 1990 . Skeletal muscle changes associated with equine myotonic dystrophy . Acta Neuropathol , 80 : 426 – 431 .
- Jamison , JM , Baird , JD , Smith-Maxie , LL and Hulland , TJ . 1987 . A congenital form of myotonia with dystrophic changes in a Quarterhorse . Equine Vet J , 19 : 353 – 358 .
- Jungbluth , H , Wallgren-Pettersson , C and Laporte , J . 2008 . Centronuclear (myotubular) myopathy . Orphanet J Rare Dis , 3 : 26
- Liquori , CL , Ricker , K Moseley , ML . 2001 . Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9 . Science , 293 : 864 – 867 .
- Lukas , Z , Falk , M , Feit , J , Soucek , O , Falkova , I , Stefancikova , L , Janousova , E , Fajkusova , L , Zaoralkova , J and Hrabalkova , R . 2012 . Sequestration of MBNL1 in tissue of patients with myotrophic dystrophy type 2 . Neuromuscul Disord , 22 : 604 – 616 .
- Lukas , Z , Kroupova , I , Bednarik , J , Falk , M , Fajkusova , L , Sedlackova , J , Valaskova , I and Vohanka , S . 2007 . Muscular biopsy in myotonic dystrophy in the era of molecular genetics . Ceska a Slovenska Neurologie a Neurochirurgie , 70 : 395 – 401 .
- Machuca-Tzili , L , Brook , D and Hilton-Jones , D . 2005 . Clinical and molecular aspects of the myotonic dystrophies: a review . Muscle Nerve , 32 : 1 – 18 .
- Montagna , P , Liguori , R , Monari , L , Strong , PN , Riva , R , Di Stassi , V , Gandini , G and Cipone , M . 2001 . Equine muscular dystrophy with myotonia . Clin Neurophysiol , 112 : 294 – 299 .
- Oldfords , A . 2007 . Hereditary myosin myopathies . Neuromuscul Disord , 17 : 355 – 367 .
- Reed , SM , Hegreberg , GA , Bayly , WM , Brown , CM , Paradis , MR and Clemmons , RM . 1988 . Progressive myotonia in foals resembling human dystrophia myotonica . Muscle Nerve , 11 : 291 – 296 .
- Romero , NB . 2010 . Centronuclear myopathies: a widening concept . Neuromuscul Disord , 20 : 223 – 228 .
- Rudolph , JA , Spier , SJ , Byrns , G , Rojas , CV , Bernoco , D and Hoffman , EP . 1992 . A sodium channel Phe to Leu mutation in periodic paralysis in Quarter horses: a common defect disseminated by selective breeding of popular sire . Nature Genetics , 2 : 144 – 147 .
- Sarli , G , Della Salda , L and Marcato , PS . 1994 . Dystrophy-like myopathy in a foal . Vet Rec , 135 : 156 – 160 .
- Selcen , D . 2011 . Myofibrillar myopathies . Neuromuscul Disord , 3 : 161 – 171 .
- Selcen , D , Ohno , K and Engel , AG . 2004 . Myofibrillar myopathy: clinical, morphological and genetic studies in 63 patients . Brain , 127 : 439 – 451 .
- Shirakawa , T , Ide , M , Taniyama , H , Tobiwatari , K , Senba , H , Oishi , H , Matsui , H and Ono , T . 1989 . Muscular dystrophy-like disease in a Thoroughbred foal . J Comp Path , 100 : 287 – 294 .
- Tominaga , K , Hayashi , YK , Goto , K , Minami , N , Nogochi , S , Nonaka , I , Miki , T and Nishino , I . 2010 . Congenital myotonic dystrophy can show congenital fiber type disproportion pathology . Acta Neuropathol , 119 : 481 – 486 .
- Vattemi , G , Tomelleri , G , Filosto , M , Savio , C , Rizzuto , N and Tonin , P . 2005 . Expression of late myogenic differentiation markers in sarcoplasmatic masses of patients with myotonic dystrophy . Neuropathol Appl Neurobiol , 31 : 45 – 52 .
- Wijnberg , ID , Owczarek-Lipska , M , Sacchetto , R , Mascarello , F , Pascoli , F , Grunberg , W , van der Kolk , JH and Drogemuller , C . 2012 . A missense mutation in the skeletal muscle chloride channel 1 (CLCN1) as candidate causal mutation for congenital myotonia in a New Forest pony . Neuromuscul Disord , 22 : 361 – 367 .