Abstract
Coxiella burnetii is the causative agent of the zoonotic disease Q fever. Since its first recognition as a disease in the 1930s, the knowledge about the agent and the disease itself has increased. This review summarizes the current knowledge on C. burnetii and Q fever, its pathogenesis, diagnosis and control. C. burnetii is a bacterium which naturally replicates inside human or animal host cells. The clinical presentation of Q fever varies per host species. C. burnetii infection in animals is mainly asymptomatic except for pregnant ruminants in which abortions and stillbirth can occur. In humans, the disease is also mainly asymptomatic, but clinical presentations include acute and chronic Q fever and the post-Q fever fatigue syndrome. Knowledge of the pathogenesis of Q fever in animals and excretion of C. burnetii in infected animals is crucial in understanding the transmission routes and risks of human infection. Our studies indicated that infected pregnant animals only excrete C. burnetii during and after parturition, independent of abortion, and that C. burnetii phase specific serology can be a useful tool in the early detection of infection. Domestic ruminants are the main reservoir for human Q fever, which has a major public health impact when outbreaks occur. In outbreaks, epidemiological source identification can only be refined by genotypic analysis of the strains involved. To control outbreaks and Q fever in domestic ruminants, vaccination with a phase 1 vaccine is effective. Future challenges are to identify factors for virulence, host susceptibility and protection.
1. History of the Q fever agent
The causative agent of Q fever was discovered simultaneously in Montana, USA, and in Queensland, Australia, in the 1930s. A laboratory infection connected the two discoveries. The first isolation of the agent causing Q fever was coincidental to research on Rocky Mountain Spotted Fever at the Rocky Mountain Laboratory in Montana, USA. In an experiment concerning 200 Dermacentor andersoni ticks collected at the Nine Mile Creek, Gordon Davis isolated an infectious agent with properties not identified before (Davis & Cox Citation1938). The agent could pass filters, was infectious for guinea pigs, Gram-negative, and had an extracellular and intracellular pleomorphic, rickettsia-like appearance (Cox Citation1938). Rolla Dyer, Director of the National Institutes of Health, USA, wanted to convince himself of the new agent described by Herald Cox and worked together with him in the laboratory. He appeared to contract the first laboratory infection with Q fever (Dyer Citation1938; McDade Citation1990). Dyer also suggested a link between the new agent and the ‘Q’ virus from Australia (Dyer Citation1938).
In Australia, Edward Derrick was assigned to investigate outbreaks of an undiagnosed febrile illness among abattoir workers in Brisbane, Queensland, Australia. These outbreaks had been occurring periodically since 1933. He first decided to determine the characteristics of the illness (Derrick Citation1983). As the illness was not previously described, it was named ‘Q’ fever ‘until fuller knowledge should allow a better name’ (Derrick Citation1983). Referring to the name, first Queensland rickettsial fever was proposed, but was regarded as derogatory to that sovereign state. ‘X-disease’ had already been appropriated in Australia, so Derrick decided that Q (for query) fever was the appropriate name (according to Macfarlane Burnet in McDade Citation1990). In order to reveal the cause of the disease, several experiments were done, but Derrick failed to detect bacteria. This led him to the (wrong) conclusion that the etiologic agent was a virus. Further studies on the virus of ‘Q’ fever were published by Macfarlane Burnet and Mavis Freeman, also indicating the rickettsia-like properties of the virus (Burnet & Freeman Citation1983).
The rickettsia-like properties of the agent resulted in the initial designation of the Q fever agent as Rickettsia diaporica (diaporica is derived from the Greek word for having the property or ability to pass through [a filter]) by the American group and as Rickettsia burnetii (after Burnet) by the Australian group (McDade Citation1990). In 1948, Philip Citation(1948) proposed a reclassification into Coxiella burneti. In this name, both Harold Cox and Frank Burnet are honoured for their contribution to the identification of the Q fever agent. C. burnetii has been phylogenetically reclassified from the order of Rickettsiales to Legionellales, based on the sequence of its 16S rRNA. In the Approved List of Bacterial Names, the Q fever agent is now listed as Coxiella burnetii (http://www.ncbi.nlm.nih.gov/books/NBK820/, access date 24 July 2012).
C. burnetii belongs to the family Coxiellaceae, together with the genus Rickettsiella. Typical for this family is the intracellular intravacuolar lifestyle in cells of invertebrate or vertebrate hosts. Closest relatives are Legionella and Francisella species (; Drancourt & Raoult Citation2005). At this time the phylogeny of the Q fever agent can be summarized as belonging to the kingdom of Bacteria, phylum of Proteobacteria, class of the Gammaproteobacteria, order of the Legionellales, family of the Coxiellaceae, with the genus Coxiella and the only species burnetii.
Figure 1. Phylogenetic relations within the phylum proteobacteria. A dendrogram based on 16s rRNA sequence comparison of C. burnetii with its closest member of the proteobacteria phylum (reprint of a part of Figure BXII.y.99 from Drancourt and Raoult Citation[2005], permission requested).
![Figure 1. Phylogenetic relations within the phylum proteobacteria. A dendrogram based on 16s rRNA sequence comparison of C. burnetii with its closest member of the proteobacteria phylum (reprint of a part of Figure BXII.y.99 from Drancourt and Raoult Citation[2005], permission requested).](/cms/asset/dea2b470-1acf-469c-9eff-9f84071afb26/tveq_a_843809_f0001_b.gif)
2. Bacteriology of C. burnetii
2.1. Phenotypic properties
C. burnetii is an obligate intracellular Gram-negative bacterium. The pleomorphic rods have a diameter of approximately 0.2–0.4 μm and are 0.4–1.0 μm in length (Drancourt & Raoult Citation2005). In the developmental cycle of C. burnetii two distinct variants have been identified: a large cell variant (LCV) and a small cell variant (SCV) (McCaul & Williams Citation1981). The LCV is the metabolically active and replicative entity of the bacterium. During the exponential growth phase generation times were calculated at approximately 11 hours (Coleman et al. Citation2004). The LCV transforms into the SCV, which is the spore-like form of C. burnetii. In this form, the bacterium is highly resistant to environmental stress, such as high temperatures, UV radiation and osmotic pressure. The resistance allows C. burnetii to survive in the environment while keeping its infectivity (McCaul & Williams Citation1981). The SCV can infect host cells, closing the developmental cycle.
Lipopolysaccharide (LPS) is demonstrated in both the C. burnetii LCV and the C. burnetii SCV, although presence of LPS is mainly associated with the SCV (Coleman et al. Citation2007). Like several other Gram-negative species, C. burnetii can display two different LPS phenotypes. The phase 1 phenotype expresses full-length LPS which corresponds to the smooth LPS of other Gram-negative bacteria (e.g. Brucella spp. and Enterobacteriaceae), while the phase 2 phenotype carries LPS that resembles the rough LPS of other Gram-negative bacteria. Phase 2 LPS lacks the O-antigenic region (Toman et al. Citation2009). Phase 1 bacteria are highly virulent and able to replicate in immunocompetent hosts. This is contrary to phase 2 bacteria which are avirulent and unable to replicate in immunocompetent animals (Moos & Hackstadt Citation1987; Andoh et al. Citation2007). During serial passage in cell culture, phase 1 C. burnetii can convert into phase 2 (Hotta et al. Citation2002). Both LPS phenotypes can be distinguished via phase-specific antibodies. Phase 1 antibodies are directed against the full-length LPS of phase 1, whereas phase 2 antibodies are directed against common surface proteins (Marrie & Raoult Citation1997). These surface proteins are also present on the surface of phase 1 C. burnetii, but seem to be shielded by the long phase 1 LPS. This may prevent binding of phase 2 antibodies to surface proteins of intact phase 1 bacteria (Hackstadt Citation1988). Unique for C. burnetii phase 1 LPS is the presence in the O-chain of galactosaminuronyl-α-glucosamide disaccharide, virenose (vir) and dihydrohydroxystreptose (strep) (Toman et al. Citation2009). Genes have been identified that might be involved in the synthesis of vir and strep. Phase variation seems to be associated with the loss of chromosomal DNA involving a large group of LPS biosynthetic genes, arranged in an apparent O-antigen cluster (Hoover et al. Citation2002). This implies a non-reversible process, but it is not known if chromosomal deletions are the only mechanism of phase variation in C. burnetii. Although LPS is an important virulence factor, the endotoxic activity of C. burnetii phase 1 and phase 2 LPS is 100–1000 times less compared to the LPS of Enterobacteriaceae (Amano et al. Citation1987).
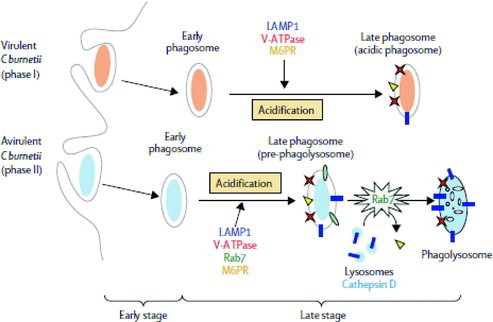
C. burnetii phase 1 LPS plays a role in both host cell internalization and survival in the phagosome. In phagocytosis of phase 1 bacteria, αvβ3-integrin and integrin-associated protein (IAP), as well as Toll-like receptor 4 (TLR4), are involved. In avirulent phase 2 bacteria, however, attachment is mediated by both αvβ3-integrin and IAP and complement receptor 3, but not by TLR4 (Capo et al. Citation1999; Honstettre et al. Citation2004; Raoult et al. Citation2005). The early phagosomes containing phase 1 bacteria acquire markers of late endosomes and lysosomes, resulting in acidification of the phagosome, but the final maturation step is inhibited by the absence of Rab7. In this acidic vacuole, the phase 1 C. burnetii escape intracellular killing and are able to replicate. Early phagosomes containing phase 2 bacteria proceed to the formation of early and late phagosomes. In the presence of Rab7, late phagosomes maturate and fuse with lysosomes forming acidic phagolysosomes. This results in effective killing of the phase 2 bacteria (Ghigo et al. Citation2002; Raoult et al. Citation2005). Thus, phase 1 C. burnetii bacteria are internalized and survive intracellular killing, whereas phase 2 bacteria are efficiently phagocytized and then killed (; Raoult et al. Citation2005).
Figure 2. C. burnetii trafficking in macrophages. Virulent phase 1 C burnetii phagocytosis leads to the formation of early phagosomes that acquire markers of late endosomes and lysosomes (LAMP1, M6PR and V-ATPase), resulting in the acidification of the phagosome. The final phagosome exhibits the features of late phagosomes, which do not acquire cathepsin D. Avirulent phase 2 C. burnetii phagocytosis leads to the formation of early and late phagosomes. Rab7 is involved in the maturation of late phagosomes, which acquire cathepsin D, forming acidic phagolysosomes (reprinted from The Lancet Infectious Diseases (Raoult et al. Citation2005). Copyright (2005), with permission from Elsevier Limited.
2.2. The C. burnetii genome
The first complete genome sequence of C. burnetii was published in 2003. Analysis of the original strain isolated from ticks by Davies and Cox in Citation1938 (called Nine Mile) revealed a circular genome of 1995,275 base pairs. Twenty-nine insertion sequence (IS) elements were identified: 21 copies of a unique IS110-related isotype, IS1111, five IS30 and three ISAs1 family elements. These IS elements are dispersed around the chromosome without apparent localized clustering (Seshadri et al. Citation2003). Analysis of multiple strains showed that the number of IS1111 elements is highly variable between strains. In an analysis of 75 strains, the copy number varied between 7 and 110 (Klee et al. Citation2006). Four extra-chromosomal plasmid types have been identified: QpH1 (36 kilo base (kb)), QpRS (39 kb), QpDG (42 kb) and QpDV (33 kb). In one strain, a plasmid seems absent (strain Scurry isolated from a human patient suffering from hepatitis). However, a DNA homologue of a segment of the QpH1 plasmid has been identified on its chromosome (Hendrix et al. Citation1991; Willems et al. Citation1997; Raoult et al. Citation2005). Initially, C. burnetii strains with specific plasmid types were associated with the development of acute or chronic Q fever in humans, but this association was found to be inconsistent (Thiele & Willems Citation1994).
The genetic heterogeneity of C. burnetii can be assessed with a number of molecular techniques. Different genotyping techniques have been described mainly based on the identification of differences between selected loci on the genome. An overview of published genotyping techniques for C. burnetii is given in . Accurate identification of the agent is important to differentiate between strains and to identify epidemiological markers. These markers are at the basis of the molecular epidemiology that enables the identification of sources of Q fever outbreaks (Arricau-Bouvery et al. Citation2006). Eventually, markers may be discovered in the C. burnetii genome that identifies C. burnetii strains posing an increased risk for infection. However, it is not known if virulence-encoding genes are located on or associated with the loci used in the current typing methods. So, to date, it is not possible to classify the virulence of strains solely based on the available genotyping methods.
Table 1. Overview of published genotyping techniques for C. burnetii and year of first publication of the technique for C. burnetii.
An important characteristic for typing systems is the discriminatory power, i.e. the ability to distinguish between unrelated strains. This is determined by the number of types defined by the test method and the relative frequencies of the types. A single numerical index of discrimination is suggested by Hunter and Gaston Citation(1988). The Hunter–Gaston Diversity Index (HGDI) is based on the probability that two unrelated strains sampled from a test population will be placed into different typing groups. By comparing the HGDI of a typing system, the discriminatory power can be compared. It is important to note that the calculated HGDI depends on the panel of strains (i.e. relatedness of the strains), so for an unbiased comparison of typing methods, preferably the same panel should be used. Despite its importance for the quality of typing systems, the discriminatory power is not assessed for most of the typing systems available for C. burnetii. For RFLP-typing, an HGDI of 0.86 is calculated (Jager et al. Citation1998). For both the MLVA typing panels 1 and 2, an HGDI of 0.92 is calculated and for the combined panels an HGDI of 0.99 is calculated (Arricau-Bouvery et al. Citation2006; Roest et al. Citation2011a). It is suggested that an HGDI of >0.90 is desirable to interpret typing results with confidence (Hunter & Gaston Citation1988), indicating the MLVA typing system as a useful typing tool for C. burnetii. The published MLVA typing method for C. burnetii is assumed to be more discriminatory than multispacer sequence typing (Svraka et al. Citation2006; Chmielewski et al. Citation2009).
2.3. Safety issues
C. burnetii is considered a biological-threat agent by its very low infectious dose and high transmissibility. It is listed as a group B bioterrorism agent by the Centres of Disease Control and Prevention in the USA (Oyston & Davies Citation2011) (http://emergency.cdc.gov/agent/agentlist.asp, access date 27 July 2012). An infection dose as low as one bacterium is reported for both guinea pigs and humans (Tigertt et al. Citation1961; Jones et al. Citation2006). For this reason, C. burnetii is considered a containment level 3 organism by the EU (directive 2000/54/EC of the European parliament and of the council). Working with containment level 3 organisms requires dedicated biosafety level 3 laboratories and animal facilities, as well as experienced and trained personnel. Emergency procedures and medical monitoring of personnel should be implemented to guarantee the safety of the people working with agents such as C. burnetii. Although a human Q fever vaccine is available in Australia, its use to prevent Q fever in laboratory employees is still under debate.
3. Isolation and cultivation of C. burnetii
3.1. Techniques
Successful isolation and cultivation of C. burnetii in laboratory animals, embryonated eggs and cell culture were already achieved in the first studies on the Q fever agent (Cox Citation1938; Davis & Cox Citation1938; Dyer Citation1938; Parker & Davis Citation1938; Burnet & Freeman Citation1983). Guinea pigs were the first laboratory animals to be used in Q fever research. These animals develop hyperthermia of over 40°C at 5–12 days post-inoculation and C. burnetii can be isolated from the spleen, indicating systemic infection (Lang Citation1990; Maurin & Raoult Citation1999). In mice, the susceptibility to infection with phase 1 C. burnetii varies between mouse strains. A/J (highly susceptible) mice are highly sensitive to the agent, resulting in a 100% morbidity and a 70% mortality, while resistant mice strains show neither morbidity nor mortality. Yet, both sensitive and resistant mice strains show a generalized C. burnetii infection, and antibody production is similar (Scott et al. Citation1987). For the isolation of strains, the resistant mice strains can be used for the isolation of C. burnetii from tissues with low Coxiella load and/or when tissues were probably contaminated with other bacteria. Tissue suspensions were intraperitoneally inoculated and after 7–21 days the mice were euthanized to isolate C. burnetii from the spleen. To minimize the use of mice, suspected samples should first be screened with a C. burnetii specific polymerase chain reaction (PCR) to assess the C. burnetii load prior to inoculation of the mice. Thus, mice were used in specific cases both for propagation of C. burnetii and as a filter for contaminants.
In embryonated eggs, C. burnetii grows almost exclusively in the yolk sac endoderm cells. This propagation technique involves the inoculation of a tissue suspension containing C. burnetii into 6–7 day old embryonated chicken eggs via the yolk sac. The yolk sac is harvested after 10–15 days (Lang Citation1990) (OIE Manual of Diagnostic Tests and Vaccines for Terrestrial Animals 2012, http://www.oie.int/fileadmin/Home/eng/Health_standards/tahm/2.01.12_Q-FEVER.pdf, access date 15 August 2013). However, this culture system is time-consuming and extensive experience is needed to harvest the yolk sac. In addition, culture results are hard to monitor and the results are comparable to cell culture isolation techniques.
The cell culture system is currently the most widely used in vitro system to isolate and cultivate C. burnetii. A number of cell lines can be used for in vitro cultures (Voth & Heinzen Citation2007). In the isolation of C. burnetii from infected tissues, filters with pore sizes of 1.2 μm and 0.45 μm were used. Although this final filter step decreases the number of Coxiella bacteria by a factor of 10 (Janse et al. Citation2011), it is necessary for some tissue material to avoid contamination of the monolayer with other bacteria. A culture medium without antibiotics is used, to avoid negative influence on the growth of the Coxiella bacteria. This method has been successfully applied to isolate Dutch outbreak strains of C. burnetii (; Roest et al. Citation2010; Roest et al. Citation2012).
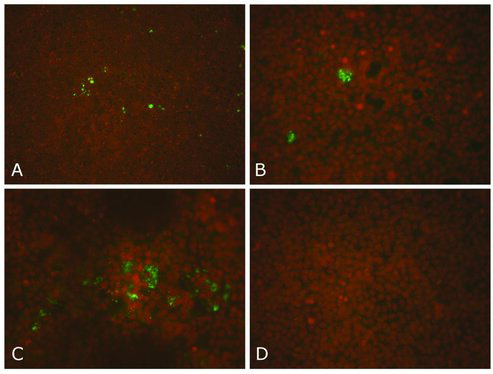
Figure 3. A–D: Dutch C. burnetii strains on the Buffalo Green Monkey (BGM) cell culture system. Results of the immunofluorescence staining of a BGM cell culture using a C. burnetii specific monoclonal antibody (MAB313-oregon green, Squarix). The presence of C. burnetii is indicated by the green dots. The nuclei of the BGM cells are stained brownish with propidium iodide (Roest et al. Citation2010). A: The first Dutch C. burnetii strain isolated from the placenta of a goat (strain X09003262-001, 100× magnification). B: close-up of (A) (400× magnification). C: C. burnetii strain isolated from the placenta of a sheep (400× magnification). D: non-infected BGM cells (negative control) (400× magnification).
A recent major improvement in the methods of isolation and cultivation of C. burnetii is the introduction of a host cell free medium based on the composition of the acidic phagosome environment in which C. burnetii replicates (Omsland et al. Citation2009; Omsland Citation2012). The major advantage of the use of a chemically defined growth environment is the absence of host cell genetic material, which is particularly helpful in the genetic analysis of C. burnetii.
3.2. Quality control in C. burnetii cultivation
A visual check on isolate purity and contamination during culturing is hampered by the intracellular nature of C. burnetii. Bacterial contamination of cell cultures is readily visible by the change in clarity of the culture medium, especially when a culture medium without antibiotics is used. However, several intracellular bacteria, such as Chlamydia spp., Simkania negevensis and Mycoplasma spp., are also able to contaminate cell cultures. To control these kind of contamination, it is advisable to implement regular checks for these contaminants in quality control (QC) protocols. To detect possible cross-contamination between cultures with different strains, regular checks on the identity of the strains by genotyping techniques should also be part of the QC.
4. Q fever in animals
4.1. Host range
C. burnetii can infect a wide range of animal species. As mentioned above, the bacterium was initially isolated from the tick Dermacentor andersoni (Davis & Cox Citation1938). Since these first reports, C. burnetii has been detected in over 40 tick species. In the Netherlands, prevalence of C. burnetii in ticks is low (0.2%, Sprong et al. Citation2012). Several bird species can also become infected with C. burnetii, as experimentally shown (Babudieri & Moscovici Citation1952; Schmatz Citation1977a, Citation1977b; Sethi et al. Citation1978). Natural infections have been reported in domestic and wild birds (To et al. Citation1998; Astobiza et al. Citation2011). In terrestrial as well as in marine wildlife, the presence of C. burnetii has been confirmed in roe deer, wild boars, rodents, European hares, Pacific harbour seals, a Steller sea lion, Northern fur seals and harbour porpoises (Lapointe et al. Citation1999; Kersh et al. Citation2010; Duncan et al. Citation2012; Kersh et al. Citation2012; Thompson et al. Citation2012). In Dutch wildlife, C. burnetii has been detected in roe deer and rats (Reusken et al. Citation2011; Rijks et al. Citation2011). C. burnetii can also be present in cats and dogs. Seroprevalences vary between 19% and 42% for cats and up to 22% for dogs (Marrie et al. Citation1988a, Citation1988b; Higgins & Marrie Citation1990; Boni et al. Citation1998; Komiya et al. Citation2003a, Citation2003b). In the Netherlands, seroprevalences in cats and dogs of 10% and 13%, respectively, have been measured (Roest et al. Citation2011c).
In domestic ruminants, C. burnetii infections are widespread. Seroprevalence levels are estimated up to 82% in cattle. In sheep and goats, average seroprevalences are slightly lower compared to cattle, with values of up to 73% (Guatteo et al. Citation2011). Prevalence of C. burnetii on cattle herd level as measured from bulk tank milk samples ranged between 32% and 94% (Kim et al. Citation2005; Angen et al. Citation2011; Astobiza et al. Citation2012). The seroprevalence and agent prevalence in Dutch cattle are comparable to what is reported in the literature. Seroprevalences in sheep and goats in the Netherlands are estimated at 2.4% and 7.8%, respectively (van den Brom et al. Citation2012a). This is in the lower range of what is internationally known. In 19% of the Dutch bulk tank milk samples from dairy sheep and in 33% of the bulk tank milk samples from dairy goats, C. burnetii DNA was detected (van den Brom et al. Citation2012b).
4.2. Pathogenesis of Q fever and immune response in animals
Little is known about the pathogenesis of Q fever in domestic animals. Under laboratory conditions, C. burnetii inoculation of both guinea pigs and mice results in a systemic infection, including pneumonia, hepatitis and splenomegaly (Scott et al. Citation1987; Russell-Lodrigue et al. Citation2006; Russell-Lodrigue et al. Citation2009). The severity of pathological changes depends on the strain. Splenomegaly is thought to be an indicator for the virulence of C. burnetii strains in guinea pigs and mice (Russell-Lodrigue et al. Citation2009). Also the inoculation route seems to influence pathogenesis. In mice, intranasal inoculation is mainly associated with pneumonia, whereas intraperitoneal inoculation is mainly associated with hepato-splenomegaly (Marrie et al. Citation1996). In inoculated pregnant mice, C. burnetii is abundantly present in both the foetal and maternal parts of the placenta (Baumgartner & Bachmann Citation1992).
In goats this is different (Sanchez et al. Citation2006; Roest et al. Citation2012). In pregnant goats, the trophoblasts of the allantochorion are target cells of C. burnetii in which multiplication occurs. C. burnetii antigens are barely detected in adjacent maternal parts of the placenta. In other maternal organs, C. burnetii DNA, but no viable bacteria, can be found at some time points during pregnancy (Sanchez et al. Citation2006). Recent research showed that the infection route does not influence the pathogenesis and that C. burnetii is not excreted during pregnancy (Roest et al. Citation2012). This hampers the detection of infected pregnant goats.
In mice, both humoral and cellular immune responses are important in limiting the infection. Macrophages and other mononuclear cells are believed to be the major target cells during C. burnetii infection (Shannon & Heinzen 2009). T-cells are suggested to be critical for clearance of C. burnetii after infection. B-cells are important for the prevention of tissue damage (Andoh et al. Citation2007). Antibodies can be detected as early as 14 days post-inoculation for anti-C. burnetii phase 2 antibodies and 21 days in the case of anti-C. burnetii phase 1 antibodies (Kishimoto et al. Citation1978). This is comparable to what was observed in goats. Upon infection, goats generate a phase 1 and phase 2 specific IgM and IgG response. After 2 weeks post-infection, a strong phase 2 specific IgM and IgG antibody response can be detected while a less pronounced IgM anti-phase 1 response is present as well. IgG anti-phase 1 antibodies start to rise at 6 weeks post-infection (Roest et al. Citation2013). This information can help in the diagnosis of Q fever, understanding herd dynamics and will be helpful in improving vaccines.
4.3. Clinical presentation
The most important clinical presentations of Q fever in animals that are relevant to its zoonotic properties are abortion and stillbirth. Field observations clearly demonstrate C. burnetii as a cause of abortion and stillbirth in goats, sheep, cattle and cats (Marrie et al. Citation1988a, Citation1988b; Lang Citation1990; van Moll et al. Citation1993; Berri et al. Citation2001; Hatchette et al. Citation2003; Rousset et al. Citation2009a, Citation2009b; Guatteo et al. Citation2011). Abortion occurs most frequently at the end of gestation, without preceding clinical symptoms (Arricau-Bouvery et al. Citation2005). In goats, abortion, stillbirth but also the birth of strong and lively kids can occur after Q fever infection of pregnant animals (Arricau-Bouvery et al. Citation2005; Roest et al. Citation2012). Experimental infections in pregnant sheep did not result in any abortions (Welsh et al. Citation1958; Martinov et al. Citation1989), while in cattle one experimental infection of a pregnant cow resulted in abortion (Lang Citation1990). The cause of these differences in pregnancy outcome after infection is unknown.
In non-pregnant animals, C. burnetii infection is virtually asymptomatic. In cattle, C. burnetii infection is associated with metritis and reproduction problems (Lang Citation1990; To et al. Citation1998). In dairy goat herds where Q fever abortions occurred, metritis can be present. Weak kids were reported with low body weight and high mortality. Rearing of apparently healthy kids can be complicated by respiratory and digestive tract disorders (Wouda & Dercksen Citation2007).
4.4. Excretion routes
Knowledge of the excretion of C. burnetii from infected animals is crucial in understanding the transmission routes and risks of human infection. Abortions in C. burnetii infected domestic ruminants are accompanied by massive excretion of the bacteria and spread into the environment. This is the most important excretion route of C. burnetii, as up to 109 organisms per gram placenta tissue are excreted (Arricau Bouvery et al. Citation2003). However, recent experiments indicate that comparable numbers of Coxiellas are also excreted during the birth of lively kids (Roest et al. Citation2012). C. burnetii has also been detected in faeces, vaginal mucus and milk of infected domestic ruminants (Berrie et al. Citation2001; Arricau-Bouvery et al. Citation2003; Guatteo et al. Citation2006; Rousset et al. Citation2009a, Citation2009b). In goat herds, both in aborting and non-aborting goats, C. burnetii DNA has been detected in faeces, vaginal mucus and/or milk (Rousset et al. Citation2009a, Citation2009b). In cattle, also variable excretion via faeces, vaginal mucus and milk has been reported, sometimes independent of an abortion history. Sixty-five per cent of cows seem to shed C. burnetii by only one route, evenly distributed over the three routes. Cows that excrete C. burnetii by all three routes seem scarce (Guatteo et al. Citation2006). Comparison of the three excretion routes in cattle, goats and sheep shows that milk shedding is more frequent in cattle and goats. Ewes shed more and longer in vaginal mucus than goats (Arricau-Bouvery et al. Citation2005). Both sheep and goats can shed C. burnetii in subsequent pregnancies (Berrie et al. Citation2002; Berrie et al. Citation2007). Based on these results, it is suggested that testing of animals based on only one biological sample may lead to false negative results. Thus, while excretion of high numbers of C. burnetii with birth products during parturition is evident, the importance of and the correlation between the excretion routes of C. burnetii via faeces, vaginal mucus and milk are much less well established. It is suggested that these inconsistencies are due to contamination of C. burnetii DNA from the environment (Roest et al. Citation2012).
4.5. Diagnostics
The occurrence of abortion is obviously not specific for Q fever. Evidence of a C. burnetii infection as a cause of the abortion requires laboratory confirmation. C. burnetii can be detected in infected placenta material via immunohistochemistry, using C. burnetii specific antibodies. When C. burnetii is the cause of abortion, high numbers of bacteria are usually observed in the trophoblasts of the allantochorion (Marrie et al. Citation1996; Wouda & Dercksen Citation2007; Roest et al. Citation2012). C. burnetii can also be detected with specific PCR tests. Specific and sensitive detection of C. burnetii can be achieved with primers and probes targeting the IS1111 element. This element has multiple copies in the genome of C. burnetii, resulting in an increased sensitivity compared to primers and probes that target single copy genes (Klee et al. Citation2006). A PCR can be used to detect C. burnetii DNA in a wide range of samples, including placenta tissues, faeces, vaginal mucus and milk (OIE Manual of Diagnostic Tests and Vaccines for Terrestrial Animals 2012, http://www.oie.int/fileadmin/Home/eng/Health_standards/tahm/2.01.12_Q-FEVER.pdf, access date 15 August 2013). Apart from histopathology and PCR, several indirect diagnostic methods of Q fever detection are available. C. burnetii specific antibodies in serum can be measured using the complement fixation test (CFT), enzyme-linked immunosorbent assay (ELISA) and immunofluorescence assay test. The CFT is most widely used in veterinary laboratories, although commercially available ELISAs to detect C. burnetii antibodies in ruminants are increasingly used. Compared to ELISA, the CFT lacks sensitivity to detect C. burnetii specific antibodies in ruminant serum (Rousset et al. Citation2007; AHAW Citation2010; Horigan et al. Citation2011). Several reports indicate that C. burnetii can be shed by cattle, sheep and goats without a detectable serological response. Although this could be due to the delay in the serological response after a primary infection, it may indicate that serology is not a useful tool to diagnose abortions at the individual level (Berri et al. Citation2001; Guatteo et al. Citation2007; Rousset et al. Citation2009a, Citation2009b). This lack of antibody response is not well explained yet, but can be caused by a false detection of Coxiella as discussed above.
Current protocols to diagnose Q fever on herd level in cattle and small ruminant herds recommend a combination of both direct and indirect techniques. It is recommended that samples from abortion material and/or from vaginal mucus of two to six animals are investigated by a PCR within 8 days after abortion. In addition, serum samples from at least 6–10 animals should be investigated, especially when PCR results are negative. These animals should preferably have delivered at least 15 days prior to sampling (Sidi-Boumedine et al. Citation2010). A PCR targeting the IS1111 genetic element is the most sensitive technique to detect C. burnetii, whereas ELISA is the most sensitive technique to detect C. burnetii specific antibodies.
4.6. Disease control
To control Q fever in domestic ruminants, two methods are available: antibiotic treatment and vaccination. In the case of acute Q fever in humans, doxycycline is the antibiotic of choice. For chronic Q fever, long-term treatment with doxycycline and hydrochloroquine is recommended. Although thorough evaluations of these therapies are lacking, they are considered to be effective (van der Hoek Citation2012). The effect of antibiotics to treat Q fever in animals is poorly studied. A few studies suggest a reduction in the abortion rate and in the number of bacteria excreted at parturition (Berri et al. Citation2005a, Citation2005b; Muskens et al. Citation2007). However, this effect could not be observed in small ruminants in the Netherlands, treated with oxytetracycline (Wouda & Dercksen Citation2007). In dairy cattle, a beneficial effect of antibiotic treatment complementary to vaccination with a phase 1 C. burnetii inactivated vaccine was observed (Taurel et al. Citation2012). Because the effect of antibiotic treatment is not clearly evidence based and prudent use of antibiotics is needed to avoid microbial resistance, it is advised that antibiotics should not be used for treatment of Q fever in animals.
Vaccination with a phase 1 C. burnetii inactivated vaccine is reported to be effective in preventing abortion, as well as in reducing bacterial shedding in goats and cattle (Hogerwerf et al. 2011; Arricau-Bouvery et al. Citation2005; Guatteo et al. Citation2011). Vaccination in goats and cattle is most effective in primiparus animals and reduces both the prevalence of shedders and the bacterial load (Guatteo et al. Citation2008; Rousset et al. Citation2009a, Citation2009b; Hogerwerf et al. Citation2011). However, vaccination might not be effective in goats or cattle that are already infected, nor in pregnant cattle (Guatteo et al. Citation2008; Rousset et al. Citation2009a, Citation2009b). Other drawbacks of the vaccine are the complicated production process, the need for dedicated BSL3 facilities for production and the impossibility to differentiate between infected and vaccinated animals. Contrary to vaccination with a phase 1 C. burnetii vaccine, the current phase 2 C. burnetii vaccine is not effective (Arricau-Bouvery et al. Citation2005). This result indicates that currently Q fever in domestic ruminants can best be prevented by vaccination with a phase 1 C. burnetii vaccine. A new generation of vaccines that are easier to produce (e.g. recombinant vaccines) may overcome the main drawbacks of the current vaccine, but these are not available yet.
5. Q fever in humans
5.1. Pathogenesis of Q fever and immune response
Humans usually acquire Q fever by inhalation of environmental C. burnetii. Alveolar macrophages and other mononuclear phagocytes are thought to be the primary target cells of the pathogen (Shannon & Heinzen 2009). In these cells, C. burnetii survives intracellular killing and is able to replicate. A bacteraemia will lead to systemic infection with involvement of the liver, spleen, lungs and bone marrow (Maurin & Raoult Citation1999). C. burnetii infection often leads to protective granulomatous lesions in these organs. The TLR 4 innate immune receptor also plays a role in the formation of these granulomas, as well as in the production of the interferon gamma (IFN-γ) and tumor necrosis factor by T-cells (Honstettre et al. Citation2004; Shannon & Heinzen Citation2009). It is suggested that IFN-γ mediates C. burnetii killing via phagosome maturation and phagosome alkalinization (Ghigo et al. Citation2002). Both anti-C. burnetii phase 2 IgM and IgG antibodies develop within 3 weeks post-infection, accompanied by low levels of anti-phase 1 IgM (Dupuis et al. Citation1985). Antibody-mediated immunity seems to be important, as infection of dendritic cells with antibody-opsonized bacteria resulted in the increased expression of maturation markers and inflammatory cytokines in mice (Shannon & Heinzen Citation2009). Thus, in humans both humoral and cellular immunity play a role in C. burnetii infection. In pregnant women, an adverse pregnancy outcome has been associated with Q fever (Maurin & Raoult Citation1999). However, no evidence for this has been found during the recent Dutch Q fever outbreak (van der Hoek et al. Citation2011a, Citation2011b).
5.2. Clinical presentation
Infection can manifest as three main clinical presentations: acute Q fever, chronic Q fever and the post-Q fever fatigue syndrome (QFS). Following exposure to C. burnetii almost 60% of the Q fever cases are asymptomatic. Among the 40% symptomatic acute Q fever patients, the majority will present a non-specific, self-limiting illness. More severe clinical symptoms include fever, headache, chills, atypical pneumonia and hepatitis (Derrick Citation1983; Maurin & Raoult Citation1999; Raoult et al. Citation2005). In the Netherlands, the recent acute Q fever outbreak showed a mortality rate of 1.2% within approximately 1 month after hospitalization of patients. All lethal cases suffered severe underlying medical conditions (Kampschreur et al. Citation2010). Acute Q fever is diagnosed in the laboratory following (i) a positive C. burnetii specific PCR, (ii) the presence of IgM phase 2 antibodies in serum accompanied by clinical symptoms, or (iii) a fourfold increase of the IgG phase 2 antibody titre. These laboratory findings are also the notification criteria in the Netherlands (Wegdam-Blans et al. Citation2010).
Chronic Q fever can develop from a primary infection in about 1%–5% of the patients and can become manifest years after the initial infection. Clinical symptoms include non-specific fatigue, fever, weight loss, night sweats and hepato-splenomegaly as well as endocarditis (Raoult et al. Citation2005; Wegdam-Blans et al. Citation2012). After the recent outbreak in the Netherlands, vascular complications appeared to be more common than endocarditis, but endocarditis may not have become apparent yet (van der Hoek Citation2012). Laboratory diagnosis of chronic Q fever includes the detection of the Q fever agent by a PCR or a high titre of C. burnetii phase 1 IgG (Wegdam-Blans et al. Citation2012).
QFS is another long-term presentation of Q fever. Contrary to chronic Q fever, C. burnetii cannot be detected in QSF patients. Furthermore, antibody levels against the bacteria are low or negligible. Symptoms of QFS include prolonged fatigue, arthralgia, myalgia, blurred vision and enlarged painful lymph nodes (Maurin & Raoult Citation1999). Acute Q fever with hospitalization was found to be a risk factor for QFS (Morroy et al. Citation2011). The cause of the development of chronic Q fever or QFS in certain individuals is still unknown.
6. Epidemiology of C. burnetii
In humans, Q fever is essentially an airborne infection resulting from the inhalation of contaminated aerosols (Benenson & Tigertt Citation1956; Tigertt et al. Citation1961). Aerosols can become contaminated with C. burnetii during abortion and parturition of infected pregnant ruminants (Welsh et al. Citation1958; Benson et al. Citation1963). Environmental contamination resulting in contaminated aerosols may also follow excretion of C. burnetii via faeces, vaginal mucus or possibly milk of animals. This contributes to the occupational hazard of Q fever. However, contaminated aerosols are able to travel up to 5 km and infect humans. Windy, warm and dry weather can facilitate this transport (Tissot-Dupont et al. Citation2004; van Steenbergen et al. Citation2007; Schimmer et al. Citation2010). Also other factors, such as vegetation and soil moisture, seem to be relevant in the dispersion of the bacteria (van der Hoek et al. Citation2011a, Citation2011b). So, direct contact with animals is not a prerequisite for acquiring Q fever, as several outbreaks demonstrated (Salmon et al. Citation1982; Tissot-Dupont et al. Citation1999; Tissot-Dupont et al. Citation2004; Gilsdorf et al. Citation2008). It is assumed that most animals also become infected via inhalation (Berri et al. Citation2005a, Citation2005b), although oral uptake in a heavily infected environment cannot be excluded.
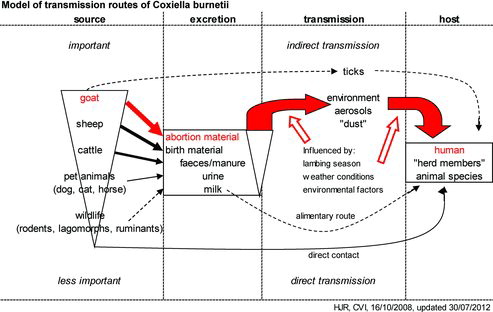
Domestic ruminants appear to be the main source of infection for human Q fever. As indicated earlier, parturition in infected pregnant goats, sheep and cattle results in massive excretion of C. burnetii into the environment. Q fever in cattle seems to result in fewer abortions, probably resulting in a lower risk. Companion animals should also be considered as a source for human Q fever, since several human outbreaks were related to parturient cats and dogs (Marrie et al. Citation1988a, Citation1988b; Pinsky et al. Citation1991; Buhariwalla et al. Citation1996; Komiya et al. Citation2003a, Citation2003b). The role of horses and wildlife as a source of Q fever for humans is not clear. In overview, the epidemiology of Q fever can be summarized in a transmission model as presented in (adapted from Roest et al. Citation2009).
Figure 4. Transmission model for Q fever. An overview of the possible transmission routes of C. burnetii from the animal reservoir to the human (and animal) hosts. The bold-facing of the arrows indicates the importance of the route and the dotted lines indicate possible contributions. The most probable transmission route of C. burnetii in the Dutch Q fever outbreak is indicated in red (adapted from Roest et al. Citation2009).
7. Q fever outbreaks
Q fever has a major public health impact when outbreaks occur. Outbreaks are reported frequently and worldwide, involving up to 415 laboratory-confirmed human cases per outbreak. Even higher numbers of human cases are reported, but the attribution to Q fever is unclear as cases are not always laboratory-confirmed (Arricau-Bouvery and Rodolakis Citation2005; AHAW Citation2010; van der Hoek Citation2012). Outbreaks are usually geographically localized and restricted to one episode. Sheep are identified as the source in the majority of the outbreaks, with goats as the ‘second best’. Therefore, the Dutch Q fever outbreak was extraordinary due to its duration (from 2007 until 2010), the number of people involved (up to 4000 human cases) and the source of infection (mainly dairy goats) (Roest et al. Citation2009; Roest et al. Citation2011b). Only a few outbreaks of Q fever have been related to infected cattle and cats (Arricau-Bouvery and Rodolakis Citation2005; AHAW Citation2010; van der Hoek Citation2012). Source identification, however, is mainly based on epidemiological examinations. In most outbreaks, confirmation of the identity of the Coxiella strain involved in both the host and the source, for example, by genotyping, is lacking. This is a major drawback in the identification of sources, as sources of Q fever are multiple and C. burnetii can be transmitted over larger distances. Thus, identification of the source and confirmation of the relation with the human disease is preferably done by genotyping of the involved C. burnetii strains.
References
- Amano K, Williams JC, Missler SR, Reinhold VN. 1987. Structure and biological relationships of Coxiella burnetii lipopolysaccharides. J Biol Chem. 262:4740–4747.
- Andoh M, Zhang G, Russell-Lodrigue KE, Shive HR, Weeks BR, Samuel JE. 2007. T cells are essential for bacterial clearance, and gamma interferon, tumor necrosis factor alpha, and B cells are crucial for disease development in Coxiella burnetii infection in mice. Infect Immunol. 75:3245–3255.
- Angen O, Stahl M, Agerholm JS, Christoffersen AB, Agger JF. 2011. Dynamics of relationship between the presence of Coxiella burnetii DNA, antibodies, and intrinsic variables in cow milk and bulk tank milk from Danish dairy cattle. J Dairy Sci. 94:5750–5759.
- Arricau-Bouvery N, Hauck Y, Bejaoui A, Frangoulidis D, Bodier CC, Souriau A, Meyer H, Neubauer H, Rodolakis A, Vergnaud G. 2006. Molecular characterization of Coxiella burnetii isolates by infrequent restriction site-PCR and MLVA typing. BMC Microbiol. 6:38.
- Arricau-Bouvery N, Rodolakis A. 2005. Is Q fever an emerging or re-emerging zoonosis? Vet Res. 36:327–349.
- Arricau-Bouvery N, Souriau A, Bodier C, Dufour P, Rousset E, Rodolakis A. 2005. Effect of vaccination with phase I and phase II Coxiella burnetii vaccines in pregnant goats. Vaccine. 23:4392–4402.
- Arricau-Bouvery N, Souriau A, Lechopier P, Rodolakis A. 2003. Experimental Coxiella burnetii infection in pregnant goats: excretion routes. Vet Res. 34:423–433.
- Astobiza I, Barral M, Ruiz-Fons F, Barandika JF, Gerrikagoitia X, Hurtado A, Garcia-Perez AL. 2011. Molecular investigation of the occurrence of Coxiella burnetii in wildlife and ticks in an endemic area. Vet Microbiol. 147:190–194.
- Astobiza I, Ruiz-Fons F, Pinero A, Barandika JF, Hurtado A, Garcia-Perez AL. 2012. Estimation of Coxiella burnetii prevalence in dairy cattle in intensive systems by serological and molecular analyses of bulk-tank milk samples. J Dairy Sci. 95:1632–1638.
- Babudieri B, Moscovici C. 1952. Experimental and natural infection of birds by Coxiella burnetii. Nature. 169:195–196.
- Baumgartner W, Bachmann S. 1992. Histological and immunocytochemical characterization of Coxiella burnetii-associated lesions in the murine uterus and placenta. Infect Immunol. 60:5232–5241.
- Benenson AS, Tigertt WD. 1956. Studies on Q fever in man. Trans Assoc Am Physicians. 69:98–104.
- Benson WW, Brock DW, Mather J. 1963. Serologic analysis of a penitentiary group using raw milk from a Q fever infected herd. Public Health Rep. 78:707–710.
- Berri M, Crochet D, Santiago S, Rodolakis A. 2005a. Spread of Coxiella burnetii infection in a flock of sheep after an episode of Q fever. Vet Rec. 157:737–740.
- Berri M, Rousset E, Champion JL, Russo P, Rodolakis A. 2007. Goats may experience reproductive failures and shed Coxiella burnetii at two successive parturitions after a Q fever infection. Res Vet Sci. 83:47–52.
- Berri M, Rousset E, Hechard C, Champion JL, Dufour P, Russo P, Rodolakis A. 2005b. Progression of Q fever and Coxiella burnetii shedding in milk after an outbreak of enzootic abortion in a goat herd. Vet Rec. 156:548–549.
- Berri M, Souriau A, Crosby M, Crochet D, Lechopier P, Rodolakis A. 2001. Relationships between the shedding of Coxiella burnetii, clinical signs and serological responses of 34 sheep. Vet Rec. 148:502–505.
- Berri M, Souriau A, Crosby M, Rodolakis A. 2002. Shedding of Coxiella burnetii in ewes in two pregnancies following an episode of Coxiella abortion in a sheep flock. Vet Microbiol. 85:55–60.
- Boni M, Davoust B, Tissot-Dupont H, Raoult D. 1998. Survey of seroprevalence of Q fever in dogs in the southeast of France, French Guyana, Martinique, Senegal and the Ivory Coast. Vet Microbiol. 64:1–5.
- Buhariwalla F, Cann B, Marrie TJ. 1996. A dog-related outbreak of Q fever. Clin Infect Dis. 23:753–755.
- Burnet FM, Freeman M. 1983. Experimental studies on the virus of ‘Q’ fever. Rev Infect Dis. 5:800–808.
- Capo C, Lindberg FP, Meconi S, Zaffran Y, Tardei G, Brown EJ, Raoult D, Mege JL. 1999. Subversion of monocyte functions by Coxiella burnetii: impairment of the cross-talk between alphavbeta3 integrin and CR3. J Immunol. 163:6078–6085.
- Chmielewski T, Sidi-Boumedine K, Duquesne V, Podsiadly E, Thiery R, Tylewska-Wierzbanowska S. 2009. Molecular epidemiology of Q fever in Poland. Pol J Microbiol. 58:9–13.
- Coleman SA, Fischer ER, Cockrell DC, Voth DE, Howe D, Mead DJ, Samuel JE, Heinzen RA. 2007. Proteome and antigen profiling of Coxiella burnetii developmental forms. Infect Immunol. 75:290–298.
- Coleman SA, Fischer ER, Howe D, Mead DJ, Heinzen RA. 2004. Temporal analysis of Coxiella burnetii morphological differentiation. J Bacteriol. 186:7344–7352.
- Cox HR. 1938. A filter-passing infectious agent isolated from ticks III. Description of organism and cultivation experiments. Public Health Rep. 53:7.
- Davis GE, Cox HR. 1938. A filter-passing infectious agent isolated from ticks I. Isolation from Dermacentor andersoni, reactions in animals, and filtration experiments. Public Health Rep. 53:9.
- Denison AM, Thompson HA, Massung RF. 2007. IS1111 insertion sequences of Coxiella burnetii: characterization and use for repetitive element PCR-based differentiation of Coxiella burnetii isolates. BMC Microbiol. 7:91.
- Derrick EH. 1983. ‘Q’ fever, a new fever entity: clinical features, diagnosis and laboratory investigation. Rev Infect Dis. 5:790–800.
- Drancourt M, Raoult D. 2005. Family Coxiellaceae. In: Garrity GM, editor. Bergey's manual of systematic bacteriology 2nd ed., Part B: the gammaproteobacteria. New York: Springer; p. 237–247.
- Duncan C, Kersh GJ, Spraker T, Patyk KA, Fitzpatrick KA, Massung RF, Gelatt T. 2012. Coxiella burnetii in northern fur seal (Callorhinus ursinus) placentas from St. Paul Island, Alaska. Vector Borne Zoonotic Dis. 12:192–195.
- Dupuis G, Peter O, Peacock M, Burgdorfer W, Haller E. 1985. Immunoglobulin responses in acute Q fever. J Clin Microbiol. 22:484–487.
- Dyer RE. 1938. A filter-passing infectious agent isolated from ticks IV. Human infection. Public Health Rep. 53:6.
- [AHAW] EFSA Panel on Animal Health and Welfare. 2010. Scientific opinion on Q fever. EFSA J. 8 (5):1595. Available from: www.efsa.europa.eu/.
- Ghigo E, Capo C, Tung CH, Raoult D, Gorvel JP, Mege JL. 2002. Coxiella burnetii survival in THP-1 monocytes involves the impairment of phagosome maturation: IFN-gamma mediates its restoration and bacterial killing. J Immunol. 169:4488–4495.
- Gilsdorf A, Kroh C, Grimm S, Jensen E, Wagner-Wiening C, Alpers K. 2008. Large Q fever outbreak due to sheep farming near residential areas, Germany, 2005. Epidemiol Infect. 136:1084–1087.
- Glazunova O, Roux V, Freylikman O, Sekeyova Z, Fournous G, Tyczka J, Tokarevich N, Kovacava E, Marrie TJ, Raoult D. 2005. Coxiella burnetii genotyping. Emerg Infect Dis. 11:1211–1217.
- Guatteo R, Beaudeau F, Berri M, Rodolakis A, Joly A, Seegers H. 2006. Shedding routes of Coxiella burnetii in dairy cows: implications for detection and control. Vet Res. 37:827–833.
- Guatteo R, Beaudeau F, Joly A, Seegers H. 2007. Coxiella burnetii shedding by dairy cows. Vet Res. 38:849–860.
- Guatteo R, Seegers H, Joly A, Beaudeau F. 2008. Prevention of Coxiella burnetii shedding in infected dairy herds using a phase I C. burnetii inactivated vaccine. Vaccine. 26:4320–4328.
- Guatteo R, Seegers H, Taurel AF, Joly A, Beaudeau F. 2011. Prevalence of Coxiella burnetii infection in domestic ruminants: a critical review. Vet Microbiol. 149:1–16.
- Hackstadt T. 1988. Steric hindrance of antibody binding to surface proteins of Coxiella burnetti by phase I lipopolysaccharide. Infect Immunol. 56:802–807.
- Hatchette T, Campbell N, Hudson R, Raoult D, Marrie TJ. 2003. Natural history of Q fever in goats. Vector Borne Zoonotic Dis. 3:11–15.
- Heinzen R, Stiegler GL, Whiting LL, Schmitt SA, Mallavia LP, Frazier ME. 1990. Use of pulsed field gel electrophoresis to differentiate Coxiella burnetti strains. Ann N Y Acad Sci. 590:504–513.
- Hendrix LR, Samuel JE, Mallavia LP. 1991. Differentiation of Coxiella burnetti isolates by analysis of restriction-endonuclease-digested DNA separated by SDS-PAGE. J Gen Microbiol. 137:269–276.
- Higgins D, Marrie TJ. 1990. Seroepidemiology of Q fever among cats in New Brunswick and Prince Edward Island. Ann N Y Acad Sci. 590:271–274.
- Hogerwerf L, van den Brom R, Roest HI, Bouma A, Vellema P, Pieterse M, Dercksen D, Nielen M. 2011. Reduction of Coxiella burnetti prevalence by vaccination of goats and sheep, The Netherlands. Emerg Infect Dis. 17:379–386.
- Honstettre A, Ghigo E, Moynault A, Capo C, Toman R, Akira S, Takeuchi O, Lepidi H, Raoult D, Mege JL. 2004. Lipopolysaccharide from Coxiella burnetii is involved in bacterial phagocytosis, filamentous actin reorganization, and inflammatory responses through Toll-like receptor 4. J Immunol. 172:3695–3703.
- Hoover TA, Culp DW, Vodkin MH, Williams JC, Thompson HA. 2002. Chromosomal DNA deletions explain phenotypic characteristics of two antigenic variants, phase II and RSA 514 (crazy), of the Coxiella burnetii Nine Mile strain. Infect Immunol. 70:6726–6733.
- Horigan MW, Bell MM, Pollard TR, Sayers AR, Pritchard GC. 2011. Q fever diagnosis in domestic ruminants: comparison between complement fixation and commercial enzyme-linked immunosorbent assays. J Vet Diagn Invest. 23:924–931.
- Hotta A, Kawamura M, To H, Andoh M, Yamaguchi T, Fukushi H, Hirai K. 2002. Phase variation analysis of Coxiella burnetii during serial passage in cell culture by use of monoclonal antibodies. Infect Immunol. 70:4747–4749.
- Huijsmans CJ, Schellekens JJ, Wever PC, Toman R, Savelkoul PH, Janse I, Hermans MH. 2011. SNP-genotyping of a Coxiella burnetii outbreak in the Netherlands. Appl Environ Microbiol. 77(6):2051–2057.
- Hunter PR, Gaston MA. 1988. Numerical index of the discriminatory ability of typing systems: an application of Simpson's index of diversity. J Clin Microbiol. 26:2465–2466.
- Jager C, Willems H, Thiele D, Baljer G. 1998. Molecular characterization of Coxiella burnetii isolates. Epidemiol Infect. 120:157–164.
- Janse I, Bossers A, Roest HIJ, Rotterdam B. 2011. Hoge Resolutie Typering van Coxiella burnetii. Bilthoven: RIVM (RIVM briefrapport / Rijksinstituut voor Volksgezondheid en Milieu 330302001/2011); p. 38.
- Jones RM, Nicas M, Hubbard AE, Reingold AL. 2006. The infectious dose of Coxiella burnetii (Q fever). Appl Biosafety. 11:10.
- Kampschreur LM, Wegdam-Blans MC, Thijsen SF, Groot CA, Schneeberger PM, Hollander AA, Schijen JH, Arents NL, Oosterheert JJ, Wever PC. 2010. Acute Q fever related in-hospital mortality in the Netherlands. Neth J Med. 68:408–413.
- Kersh GJ, Lambourn DM, Raverty SA, Fitzpatrick KA, Self JS, Akmajian AM, Jeffries SJ, Huggins J, Drew CP, Zaki SR, Massung RF. 2012. Coxiella burnetii infection of marine mammals in the Pacific Northwest, 1997–2010. J Wildl Dis. 48:201–206.
- Kersh GJ, Lambourn DM, Self JS, Akmajian AM, Stanton JB, Baszler TV, Raverty SA, Massung RF. 2010. Coxiella burnetii infection of a Steller sea lion (Eumetopias jubatus) found in Washington State. J Clin Microbiol. 48:3428–3431.
- Kim SG, Kim EH, Lafferty CJ, Dubovi E. 2005. Coxiella burnetii in bulk tank milk samples, United States. Emerg Infect Dis. 11:619–621.
- Kishimoto RA, Rozmiarek H, Larson EW. 1978. Experimental Q fever infection in congenitally athymic nude mice. Infect Immunol. 22:69–71.
- Klee SR, Tyczka J, Ellerbrok H, Franz T, Linke S, Baljer G, Appel B. 2006. Highly sensitive real-time PCR for specific detection and quantification of Coxiella burnetii. BMC Microbiol. 6:2.
- Komiya T, Sadamasu K, Kang MI, Tsuboshima S, Fukushi H, Hirai K. 2003a. Seroprevalence of Coxiella burnetii infections among cats in different living environments. J Vet Med Sci. 65:1047–1048.
- Komiya T, Sadamasu K, Toriniwa H, Kato K, Arashima Y, Fukushi H, Hirai K, Arakawa Y. 2003b. Epidemiological survey on the route of Coxiella burnetii infection in an animal hospital. J Infect Chemother. 9:151–155.
- Lang GH. 1990. Coxiellosis (Q fever) in animals. In: Marrie TJ, editor. Q fever, volume I: the disease. Boca Raton (FL): CRC Press; p. 23–48.
- Lapointe JM, Gulland FM, Haines DM, Barr BC, Duignan PJ. 1999. Placentitis due to Coxiella burnetii in a Pacific harbor seal (Phoca vitulina richardsi). J Vet Diagn Invest. 11:541–543.
- McCaul TF, Williams JC. 1981. Developmental cycle of Coxiella burnetii: structure and morphogenesis of vegetative and sporogenic differentiations. J Bacteriol. 147:1063–1076.
- McDade JE. 1990. Historical aspects of Q fever. In: Marrie TJ, editor. Q fever Volume I: the disease. Boca Raton (FL): CRC Press; p. 5–21.
- Marrie TJ, Durant H, Williams JC, Mintz E, Waag DM. 1988a. Exposure to parturient cats: a risk factor for acquisition of Q fever in Maritime Canada. J Infect Dis. 158:101–108.
- Marrie TJ, MacDonald A, Durant H, Yates L, McCormick L. 1988b. An outbreak of Q fever probably due to contact with a parturient cat. Chest. 93:98–103.
- Marrie TJ, Raoult D. 1997. Q fever-a review and issues for the next century. Int J Antimicrob Agents. 8:145–161.
- Marrie TJ, Stein A, Janigan D, Raoult D. 1996. Route of infection determines the clinical manifestations of acute Q fever. J Infect Dis. 173:484–487.
- Martinov SP, Neikov P, Popov GV. 1989. Experimental Q fever in sheep. Eur J Epidemiol. 5:428–431.
- Maurin M, Raoult D. 1999. Q fever. Clin Microbiol Rev. 12:518–553.
- Moos A, Hackstadt T. 1987. Comparative virulence of intra- and inter-strain lipopolysaccharide variants of Coxiella burnetii in the guinea pig model. Infect Immunol. 55:1144–1150.
- Morroy G, Peters JB, van Nieuwenhof M, Bor HH, Hautvast JL, van der Hoek W, Wijkmans CJ, Vercoulen JH. 2011. The health status of Q-fever patients after long-term follow-up. BMC Infect Dis. 11:97.
- Muskens J, Mars MH, Franken P. 2007. [Q fever: an overview]. Tijdschr Diergeneeskd. 132:912–917.
- Omsland A. 2012. Axenic growth of Coxiella burnetii. Adv Exp Med Biol. 984:215–229.
- Omsland A, Cockrell DC, Howe D, Fischer ER, Virtaneva K, Sturdevant DE, Porcella SF, Heinzen RA. 2009. Host cell-free growth of the Q fever bacterium Coxiella burnetii. Proc Natl Acad Sci USA. 106:4430–4434.
- Oyston PC, Davies C. 2011. Q fever: the neglected biothreat agent. J Med Microbiol. 60:9–21.
- Parker RR, Davis GE. 1938. A filter passing infectious agent isolated from ticks II. Transmission by Dermacentor andersoni. Public Health Rep. 53:4.
- Philip CB. 1948. Comments on the name of the Q fever organism. Public Health Rep. 63:1.
- Pinsky RL, Fishbein DB, Greene CR, Gensheimer KF. 1991. An outbreak of cat-associated Q fever in the United States. J Infect Dis. 164:202–204.
- Raoult D, Marrie TJ, Mege JL. 2005. Natural history and pathophysiology of Q fever. Lancet Infect Dis. 5:219–226.
- Reusken C, van der Plaats R, Opsteegh M, de Bruin A, Swart A. 2011. Coxiella burnetii (Q fever) in Rattus norvegicus and Rattus rattus at livestock farms and urban locations in the Netherlands: could Rattus spp. represent reservoirs for (re)introduction? Prev Vet Med. 101:124–130.
- Rijks JM, Roest HI, van Tulden PW, Kik MJ, IJzer J, Grone A. 2011. Coxiella burnetii infection in roe deer during Q fever epidemic, the Netherlands. Emerg Infect Dis. 17:2369–2371.
- Roest HI, Post J, van Gelderen B, van Zijderveld FG, Rebel JM. 2013. Q fever in pregnant goats: humoral and cellular immune responses. Vet Res. 44:67.
- Roest HI, Ruuls RC, Tilburg JJ, Nabuurs-Franssen MH, Klaassen CH, Vellema P, van den Brom R, Dercksen D, Wouda W, Spierenburg MA. et al. 2011a. Molecular epidemiology of Coxiella burnetii from ruminants in Q fever outbreak, the Netherlands. Emerg Infect Dis. 17:668–675.
- Roest HIJ, Dinkla A, Frangoulidis D, Wouda W, Vellema P, Horrevorts A, Nabuurs-Franssen MH, Sturm P, Klaassen CHW, van Zijderveld FG. 2010. Isolation of Coxiella burnetii strains from clinical material from humans and small ruminants originating from the Netherlands. Poster session presented at: Voorjaarsvergadering van de NVMM, 2010 April 20–21; Papendal, Arnhem, the Netherlands.
- Roest HIJ, Hogerwerf L, van der Brom R, Oomen T, van Steenbergen JE, Nielen M, Vellema P. 2011b. [Q fever in the Netherlands: current status, results from veterinary research and expectations of the coming years]. Tijdschr Diergeneeskd. 136:340–343.
- Roest HIJ, Tilburg JJ, van der Hoek W, Vellema P, van Zijderveld FG, Klaassen CH, Raoult D. 2011c. The Q fever epidemic in The Netherlands: history, onset, response and reflection. Epidemiol Infect. 139:1–12.
- Roest HIJ, van Gelderen B, Dinkla A, Frangoulidis D, van Zijderveld FG, Rebel J, van Keulen L. 2012. Q fever in pregnant goats: pathogenesis and excretion of Coxiella burnetii. PLoS ONE. 7:14.
- Roest HIJ, van Steenbergen J, Wijkmans C, van Duijnhoven Y, Stenvers O, Oomen T, Vellema P. 2009. [Q fever in 2008 in the Netherlands and the expectations of 2009]. Tijdschr Diergeneeskd. 134:300–303.
- Rousset E, Berri M, Durand B, Dufour P, Prigent M, Delcroix T, Touratier A, Rodolakis A. 2009a. Coxiella burnetii shedding routes and antibody response after outbreaks of Q fever-induced abortion in dairy goat herds. Appl Environ Microbiol. 75:428–433.
- Rousset E, Durand B, Berri M, Dufour P, Prigent M, Russo P, Delcroix T, Touratier A, Rodolakis A, Aubert M. 2007. Comparative diagnostic potential of three serological tests for abortive Q fever in goat herds. Vet Microbiol. 124:286–297.
- Rousset E, Durand B, Champion JL, Prigent M, Dufour P, Forfait C, Marois M, Gasnier T, Duquesne V, Thiery R, Aubert MF. 2009b. Efficiency of a phase 1 vaccine for the reduction of vaginal Coxiella burnetii shedding in a clinically affected goat herd. Clin Microbiol Infect. 15(2):188–189.
- Russell-Lodrigue KE, Andoh M, Poels MW, Shive HR, Weeks BR, Zhang GQ, Tersteeg C, Masegi T, Hotta A, Yamaguchi T, et al. 2009. Coxiella burnetii isolates cause genogroup-specific virulence in mouse and guinea pig models of acute Q fever. Infect Immunol. 77:5640–5650.
- Russell-Lodrigue KE, Zhang GQ, McMurray DN, Samuel JE. 2006. Clinical and pathologic changes in a guinea pig aerosol challenge model of acute Q fever. Infect Immunol. 74:6085–6091.
- Salmon MM, Howells B, Glencross EJ, Evans AD, Palmer SR. 1982. Q fever in an urban area. Lancet. 1:1002–1004.
- Sanchez J, Souriau A, Buendia AJ, Arricau-Bouvery N, Martinez CM, Salinas J, Rodolakis A, Navarro JA. 2006. Experimental Coxiella burnetii infection in pregnant goats: a histopathological and immunohistochemical study. J Comp Pathol. 135:108–115.
- Schimmer B, Ter Schegget R, Wegdam M, Zuchner L, de Bruin A, Schneeberger PM, Veenstra T, Vellema P, van der Hoek W. 2010. The use of a geographic information system to identify a dairy goat farm as the most likely source of an urban Q-fever outbreak. BMC Infect Dis. 10:69.
- Schmatz HD, Busche R, Krauss H, Thiel N. 1977b. [The behavior of the Q-fever agent, Coxiella burnetii, in birds. 3. Experimental infection of quail]. Dtsch Tierarztl Wochenschr. 84:375–376.
- Schmatz HD, Busche R, Thiel N, Sailer J. 1977a. [Behavior of the Q fever vector Coxiella burnetii in birds. 2. Experimental infection of pigeons]. Dtsch Tierarztl Wochenschr. 84:60–63.
- Scott GH, Williams JC, Stephenson EH. 1987. Animal models in Q fever: pathological responses of inbred mice to phase I Coxiella burnetii. J Gen Microbiol. 133:691–700.
- Sekeyova Z, Roux V, Raoult D. 1999. Intraspecies diversity of Coxiella burnetii as revealed by com1 and mucZ sequence comparison. FEMS Microbiol Lett. 180:7.
- Seshadri R, Paulsen IT, Eisen JA, Read TD, Nelson KE, Nelson WC, Ward NL, Tettelin H, Davidsen TM, Beanan MJ, et al. 2003. Complete genome sequence of the Q-fever pathogen Coxiella burnetii. Proc Natl Acad Sci USA. 100:5455–5460.
- Sethi MS, Singh B, Yadav MP. 1978. Experimental infection of Coxiella burnetii in chicken: clinical symptoms, serologic response, and transmission through egg. Avian Dis. 22:391–395.
- Shannon JG, Heinzen RA. 2009. Adaptive immunity to the obligate intracellular pathogen Coxiella burnetii. Immunol Res. 43:138–148.
- Sidi-Boumedine K, Duquesne V, Fernandes I, Marro S, Thiery R. 2009. Evaluation of randomly amplified polymorphic DNA (RAPD) for discrimination of Coxiella burnetii ruminant strains isolated in France. Clin Microbiol Infect. 2 (Suppl.):194–195.
- Sidi-Boumedine K, Rousset E, Henning K, Ziller M, Niemczuck K, Roest HIJ, Thiery R. 2010. Development of harmonised schemes for the monitoring and reporting of Q fever in animals in the European Union. 2010. Parma: EFSA.
- Sprong H, Tijsse-Klasen E, Langelaar M, De Bruin A, Fonville M, Gassner F, Takken W, van Wieren S, Nijhof A, Jongejan F, et al. 2012. Prevalence of Coxiella burnetii in ticks after a large outbreak of Q fever. Zoonoses Public Health. 59:69–75.
- Svraka S, Toman R, Skultety L, Slaba K, Homan WL. 2006. Establishment of a genotyping scheme for Coxiella burnetii. FEMS Microbiol Lett. 254:268–274.
- Taurel AF, Guatteo R, Joly A, Beaudeau F. 2012. Effectiveness of vaccination and antibiotics to control Coxiella burnetii shedding around calving in dairy cows. Vet Microbiol. 159:432–437.
- Thiele D, Willems H. 1994. Is plasmid based differentiation of Coxiella burnetii in ‘acute’ and ‘chronic’ isolates still valid? Eur J Epidemiol. 10:427–434.
- Thompson M, Mykytczuk N, Gooderham K, Schulte-Hostedde A. 2012. Prevalence of the bacterium Coxiella burnetii in wild rodents from a Canadian Natural Environment Park. Zoonoses Public Health. 59(8):553–560.
- Tigertt WD, Benenson AS, Gochenour WS. 1961. Airborne Q fever. Bacteriol Rev. 25:285–293.
- Tissot-Dupont H, Amadei MA, Nezri M, Raoult D. 2004. Wind in November, Q fever in December. Emerg Infect Dis. 10:1264–1269.
- Tissot-Dupont H, Torres S, Nezri M, Raoult D. 1999. Hyperendemic focus of Q fever related to sheep and wind. Am J Epidemiol. 150:67–74.
- To H, Sakai R, Shirota K, Kano C, Abe S, Sugimoto T, Takehara K, Morita C, Takashima I, Maruyama T, et al. 1998. Coxiellosis in domestic and wild birds from Japan. J Wildl Dis. 34:310–316.
- Toman R, Skultety L, Ihnatko R. 2009. Coxiella burnetii glycomics and proteomics–tools for linking structure to function. Ann N Y Acad Sci. 1166:67–78.
- van den Brom R, Moll L, van Schaik G, Vellema P. 2012a. Demography of Q fever seroprevalence in sheep and goats in The Netherlands in 2008. Prev Vet Med. 109(1–2):76–82.
- van den Brom R, van Engelen E, Luttikholt S, Moll L, van Maanen K, Vellema P. 2012b. Coxiella burnetii in bulk tank milk samples from dairy goat and dairy sheep farms in The Netherlands in 2008. Vet Rec. 170:310.
- van der Hoek W. 2012. The 2007–2010 Q fever epidemic in the Netherlands: risk factors and risk groups. Utrecht: University of Utrecht.
- van der Hoek W, Hunink J, Vellema P, Droogers P. 2011a. Q fever in The Netherlands: the role of local environmental conditions. Int J Environ Health Res. 21:441–451.
- van der Hoek W, Meekelenkamp JC, Leenders AC, Wijers N, Notermans DW, Hukkelhoven CW. 2011b. Antibodies against Coxiella burnetii and pregnancy outcome during the 2007–2008 Q fever outbreaks in the Netherlands. BMC Infect Dis. 11:44.
- van Moll P, Baumgartner W, Eskens U, Hanichen T. 1993. Immunocytochemical demonstration of Coxiella burnetii antigen in the fetal placenta of naturally infected sheep and cattle. J Comp Pathol. 109:295–301.
- van Steenbergen JE, Morroy G, Groot CA, Ruikes FG, Marcelis JH, Speelman P. 2007. [An outbreak of Q fever in The Netherlands–possible link to goats]. Ned Tijdschr Geneeskd. 151:1998–2003.
- Voth DE, Heinzen RA. 2007. Lounging in a lysosome: the intracellular lifestyle of Coxiella burnetii. Cell Microbiol. 9:829–840.
- Wegdam-Blans MC, Kampschreur LM, Delsing CE, Bleeker-Rovers CP, Sprong T, van Kasteren ME</given-names>, Notermans DW, Renders NH, Bijlmer HA, Lestrade PJ, et al. 2012. Chronic Q fever: review of the literature and a proposal of new diagnostic criteria. J Infect. 64:247–259.
- Wegdam-Blans MC, Nabuurs-Franssen MN, Horrevorts AM, Peeters MF, Schneeberger PM, Bijlmer HA. 2010. [Laboratory diagnosis of acute Q fever]. Ned Tijdschr Geneeskd. 154: A2388.
- Welsh HH, Lennette EH, Abinanti FR, Winn JF. 1958. Air-borne transmission of Q fever: the role of parturition in the generation of infective aerosols. Ann N Y Acad Sci. 70:528–540.
- Willems H, Ritter M, Jager C, Thiele D. 1997. Plasmid-homologous sequences in the chromosome of plasmidless Coxiella burnetii Scurry Q217. J Bacteriol. 179:3293–3297.
- Wouda W, Dercksen DP. 2007. [Abortion and stillbirth among dairy goats as a consequence of Coxiella burnetii]. Tijdschr Diergeneeskd. 132:908–911.
- Zhang GQ, To H, Yamaguchi T, Fukushi H, Hirai K. 1997. Differentiation of Coxiella burnetii by sequence analysis of the gene (com1) encoding a 27-kDa outer membrane protein. Microbiol Immunol. 41:871–877.