
Abstract
Around the globe, chemical compounds are used to treat or repel pests and plagues that pose a threat to food and feed production. From epidemiological studies, it is known that there is a link between exposure to certain chemical classes of these so-called pesticides and the prevalence of neurodegenerative disorders such as Parkinson's disease in humans. However, which particular compound(s) account for this link or what underlying mechanisms are involved is still largely unresolved.
The degenerative process in Parkinson's disease is largely limited to the dopaminergic neurons in the basal ganglia. Cellular mechanisms that are implicated in parkinsonian neurodegeneration include mitochondrial dysfunction, oxidative stress, disturbance of intracellular calcium homeostasis and endoplasmic reticulum (ER) stress. A major characteristic that distinguishes the dopaminergic neurons in the basal ganglia from other dopaminergic neurons is a particular reliance on intracellular calcium for spontaneous activity. Considering the energy consuming nature of maintenance of the intracellular calcium homeostasis and its involvement in life and death of a neuron, this may explain the specific vulnerability of this neuronal population. Despite a large variation in primary mechanism of action it has been demonstrated that pesticides from different classes disturb intracellular calcium homeostasis, thus interfering with intracellular calcium signalling. This relates to altered dopaminergic signalling, disturbed protein homeostasis and increased oxidative stress. Therefore, effects of (mixtures of) pesticides on the intracellular calcium homeostasis may play a role in the development of Parkinson's disease in humans.
Although human exposure to pesticides via e.g. food often occurs in complex mixtures, (human) risk assessment is largely based on the assessment of single compounds. The discovery of common modes of action across different classes of pesticides therefore underpins the urgency of development of new models and approaches in risk assessment.
1. Pesticides
1.1. Introduction
Ever since people culture crops for food and feed production, people have been struggling with plagues and pests. In an attempt to assure a stable production of food and feed, humans used and designed numerous pesticides to treat animals and crops and destroy specific plagues. The first recorded use of (natural) compounds as pesticides dates back more than three millennia, to the use of sulphur as fumigant in the time of Homer around 1000 BC (Whitacre et al. Citation2004). Still, sulphur is among the most widely used fungicides in vineyards around the world.
Systematized development of synthetic chemicals for crop protection started in the 1930s and resulted in the development of a number of still well-known chemicals, such as dichlorodiphenyltrichloroethane (DDT) and 2,4-dichlorophenoxyaceticacid (2,4-D). After the Second World War, the field of agrochemicals developed quickly and a wide array of compounds has been introduced for pesticidal use.
Primary classification of pesticides is based on and named after the pesticidal target such as herbicides, insecticides, fungicides and rodenticides. Within these classes, further classification is based on similarities in chemical structure. Well-known sub-classes include organochlorine insecticides, phenoxyacetic herbicides and azole fungicides. Insecticides and fungicides can have an agricultural and/or pharmaceutical application. Examples of compounds with a double use are the organochlorine insecticide lindane (Nolan et al. Citation2012) and the azole fungicides (Stevens Citation2012), which are used as pharmaceutical in both veterinary and human medicine, but are also applied in agriculture as pesticides.
1.2. Toxicity of pesticides in humans
Although it seems self-explanatory, it is important to note that all pesticides possess an inherent degree of toxicity to some living organism; otherwise they would be of no practical use. In general, the target-species and the main pathway of toxicity are believed to determine the risk to humans. In other words, a herbicide targeting a plant-specific process that is absent in animals is perceived more safe than an insecticide targeting the nervous system of insects. From many insecticides it is known that they are indeed also neurotoxic in mammals, for example, organochlorine insecticides (Raymond-Delpech et al. Citation2005; Hatcher et al. Citation2008).
However, for many pesticides that presumably target species-specific pathways that seem not relevant to humans, such as herbicides and fungicides, this is less obvious. From epidemiological studies it is now known that a relationship exists between exposure to certain classes of pesticides and the occurrence of Parkinson's disease (PD) (Rajput & Birdi Citation1997; Elbaz & Tranchant Citation2007; Freire & Koifman Citation2012; Mostafalou & Abdollahi Citation2013), although the underlying neurotoxic mechanisms are often unknown.
2. Neurotransmission
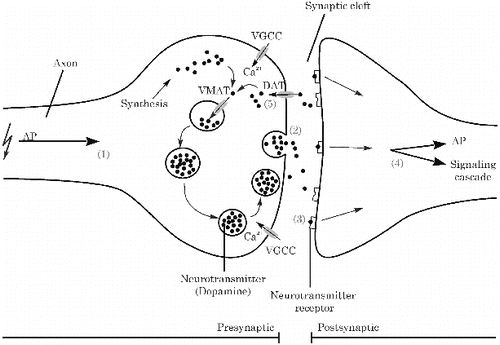
Communication in the (human) central nervous system is based on the transmission of a (chemical) signal from one cell to another (). A neuron contains many dendrites to receive input from neighbouring cells. Upon activation of its dendrites, a neuron generates an action potential (AP) via opening of voltage-gated sodium and potassium channels. This AP will travel along the axon to reach the synapse (1). In the synapse, the electrical signal is translated to a chemical signal via opening of voltage-gated calcium channels (VGCCs) and release of neurotransmitter from the presynaptic cell (2). These neurotransmitters are chemical signalling molecules that, following release into the synaptic cleft, bind to receptors on the membrane of the postsynaptic cell (3). In the postsynaptic cell, the chemical signal is then either translated again to an AP that will travel to the cell body of the postsynaptic cell or it translates to activation of intracellular signalling pathways (4). Signal transduction in the synaptic cleft is terminated by either (enzymatic) degradation or reuptake with transporters (5) of the neurotransmitter (for review see Westerink Citation2006).
Figure 1. Schematic overview of neurotransmission between a presynaptic (left) and postsynaptic (right) cell. AP: action potential, VMAT: vesicular monoamine transporter, VGCC: voltage-gated calcium channel, DAT: (membrane) dopamine transporter.
2.1. Dopaminergic neurotransmission
Within the brain, several subtypes of cells are available in different regions. These different cell types can utilize different neurotransmitters, such as acetylcholine (ACh), glutamate, GABA and dopamine (DA). DA is synthesized intracellularly by tyrosine hydroxylase-mediated conversion of tyrosine to the DA-precursor l-3,4-dihydroxyphenylalanine (l-DOPA) and subsequent conversion to DA by aromatic amino acid decarboxylase (for review see Westerink Citation2006). Upon synthesis, DA is stored in vesicles by active transport through the vesicular monoamine transporter (VMAT). Dopaminergic neurotransmission is achieved through fusion of a DA-filled vesicle with the plasma membrane, releasing its contents in the synaptic cleft. This process of neurotransmitter release is called exocytosis. Once released in the synaptic cleft, DA binds to DA receptors present on both the pre- and postsynaptic membrane. Two families of G-protein-coupled DA receptors can be distinguished: D1- and D2-like (auto) receptors (for review see Neve et al. Citation2004). Activation of D1 receptors is linked to feedback loops regulating VGCCs and potassium channels, via regulation of PKA- and PKC-mediated protein phosphorylation and activation of calcium-binding proteins. Activation of D2 receptors largely results in less phosphorylation activity and an inhibitory effect on VGCCs and a reduction in dopaminergic neurotransmission (Neve et al. Citation2004). Thus, the D1 and D2 receptors act in concert to balance dopaminergic signalling.
Extracellular DA is broken down by Catechol-O-methyltransferase (COMT) to terminate neurotransmission. Alternatively, DA is recycled from the synaptic cleft by transport through the dopamine transporter (DAT) making DA available for either storage in vesicles or enzymatic degradation by monoamine oxidase (MAO).
Dopaminergic neurotransmission is involved in a large variety of human behaviours, including (psycho)motor function, memory, motivation, reward and addiction. Areas containing most dopaminergic neurons can be found in a small number of nuclei, mainly located in the forebrain and the basal ganglia. Among the nuclei in the basal ganglia, the substantia nigra (SN), the ventral tegmental area (VTA) and the striatum (ST) contain the most dopaminergic neurons (Obeso et al. Citation2008). The basal ganglia and forebrain are innervated by three distinct major dopaminergic pathways: the nigrostriatal, mesocortical and mesolimbic pathways (). The nigrostriatal pathway arises in the SN pars compacta (SNpc) and projects to the striatum. Spontaneous and continuing rhythmic activity (i.e. pacemaking) of this dopaminergic innervation of the striatum tightly regulates DA levels in the ST (Guzman et al. Citation2009). This pathway is involved in facilitation of voluntary movement as well as inhibition of unwanted movement (Obeso et al. Citation2008).
Figure 2. Schematic view of the mesocortical, mesolimbic and nigrostriatal dopaminergic pathways in the human brain (modified from http://www.peoi.org chapter 8, section B).
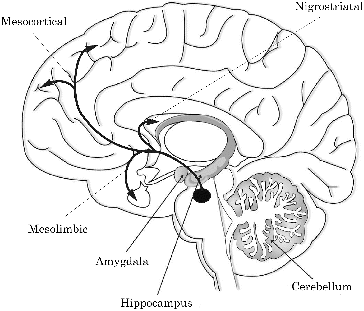
The mesocortical pathway arises from the VTA, projects to the prefrontal cortex (PFC) and is critically involved in normal cognitive function. The mesolimbic pathway also arises from the VTA, but innervates the nucleus accumbens, hippocampus, amygdala and PFC. This pathway is involved in e.g. the reward circuitry and is therefore important in conditions such as addiction.
In general, dopaminergic neurotransmission in the SN and ST is regulated by excitatory input from glutamatergic neurons, whereas inhibitory input is generated by GABA-ergic neurons. Depending on the type of input, dopaminergic neurotransmission increases (excitatory input) or decreases (inhibitory input). Excitatory input generally results in a depolarization of the membrane and subsequent Ca2+-influx that triggers exocytosis. In contrast, inhibitory GABA-ergic input results in hyperpolarization of the membrane, thereby reducing the chance of generation of an AP and consequently reducing the chance of VGCC opening and Ca2+-influx, thus reducing the chance of exocytosis.
2.2. Intracellular Ca2+ homeostasis
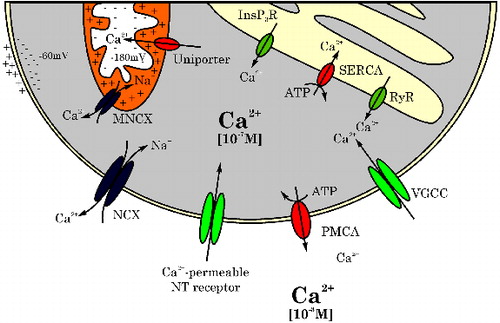
As all neurotransmission, dopaminergic neurotransmission relies heavily on calcium signalling (for review, see Barclay et al. Citation2005; Garcia et al. Citation2006; Westerink Citation2006). However, Ca2+ plays pivotal roles in many additional inter- and intraneuronal processes, including neurodevelopment (Pravettoni et al. Citation2000) and neurodegeneration (Mattson Citation2012). Therefore, the intracellular Ca2+ concentration ([Ca2+]i; typically ∼10−7 M) is tightly regulated by a system of VGCCs, sensors and pumps located in the plasma membrane and in the membrane of intracellular stores, in particular mitochondria (MNCX, uniporter) and the endoplasmic reticulum (ER: InsP3R, SERCA, RyR) (). As a roughly 10.000x Ca2+ gradient exists between the intra- and extracellular space (), maintenance of the intracellular [Ca2+]i is an essential but energy consuming process.
Figure 3. Schematic representation of channels and receptors in the cell membrane and the membranes of intracellular stores implicated in maintenance of the intracellular calcium homeostasis. NCX: Na+/Ca2+ exchanger, NT: neurotransmitter receptor, PMCA: plasma membrane Ca2+-ATPase, VGCC: voltage-gated calcium channel, RyR: ryanodine receptor, SERCA: sarco/endoplasmic reticulum Ca2+-ATPase, InsP3R: inositol triphosphate receptor, MNCX: mitochondrial Na+/Ca2+ exchanger.
3. Neurodegeneration and Parkinson's disease (PD)
Loss of neurons in the central nervous system (CNS) is an inevitable process that every individual experiences with ageing. Whenever this decline in neurons leads to functional impairment of e.g. cognition or motor control, the diagnosis neurodegeneration is evident. PD, one of the best-known neurodegenerative disorders, is named after James Parkinson who first described the symptoms of so-called shaking palsy in ‘modern’ medical literature back in 1817 (republication: Parkinson Citation2002). In developed countries, PD is the second most common neurodegenerative disorder after Alzheimer's disease and affects more than 1% of the population over the age of 60 (de Lau & Breteler Citation2006). The large majority (90%–95%) of PD cases is not related to inheritable genetic mutations and is therefore considered idiopathic PD (Bartels & Leenders Citation2009; Lees et al. Citation2009). Idiopathic PD is characterized by bradykinesia, muscle rigidity and resting tremor (Hughes et al. Citation1992; Lees et al. Citation2009; Brooks Citation2012). Parkinsonian neurodegeneration is characterized by selective degeneration of dopaminergic cells in the basal ganglia leading to reduced feedback in the neuronal circuitry involved in voluntary movement. According to a long-established scientific view, PD becomes clinically manifest when at least 70% of the striatal dopamine levels and 50% of the dopaminergic neurons in the nigrostriatal pathway () is lost (Braak et al. Citation2003; Lees et al. Citation2009). Although more brain areas are affected, degeneration of the nigrostriatal pathway reduces the amount of dopaminergic input in the ST, resulting in decreased DA levels and loss of function (Blandini et al. Citation2000; Rice et al. Citation2011). This is held responsible for the clinical observation of loss of (voluntary) movement control.
4. Aetiology of PD and related disorders
Idiopathic PD and parkinsonisms are considered as multifactorial and complex disorders with limited involvement of (inheritable) genetic defects (Bartels & Leenders Citation2009).
The exact aetiology of PD and related parkinsonisms is unknown although several hypotheses exist. These include excess oxidative stress due to high intrinsic production of oxidative molecules (H2O2, quinones, etc.) as a consequence of dopamine turnover, and accelerated ageing of mitochondria in dopaminergic cells due to the reliance on energy-consuming mechanisms for neurotransmission (Schapira & Jenner Citation2011; Surmeier et al. Citation2011). In addition, changes in intracellular Ca2+-homeostasis leading to impaired dopamine handling, hampered neurotransmission and dysfunction of ER and mitochondria are implicated (Bezprozvanny Citation2009; Surmeier et al. Citation2011; Mattson Citation2012). Furthermore, activation of glial cells and astrocytes (microgliosis, astrocytosis) by both exogenous (e.g. environmental factors) and endogenous (e.g. systemic inflammation, brain trauma) factors is demonstrated to contribute to the pathophysiology of PD (McGeer & McGeer Citation2004; Taylor et al. Citation2013). Finally, PD and parkinsonisms are characterized by formation of intracellular α-synuclein-rich protein inclusions called Lewy bodies. The particular involvement of α-synuclein is underlined by the observation that mutations leading to overexpression or hampered intracellular handling of α-synuclein are linked to parkinsonism (Venda et al. Citation2010; Bezard & Przedborski Citation2011).
5. The role of pesticides in PD
In the early 1980s, it was observed that accidental exposure of intravenous drug abusers to synthetic drugs contaminated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) caused acute parkinsonism (Langston et al. Citation1983; Langston Citation1996; Venda et al. Citation2010; Bezard & Przedborski Citation2011). Soon thereafter, the close chemical resemblance between the active metabolite of MPTP, the cation 1-methyl-4-phenylpyridinium (MPP+), and the well-known and widely used herbicide paraquat, raised the question about the role of pesticides and other environmental factors in the pathophysiology of PD (Barbeau Citation1984). By now, epidemiological studies have demonstrated an increased risk for the development of PD related to pesticide exposure (Ascherio et al. Citation2006; Brown et al. Citation2006; van der Mark et al. Citation2011; Mostafalou & Abdollahi Citation2013; Pezzoli & Cereda Citation2013). However, considerable heterogeneity in the available studies precludes conclusiveness as to which class of pesticides is involved (van der Mark et al. Citation2011). Available epidemiological studies point towards an increased risk associated with insecticides (in particular organochlorine insecticides; Elbaz et al. Citation2009; Freire & Koifman Citation2012) and herbicides (Pezzoli & Cereda Citation2013). Interestingly, the epidemiological evidence for the involvement of organochlorine insecticides is underlined by biomarkers of exposure in serum (Weisskopf et al. Citation2010) and post mortem detection of elevated levels of particular organochlorine insecticides in brains of PD patients (Corrigan et al. Citation1996; Corrigan et al. Citation2000).
6. Proposed mechanisms of pesticide-induced PD
The first mechanisms by which pesticides can induce dopaminergic neurodegeneration were revealed in research on the toxicity of MPP+ and paraquat. Acute exposure to paraquat was reported to cause oxidative stress via mitochondrial dysfunction and ER stress in dopaminergic cells from the SNpc, ultimately resulting in misfolded proteins and degeneration (Chinta & Andersen Citation2008; Chinta et al. Citation2008). Therefore, paraquat is considered as a model compound for pesticide-induced PD.
Another model compound for PD is the insecticide rotenone, naturally occurring in several plant species (Betarbet et al. Citation2000). Rotenone administration in rodents results in degeneration of the nigral dopaminergic pathway, characterized by α-synuclein aggregation and reactive astrogliosis (Betarbet et al. Citation2000; Sherer et al. Citation2003a; Sherer et al. Citation2003b). The mechanism of toxicity is pinpointed to uncoupling of complex I of the mitochondrial phosphorylation (Radad et al. Citation2006).
Following the discovery of MPTP, paraquat and rotenone causing parkinsonism in vivo, early theories on the particular vulnerability of the SNpc focussed on a high oxidative burden in dopaminergic cells in general as a result of DA handling and oxidation of cytosolic DA. Similarly, mitochondrial dysfunction (e.g. uncoupling of mitochondrial complexes I and III; (Bywood & Johnson Citation2003) has been indicated as an important pathway in the pathophysiology of PD. However, it is debatable whether the high oxidative burden and mitochondrial function are causative factors since these are not specific for dopaminergic neurons in SNpc. Consequently, SNpc dopaminergic neurons must have distinct (functional) properties that, upon disturbance or malfunction, lead to selective degeneration of the SNpc and the development of idiopathic PD. One of the things that distinguishes dopaminergic neurons in the SNpc from other dopaminergic neurons in the CNS and the basal ganglia is a constant level of spontaneous activity; pacemaking (Guzman et al. Citation2009; Imtiaz Citation2012). Dopaminergic neurons in the SNpc generate APs on a rhythmic basis, thereby providing a constant dopaminergic input from the SN to the striatum. This process is held responsible for the maintenance of DA levels in the striatum and basal ganglia (Imtiaz Citation2012) and in that sense for the fine-tuning of neurotransmission. Pacemaking activity in the SNpc involves a particular subtype of the dihydropyridine-sensitive high-voltage activated (HVA) Cav1.3 (L-type) VGCCs (Chan et al. Citation2007; Guzman et al. Citation2009; Putzier et al. Citation2009). In contrast, pacemaking activity in dopaminergic neurons in the neighbouring VTA largely relies on sodium channels (Chan et al. Citation2007; Imtiaz Citation2012). Moreover, influx of Ca2+ through Cav1.3 VGCCs increases DA metabolism, which leads to elevated cytosolic DA concentrations, thus creating an increased level of mitochondrial oxidative stress that is specific for SNpc neurons (Mosharov et al. Citation2009). Notably, other brain areas that experience cell loss parallel to the SNpc in PD such as the locus coeruleus and the hypothalamus (Braak et al. Citation2003) also display autonomous Ca2+-dependent pacemaking activity (for review see Surmeier et al. Citation2011). The Ca2+-dependence of autonomous pacemaking thus provides a plausible explanation for the specific vulnerability of dopaminergic neurons in the SNpc. Therefore, it is worrisome that several pesticides from different chemical classes are able to block VGCCs in either a selective (Cav1.3 inhibition by lindane: Heusinkveld et al. Citation2010) or non-selective (dieldrin: Heusinkveld & Westerink Citation2012; azole fungicides: Heusinkveld et al. Citation2013; organophosphate insecticides: Meijer et al. Citation2014a) manner. Accordingly, disturbance of the intracellular calcium homeostasis may provide part of an adverse outcome pathway that connects pesticide exposure with neurodegeneration.
Dopaminergic neurotransmission in the basal ganglia is regulated by a network that primarily consists of excitatory input from glutamatergic neurons and inhibitory input from GABA-ergic neurons. Misbalance in excitatory- and inhibitory input in the neuronal network of the basal ganglia is implicated in PD (Dexter & Jenner Citation2013). Importantly, chemical dopaminergic degeneration can be prevented by inhibition of glutamate receptors (Turski et al. Citation1991), suggesting the involvement of excitotoxicity caused by glutamatergic over-excitation in the pathophysiology of PD (Dexter & Jenner Citation2013). However, excitotoxicity is considered merely a consequence of the degenerative process that is responsible for the disease progression (Blandini et al. Citation2000). Notably, inhibitory input from GABA-ergic neurons is responsible for negative regulation of glutamatergic stimulation from the subthalamic nucleus (STN). It is therefore likely that chemical interference with GABA-ergic neurotransmission, as observed for lindane and dieldrin (Vale et al. Citation2003) as well as for MPP+ (Wu et al. Citation2002) and dinitrophenolic herbicides (Heusinkveld et al., unpublished observations), can modulate the negative feedback, thereby leading to glutamatergic overstimulation and excitotoxicity.
PD is often referred to as a proteinopathy because of the distinctive role of proteins and protein depositions in its pathophysiology. It is known that ER-stress is related to misfolding of proteins, including α-synuclein, whereas excess oxidative stress and mitochondrial dysfunction is related to hampered protein degradation resulting in fibrilization and aggregation of proteins in the cytoplasm (Nath et al. Citation2011; Goodwin et al. Citation2013). This is a dangerous process as pathogenic α-synuclein, which can be transferred from one cell to another in a prion-like manner (Dunning et al. Citation2012; Yasuda et al. Citation2013), is related to a change in intracellular DA handling, increased mitochondrial and ER-stress and cell death (Goodwin et al. Citation2013; Wilkaniec et al. Citation2013). A local change in α-synuclein production, as observed in vitro with dinitrophenolic herbicides (Heusinkveld et al., unpublished observations), following a toxic insult may thus result in the onset of a cascade that results in spreading of pathogenic proteins through the brain.
It has been demonstrated extensively in vitro as well as in vivo in animals and humans that neurodegenerative disorders (including PD) are associated with activation of glia and astrocytes resulting in inflammation (Purisai et al. Citation2007; Tansey & Goldberg Citation2010; Taylor et al. Citation2013). The notion that the immune system is involved in the aetiology of PD is underlined by the observation that non-aspirin NSAIDs are neuroprotective (Gagne & Power Citation2010). Additionally, peripheral inflammation is observed to influence neurodegeneration-associated brain inflammation (Hernández-Romero et al. Citation2012).
Considering the wide array of above mentioned factors, the dopaminergic system of the basal ganglia is best described by a delicately balanced, complex system with special features that render the SNpc dopaminergic neurons more vulnerable than others. The role of environmental factors, including pesticides, in idiopathic PD is therefore probably best explained by a multi-hit model in which no single factor or compound is causative but a complex interplay between exposure to environmental factors, individual genetic susceptibility, and specific vulnerability of the dopaminergic pathways in the basal ganglia determines the disease process.
7. Human risk assessment
Whether or not exposure to a certain pesticide poses a risk to the human population depends on the likelihood and degree of internal exposure, which is largely determined by the route of exposure and the fate (e.g. metabolism) of the compound upon exposure. The most likely exposure scenario for the general population is oral exposure through ingestion of food that contains pesticide residues (Oates & Cohen Citation2011). This comprises exposure to pesticides that are currently in use in food- and feed production (e.g. azole fungicides) as well as persistent compounds (e.g. lindane, dieldrin) that have accumulated in the food chain. A different situation occurs in the case of occupational exposure to pesticides. This comprises agricultural and greenhouse operators and workers, but also people working in the veterinary or human medical practice where pesticides are applied as pharmaceutical agents. In these occupational settings, also dermal and inhalational exposure are important although to a yet unknown extent (see e.g. Ramwell et al. Citation2005).
Exposure of the brain to a pesticide depends merely on physicochemical properties that determine the ADME (absorption, distribution, metabolism and excretion) characteristics and thus the pesticide concentration in the systemic circulation and the brain. It is important to have information on these characteristics as some pesticides (e.g. azole fungicides) are known to interfere with metabolism, which may in turn influence circulating pesticide concentrations.
Brain exposure is also determined by the ability of a pesticide to cross the blood–brain barrier (BBB). Although the BBB protects the brain against a plethora of potential noxious insults such as proteins and infectious agents, animal models revealed that many chemicals including MPTP and several pesticides simply diffuse through the BBB because of their lipophilicity (Cicchetti et al. Citation2009). In addition, some pesticides (e.g. paraquat) are actively transported over the BBB by means of for instance the neutral amino acid transporter (Shimizu et al. Citation2001). Therefore, whether or not the brain is exposed depends largely on the physicochemical properties of the pesticide or whether the pesticide can act as a suitable substrate for a transporter.
Due to their persistence and bioaccumulation in the food chain, the organochlorine insecticides lindane and dieldrin are still detectable in human blood at picomolar to nanomolar levels (). Notably, increased serum levels of dieldrin have been related to the risk for PD (Weisskopf et al. Citation2010). Furthermore, compared to controls or patients with Alzheimer's disease, increased levels of lindane and dieldrin (up to ∼1 μg/g l.w.) have been detected in brain tissue of PD patients (Fleming et al. Citation1994; Corrigan et al. Citation2000).
Table 1. Human exposure data.
Average serum levels of dieldrin and lindane in developed countries (see e.g. Koppen et al. Citation2002; Thomas et al. Citation2006; Xu et al. Citation2010 and ) indicate that a margin of safety exists between the concentrations at which neurotoxic effects are observed in vitro () and actual exposure. However, the lipophilicity renders it questionable whether serum levels truly represent internal exposure and exposure of the brain in particular. This was already underlined by a pharmacokinetic study from the 1970s with dieldrin (Walsh & Fink Citation1972) in which the authors demonstrated comparable uptake kinetics in adipose tissue and the brain, whereas dieldrin disappeared from the blood quickly. Therefore, dieldrin concentrations in fatty tissue may be a more reliable representation of actual internal (brain) dieldrin exposure than serum concentrations. Consequently, the detection of dieldrin at concentration around 130 ng/g l.w. (corresponding to ∼270 nM, calculated using average physiological values) in adipose tissue of Spanish children (Lopez-Espinosa et al. Citation2008) is worrying. In particular because it has been demonstrated that perinatal exposure of rodents to organochlorine insecticides is related to persistent changes in dopaminergic neurotransmission (Richardson et al. Citation2006) and the development of PD later in life (see e.g. Barlow et al. Citation2007).
Table 2. In vitro effect concentrations.
The available data on human internal exposure to dinitrophenolic herbicides are limited to DNOC but clearly indicate that internal exposure in agricultural workers has been high (). Considering the lipophilicity of the other dinitrophenolic herbicides (log K ow ≥3.6), it can be expected that comparable levels are reached upon exposure to other dinitrophenolic herbicides. In addition, these compounds pass biological membranes with relative ease (Gibson & Rao Citation1973). Considering this, the in vitro effect concentrations on parameters of dopaminergic neurodegeneration and GABA-ergic neurotransmission appear to be comparable to exposure levels in humans ( and ). In fact, even when considering that brain exposure may be limited to a fraction of the blood levels (Gibson & Rao Citation1973), there appears to have been a very small (if at all) margin of safety in agricultural workers. Therefore, exposure to dinitrophenolic herbicides has likely resulted in exposure levels comparable to effect concentrations for the observed in vitro effects. Thus, exposure may be related to dopaminergic neurodegeneration observed in agricultural workers exposed to these compounds.
Human exposure data on azole fungicides are largely lacking, rendering proper risk assessment hardly doable. However, residue levels of imazalil, tebuconazole and cyproconazole on food products are often >10 μg/kg (Nougadère et al. Citation2012). Therefore, human exposure to azole fungicides appears very likely, though it is unclear how residue levels of these pesticides relate to human internal exposure as this depends on numerous factors, including for example food processing before consumption.
An animal-based PBPK study with triadimefon indicated that upon exposure triadimefon is spread throughout the body, including the brain (Crowell et al. Citation2011). Using the pharmacokinetic data obtained in this study, the authors extrapolated a human equivalent dose (HED) for the rat NOAEL for neurotoxicity. The calculated HED turned out to be only 25-fold higher than the human oral reference dose (RfD), indicating that current safety margins for environmental exposure to triadimefon may not be sufficient.
Additional concern arises from the observation that different pesticides and classes of pesticides can act on the same endpoint (e.g. inhibition of VGCC and GABA-ergic neurotransmission). We previously demonstrated additive effects on VGCC inhibition in mixtures of lindane and dieldrin (Heusinkveld & Westerink Citation2012) as well as mixtures of azole fungicides (Heusinkveld et al. Citation2013). Considering the fact that these compounds share a target, it appears thus realistic that additivity may also apply for mixtures of these different classes of compounds.
Human risk assessment of pesticides so far is based on effects induced by single compounds after short-term or acute exposure to relatively high doses. However, people experience chronic low-doses exposure to a mixture of chemicals, among which many pesticides. According to the usual approach, exposure to such a complex mixture of chemicals will not pose a risk when all individual compounds are below their individual effect level. However, as is known from research on endocrine disrupting chemicals, unexpected effects may occur due to chemical interactions in the body (Kortenkamp Citation2008). Therefore, the biggest and yet unanswered question is whether safety margins currently applied are providing enough safety to assure that even complex mixtures of compounds are not evoking adverse effects.
8. Conclusions and recommendations for future research
In conclusion, although epidemiological research provides evidence that pesticides play a role in idiopathic PD, the underlying mechanisms remain elusive. Recent findings add to the body of evidence that pesticides from different classes and with varying chemical structures can induce neurotoxicity in vitro (Heusinkveld et al. Citation2010; Heusinkveld & Westerink Citation2012; Heusinkveld et al. Citation2013; Meijer et al. Citation2014a). In many cases, neurotoxicity is due to mechanisms that are unrelated to the primary pesticidal mode of action. The neurotoxic effects observed comprise effects on hallmarks of neurodegeneration (α-synuclein, apoptosis) as well as effects on parameters (Ca2+ homeostasis and VGCC functioning, DA handling, etc.) that presumably determine the enhanced sensitivity of SNpc neurons in vivo towards insults from environmental factors including pesticides.
The elucidation of the potential role of pesticides in PD aetiology requires detailed knowledge about key processes determining the onset and progression of the disease. However, this knowledge is still largely absent. As a result, it still remains to be determined which particular effects observed in vitro or in vivo are truly predictive for the development of PD in humans.
It is generally accepted that a long latency period exists between the first neurobiological changes and the occurrence of clinical symptoms in idiopathic PD (Schapira Citation2009). Nevertheless, the majority of the research conducted in vitro as well as in vivo is based on (relatively) short-term exposures to concentrations of compounds that are often much higher than is realistic for the human situation. Hence, it is debatable to what extent the effects observed at these levels of exposure and exposure paradigms are predictive for the role of a chemical in idiopathic PD. Accordingly, compounds such as MPTP, paraquat and rotenone that are related to fast development of PD in vivo may not be entirely explanatory for the role of pesticides and environmental factors in idiopathic PD.
From the foregoing it is clear that more research is necessary. Research in chemical-induced neurotoxicity is for obvious reasons increasingly performed in in vitro systems. However, as has been demonstrated earlier, techniques and models may suffer from shortcomings and pitfalls (Westerink & Hondebrink Citation2010; Heusinkveld & Westerink Citation2011; Westerink Citation2013; Meijer et al. Citation2014b). Therefore, thorough characterization and verification of existing models and techniques as well as development of novel models are required.
Ideally, a research model is as complete, complex and non-manipulated as possible (de Groot et al. Citation2013). Cell- or animal models with e.g. induced overexpression of human α-synuclein or knock-out models can be valuable for assessment of the effect of chemicals on specific processes. However, research on the neurodegenerative properties of compounds in animals or cell lines bearing genetic defects as observed in familial PD should be interpreted with care. As the genetic background of idiopathic PD is still diffuse, it can be debated whether these models provide relevant information with regard to the role of e.g. pesticides in the occurrence of idiopathic PD. It is therefore possibly more relevant to study the involvement of chemical exposure in relation to early pathological signs of PD in (healthy) wild-type animals or cell lines, than to investigate the effect of a chemical on an animal or cell line bearing a genetic defect that renders it more vulnerable.
In addition, new and promising non-mammalian models, such as Caenorhabditis elegans (Estevez et al. Citation2012), need to be explored. However, thorough characterization of its strengths (e.g. limited number of good visible neurons, integrated model system) and weaknesses (e.g. no BBB, different metabolism, different route of exposure) is required to make it a successful model.
Furthermore, the role of activation of the immune system in the CNS has to be examined. There is increasing evidence that the immune system and inflammatory processes play a (key) role in the onset and/or progression of (parkinsonian) neurodegeneration (Rodríguez & Verkhratsky Citation2010; Tansey & Goldberg Citation2010; Taylor et al. Citation2013). However, it remains to be determined whether for instance astrogliosis following pesticide exposure is cause or consequence. Research on the involvement of the immune system in vitro requires advanced co-cultures of neurons and astroglial cells with conditioned media to determine whether glial activation triggers changes in neuronal function or the other way around.
As the BBB has a key-protective role in the CNS, the role of BBB (mal-)function in neurodegenerative processes also needs to be taken into account (Obermeier et al. Citation2013). In general, in vitro models do not take into account the fact that a BBB may prevent or aggravate toxicity as it may repel or concentrate toxicants. Therefore, to be able to reliably extrapolate from in vitro to in vivo the role of the BBB should be known and taken into account.
A well-known route for chemicals to largely by-pass the BBB is via the olfactory nerve. This has already been demonstrated for transport of nanoparticles and metals via the olfactory nerve into the brain (Oberdorster et al. Citation2004; Elder et al. Citation2006; Lucchini et al. Citation2012). As the early stages in PD are characterized by a loss of olfaction (Braak et al. Citation2003), it remains to be determined whether or not the first event in the process(es) leading to PD can be related to transport of chemicals via or degeneration of the olfactory tract. Agricultural workers experience inhalational and olfactory exposure in addition to dermal and oral exposure in their occupational situation. Therefore, when olfactory exposure can give rise to neurotoxicity or degeneration, it needs to be determined whether health protection by measures of occupational hygiene provides an adequate level of protection.
Detailed human exposure data are one of the requirements for reliable risk assessment. Although monitoring programmes exist for a plethora of compounds, including many phased-out but persistent organic pollutants such as lindane and dieldrin, hardly any human exposure data are available for many pesticides that are currently in use (e.g. azole fungicides). In addition, less obvious but nonetheless relevant routes of exposure need to be assessed such as (occupational) exposure to pesticides with a (veterinary) pharmaceutical application. More elaborate monitoring data on human exposure is thus needed to avoid inadequate assessment of the risk posed by particular compounds. In addition, as discussed above, current risk assessment is based on individual compounds, rendering it elusive whether the current approach is granting enough margin of safety in case of complex mixtures. However, the lack of human exposure data and the absence of (software) tools to realistically estimate internal exposure and the according risk of a complex mixture, hamper reliable risk assessment of mixtures.
Taking everything into account, the immense jigsaw provided by the question whether pesticide exposure is related to neurodegeneration is still far from finalized. Finding the core-pieces that speed up finalization of the jigsaw will likely require innovative in vitro and in vivo testing strategies that identify truly predictive processes determining chemical-induced neurodegeneration. Nevertheless, recent research added some new pieces to the puzzle, demonstrating that several classes of pesticides can induce in vitro (dopaminergic) neuronal dysfunction that may lead to degeneration.
Acknowledgements
We gratefully acknowledge Dr PCG Nijssen for helpful discussions.
Disclosure
No potential conflict of interest was reported by the authors.
Funding
This work was funded by the Dutch Internationaal Parkinson Funds and the EU-funded project ACROPOLIS [grant number KBBE-245163].
References
- Ascherio A, Honglei C, Weisskopf MG, O’Reilly E, McCullough ML, Calle EE, Schwarzschild MA, Thun MJ. 2006. Pesticide exposure and risk for Parkinson's disease. Ann Neurol. 60:197–203.
- Barbeau A. 1984. Manganese and extrapyrimidal disorders (a critical review and tribute to Dr. George C. Cotzias). Neurotoxicology. 5:13–35.
- Barclay JW, Morgan A, Burgoyne RD. 2005. Calcium-dependent regulation of exocytosis. Cell Calcium. 38:343–353.
- Barlow BK, Cory-Slechta DA, Richfield EK, Thiruchelvam M. 2007. The gestational environment and Parkinson's disease: evidence for neurodevelopmental origins of a neurodegenerative disorder. Reprod Toxicol. 23:457–470.
- Bartels AL, Leenders KL. 2009. Parkinson's disease: the syndrome, the pathogenesis and pathophysiology. Cortex. 45:915–921.
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre TJ. 2000. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 3:1301–1306.
- Bezard E, Przedborski S. 2011. A tale on animal models of Parkinson's disease. Move Disord. 26:993–1002.
- Bezprozvanny I. 2009. Calcium signaling and neurodegenerative diseases. Trends Mol Med. 15:89–100.
- Blandini F, Nappi G, Tassorelli C, Martignoni E. 2000. Functional changes of the basal ganglia circuitry in Parkinson's disease. Prog Neurobiol. 62:63–88.
- Botella B, Crespo J, Rivas A, Cerrillo I, Olea-Serrano MF, Olea N. 2004. Exposure of women to organochlorine pesticides in Southern Spain. Environ Res. 96:34–40.
- Braak H, Tredici KD, Rnb U, de Vos RAI, Jansen Steur ENH, Braak E. 2003. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 24:197–211.
- Brooks DJ. 2012. Parkinson's disease: diagnosis. Parkinsonism Relat Disord. 18(Supplement 1):S31–S33.
- Brown TP, Rumsby PC, Capleton AC, Rushton L, Levy LS. 2006. Pesticides and Parkinson's disease – is there a link? Environ Health Perspect. 114:156–164.
- Butler Walker J, Seddon L, McMullen E, Houseman J, Tofflemire K, Corriveau A, Weber J-P, Mills C, Smith S, Van Oostdam J. 2003. Organochlorine levels in maternal and umbilical cord blood plasma in Arctic Canada. Sci Total Environ. 302:27–52.
- Bywood PT, Johnson SM. 2003. Mitochondrial complex inhibitors preferentially damage substantia nigra dopamine neurons in rat brain slices. Exp Neurol. 179:47–59.
- Carreño J, Rivas A, Granada A, Jose Lopez-Espinosa M, Mariscal M, Olea N, Olea-Serrano F. 2007. Exposure of young men to organochlorine pesticides in Southern Spain. Environ Res. 103:55–61.
- Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, Meredith GE, Surmeier DJ. 2007. Rejuvenation’ protects neurons in mouse models of Parkinson's disease. Nature. 447:1081–1086.
- Chinta SJ, Andersen JK. 2008. Redox imbalance in Parkinson's disease. Biochim Biophys Acta. 1780:1362–1367.
- Chinta SJ, Rane A, Poksay KS, Bredesen DE, Andersen JK, Rao RV. 2008. Coupling endoplasmic reticulum stress to cell death program in dopaminergic cells: effect of paraquat. Neuromolecular Med. 10:333–342.
- Cicchetti F, Drouin-Ouellet J, Gross RE. 2009. Environmental toxins and Parkinson's disease: what have we learned from pesticide-induced animal models? Trends Pharmacol Sci. 30:475–483.
- Corrigan FM, French M, Murray L. 1996. Organochlorine compounds in human brain. Human Exp Toxicol. 15:262–264.
- Corrigan FM, Wienburg CL, Shore RF, Daniel SE, Mann D. 2000. Organochlorine insecticides in substantia nigra in Parkinson's disease. J Toxicol Environ Health A. 59:229–234.
- Crowell SR, Henderson WM, Kenneke JF, Fisher JW. 2011. Development and application of a physiologically based pharmacokinetic model for triadimefon and its metabolite triadimenol in rats and humans. Toxicol Lett. 205:154–162.
- Cruz S, Lino C, Silveira MI. 2003. Evaluation of organochlorine pesticide residues in human serum from an urban and two rural populations in Portugal. Sci Total Environ. 317:23–35.
- de Groot MW, Westerink RH, Dingemans MM. 2013. Don't judge a neuron only by its cover: neuronal function in in vitro developmental neurotoxicity testing. Toxicol Sci. 132:1–7.
- de Lau LM, Breteler MM. 2006. Epidemiology of Parkinson's disease. Lancet Neurol. 5:525–535.
- Dexter DT, Jenner P. 2013. Parkinson disease: from pathology to molecular disease mechanisms. Free Radical Biol Med. 62:132–144.
- Dirtu AC, Cernat R, Dragan D, Mocanu R, van Grieken R, Neels H, Covaci A. 2006. Organohalogenated pollutants in human serum from Iassy, Romania and their relation with age and gender. Environ Int. 32:797–803.
- Dunning CJR, Reyes JF, Steiner JA, Brundin P. 2012. Can Parkinson's disease pathology be propagated from one neuron to another? Prog Neurobiol. 97:205–219.
- Elbaz A, Clavel J, Rathouz PJ, Moisan F, Galanaud J-P, Delemotte B, Alpérovitch A, Tzourio C. 2009. Professional exposure to pesticides and Parkinson disease. Ann Neurol. 66:494–504.
- Elbaz A, Tranchant C. 2007. Epidemiologic studies of environmental exposures in Parkinson's disease. J Neurol Sci. 262:37–44.
- Elder A, Gelein R, Silva V, Feikert T, Opanashuk L, Carter J, Potter R, Maynard A, Ito Y, Finkelstein J, Oberdörster G. 2006. Translocation of inhaled ultrafine manganese oxide particles to the central nervous system. Environ Health Perspect. 114:1172–1178.
- Estevez AO, Mueller CL, Morgan KL, Szewczyk NJ, Teece L, Miranda-Vizuete A, Estevez M. 2012. Selenium induces cholinergic motor neuron degeneration in Caenorhabditis elegans. Neurotoxicology. 33:1021–1032.
- Fleming L, Mann JB, Bean J, Briggle T, Sanchez-Ramos JR. 1994. Parkinson's disease and brain levels of organochlorine pesticides. Ann Neurol. 36:100–103.
- Freire C, Koifman S. 2012. Pesticide exposure and Parkinson's disease: epidemiological evidence of association. Neurotoxicology. 33:947–971.
- Gagne JJ, Power MC. 2010. Anti-inflammatory drugs and risk of Parkinson disease: a meta-analysis. Neurology. 74:995–1002.
- Garcia AG, Garcia-De-Diego AM, Gandia L, Borges R, Garcia-Sancho J. 2006. calcium signaling and exocytosis in adrenal chromaffin cells. Physiol Rev. 86:1093–1131.
- Gibson JE, Rao KS. 1973. Disposition of 2-sec-butyl-4,6-dinitrophenol (dinoseb) in pregnant mice. Fd Cosmet Toxical. 11:45–52.
- Goodwin J, Nath S, Engelborghs Y, Pountney DL. 2013. Raised calcium and oxidative stress cooperatively promote alpha-synuclein aggregate formation. Neurochem Int. 62:703–711.
- Guzman JN, Sánchez-Padilla J, Chan CS, Surmeier DJ. 2009. Robust pacemaking in substantia nigra dopaminergic neurons. J Neurosci. 29:11011–11019.
- Harvey DG, Bidstrup PL, Bonnell JAL. 1951. Poisoning by Dinitro-ortho-cresol: some observations on the effects of Dinitro-ortho-cresol administered by mouth to human volunteers. Br Med J. 2:13–16.
- Hatcher JM, Delea KC, Richardson JR, Pennell KD, Miller GW. 2008. Disruption of dopamine transport by DDT and its metabolites. Neurotoxicology. 29:682–690.
- Hernández-Romero MC, Delgado-Cortés MJ, Sarmiento M, de Pablos RM, Espinosa-Oliva AM, Argüelles S, Bández MJ, Villarán RF, Mauriño R, Santiago M, et al. 2012. Peripheral inflammation increases the deleterious effect of CNS inflammation on the nigrostriatal dopaminergic system. Neurotoxicology. 33:347–360.
- Heusinkveld HJ, Molendijk J, van den Berg M, Westerink RHS. 2013. Azole fungicides disturb intracellular Ca2+ in an additive manner in dopaminergic PC12 cells. Toxicol Sci. 134:374–381.
- Heusinkveld HJ, Thomas GO, Lamot I, van den Berg M, Kroese ABA, Westerink RHS. 2010. Dual actions of lindane (γ-hexachlorocyclohexane) on calcium homeostasis and exocytosis in rat PC12 cells. Toxicol Appl Pharmacol. 248:12–19.
- Heusinkveld HJ, Westerink RHS. 2011. Caveats and limitations of plate reader-based high-throughput kinetic measurements of intracellular calcium levels. Toxicol Appl Pharmacol. 255:1–8.
- Heusinkveld HJ, Westerink RHS. 2012. The organochlorine insecticides lindane, dieldrin and their binary mixture disturb calcium homeostasis in dopaminergic PC12 cells. Environ Sci Technol. 46:1842–1848.
- Heuts FJM. 1993. Biological Monitoring of DNOC in applicators after use as Haulmkiller in seed potatoes. Rotterdam ( unpublished report prepared for Elf Atochem Agri bv).
- Hughes AJ, Daniel SE, Kilford L, Lees AJ. 1992. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 55:181–184.
- Imtiaz M. 2012. Calcium oscillations and pacemaking. In: Islam MS, editor. Calcium signaling, Vol. 740. Netherlands: Springer; p. 511–520.
- Jimenez-Torres M, Campoy-Folgoso C, Canabate-Reche F, Rivas-Velasco A, Cerrillo-Garcia I, Mariscal-Arcas M, Olea-Serrano F. 2006. Organochlorine pesticides in serum and adipose tissue of pregnant women in Southern Spain giving birth by cesarean section. Sci Total Environ. 372:32–38.
- Koppen G, Covaci A, Van Cleuvenbergen R, Schepens P, Winneke G, Nelen V, van Larebeke N, Vlietinck R, Schoeters G. 2002. Persistent organochlorine pollutants in human serum of 50–65 years old women in the Flanders Environmental and Health Study (FLEHS). Part 1: concentrations and regional differences. Chemosphere. 48:811–825.
- Kortenkamp A. 2008. Low dose mixture effects of endocrine disrupters: implications for risk assessment and epidemiology. Int J Androl. 31:233–240.
- Langston JW. 1996. The etiology of Parkinson's disease with emphasis on the MPTP story. Neurology. 47:S153–160.
- Langston JW, Ballard P, Tetrud JW, Irwin I. 1983. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 219:979–980.
- Lees AJ, Hardy J, Revesz T. 2009. Parkinson's disease. The Lancet. 373:2055–2066.
- Lopez-Espinosa MJ, Lopez-Navarrete E, Rivas A, Fernandez MF, Nogueras M, Campoy C, Olea-Serrano F, Lardelli P, Olea N. 2008. Organochlorine pesticide exposure in children living in southern Spain. Environ Res. 106:1–6.
- Lucchini RG, Dorman DC, Elder A, Veronesi B. 2012. Neurological impacts from inhalation of pollutants and the nose– brain connection. Neurotoxicology. 33:838–841.
- Mattson MP. 2012. Parkinson's disease: don't mess with calcium. J Clin Invest. 122:1195–1198.
- McGeer PL, McGeer EG. 2004. Inflammation and neurodegeneration in Parkinson's disease. Parkinsonism Relat Disord. 10:S3–S7.
- Meijer M, Dingemans MM, van den Berg M, Westerink RH. 2014a. Inhibition of voltage-gated calcium channels as common mode of action for (mixtures of) distinct classes of insecticides. Toxicol Sci. 141:103–111.
- Meijer M, Hendriks HS, Heusinkveld HJ, Langeveld WT, Westerink RH. 2014b. Comparison of plate reader-based methods with fluorescence microscopy for measurements of intracellular calcium levels for the assessment of in vitro neurotoxicity. Neurotoxicology. 45C:31–37.
- Mosharov EV, Larsen KE, Kanter E, Phillips KA, Wilson K, Schmitz Y, Krantz DE, Kobayashi K, Edwards RH, Sulzer D. 2009. interplay between cytosolic dopamine, calcium, and α-synuclein causes selective death of substantia nigra neurons. Neuron. 62:218–229.
- Mostafalou S, Abdollahi M. 2013. Pesticides and human chronic diseases: evidences, mechanisms, and perspectives. Toxicol Appl Pharmacol. 268:157–177.
- Mustafa MD, Pathak R, Tripathi A, Ahmed R, Guleria K, Banerjee B. 2010. Maternal and cord blood levels of Aldrin and Dieldrin in Delhi population. Environ Monit Assess. 171:633–638.
- Nath S, Goodwin J, Engelborghs Y, Pountney DL. 2011. Raised calcium promotes α-synuclein aggregate formation. Mol Cellular Neurosci. 46:516–526.
- Neve KA, Seamans JK, Trantham-Davidson H. 2004. Dopamine receptor signalling. J Receptors Signal Transduction. 24:165–205.
- Nolan K, Kamrath J, Levitt J. 2012. Lindane toxicity: a comprehensive review of the medical literature. Pediatr Dermatol. 29:141–146.
- Nougadère A, Sirot V, Kadar A, Fastier A, Truchot E, Vergnet C, Hommet F, Baylé J, Gros P, Leblanc J-C. 2012. Total diet study on pesticide residues in France: levels in food as consumed and chronic dietary risk to consumers. Environ Int. 45:135–150.
- Oates L, Cohen M. 2011. Assessing diet as a modifiable risk factor for pesticide exposure. Int J Environ Res Public Health. 8:1792–1804.
- Oberdorster G, Sharp Z, Atudorei V, Elder A, Gelein R, Kreyling W, Cox C. 2004. Translocation of inhaled ultrafine particles to the brain. Inhal Toxicol. 16:437–445.
- Obermeier B, Daneman R, Ransohoff RM. 2013. Development, maintenance and disruption of the blood-brain barrier. Nat Med. 19:1584–1596.
- Obeso JA, Rodríguez-Oroz MC, Benitez-Temino B, Blesa FJ, Guridi J, Marin C, Rodriguez M. 2008. Functional organization of the basal ganglia: therapeutic implications for Parkinson's disease. Move Disord. 23:S548–559.
- Parkinson J. 2002. An essay on the shaking palsy. Neuropsychiatry Classics. 14:223–236.
- Pezzoli G, Cereda E. 2013. Exposure to pesticides or solvents and risk of Parkinson disease. Neurology. 80:2035–2041.
- Prachar V, Veningerová M, Uhnák J. 1993. Levels of polychlorinated biphenyls and some other organochlorine compounds in breast milk samples in Bratislava. Sci Total Environ. 134:237–242.
- Pravettoni E, Bacci A, Coco S, Forbicini P, Matteoli M, Verderio C. 2000. Different localizations and functions of L-type and N-type calcium channels during development of hippocampal neurons. Dev Biol. 227:581–594.
- Purisai MG, McCormack AL, Cumine S, Li J, Isla MZ, Di Monte DA. 2007. Microglial activation as a priming event leading to paraquat-induced dopaminergic cell degeneration. Neurobiol Dis. 25:392–400.
- Putzier I, Kullmann PHM, Horn JP, Levitan ES. 2009. Cav1.3 channel voltage dependence, Not Ca2+ selectivity, drives pacemaker activity and amplifies bursts in nigral dopamine neurons. J Neurosci. 29:15414–15419.
- Radad K, Rausch WD, Gille G. 2006. Rotenone induces cell death in primary dopaminergic culture by increasing ROS production and inhibiting mitochondrial respiration. Neurochem Int. 49:379–386.
- Rajput AH, Birdi S. 1997. Epidemiology of Parkinson's disease. Parkinsonism Relat Disord. 3:175–186.
- Ramwell CT, Johnson PD, Boxall ABA, Rimmer DA. 2005. Pesticide residues on the external surfaces of field crop sprayers: occupational exposure. Ann Occup Hyg. 49:345–350.
- Raymond-Delpech V, Matsuda K, Sattelle BM, Rauh JJ, Sattelle DB. 2005. Ion channels: molecular targets of neuroactive insecticides. Invertebrate Neurosci. 5:119–133.
- Rice ME, Patel JC, Cragg SJ. 2011. Dopamine release in the basal ganglia. Neuroscience. 198:112–137.
- Richardson JR, Caudle WM, Wang M, Dean ED, Pennell KD, Miller GW. 2006. Developmental exposure to the pesticide dieldrin alters the dopamine system and increases neurotoxicity in an animal model of Parkinson's disease. Faseb J. 20:1695–1697.
- Rivas A, Cerrillo I, Granada A, Mariscal-Arcas M, Olea-Serrano F. 2007. Pesticide exposure of two age groups of women and its relationship with their diet. Sci Total Environ. 382:14–21.
- Rodríguez J, Verkhratsky A. 2010. Neuroglial roots of neurodegenerative diseases? Mol Neurobiol. 43:87–96.
- Schapira AH, Jenner P. 2011. Etiology and pathogenesis of Parkinson's disease. Move Disord. 26:1049–1055.
- Schapira AHV. 2009. Neurobiology and treatment of Parkinson's disease. Trends Pharmacol Sci. 30:41–47.
- Sherer TB, Betarbet R, Kim J-H, Greenamyre JT. 2003a. Selective microglial activation in the rat rotenone model of Parkinson's disease. Neurosci Lett. 341:87–90.
- Sherer TB, Kim J-H, Betarbet R, Greenamyre JT. 2003b. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and α-synuclein aggregation. Exp Neurol. 179:9–16.
- Shimizu K, Ohtaki K, Matsubara K, Aoyama K, Uezono T, Saito O, Suno M, Ogawa K, Hayase N, Kimura K, Shiono H. 2001. Carrier-mediated processes in blood–brain barrier penetration and neural uptake of paraquat. Brain Res. 906:135–142.
- Stevens DA. 2012. Advances in systemic antifungal therapy. Clin Dermatol. 30:657–661.
- Subramanian A, Ohtake M, Kunisue T, Tanabe S. 2007. High levels of organochlorines in mothers’ milk from Chennai (Madras) city, India. Chemosphere. 68:928–939.
- Surmeier DJ, Guzman JN, Sanchez-Padilla J, Schumacker PT. 2011. The role of calcium and mitochondrial oxidant stress in the loss of substantia nigra pars compacta dopaminergic neurons in Parkinson's disease. Neuroscience. 198:221–231.
- Tansey MG, Goldberg MS. 2010. Neuroinflammation in Parkinson's disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis. 37:510–518.
- Taylor JM, Main BS, Crack PJ. 2013. Neuroinflammation and oxidative stress: co-conspirators in the pathology of Parkinson's disease. Neurochem Int. 62:803–819.
- Thomas GO, Wilkinson M, Hodson S, Jones KC. 2006. Organohalogen chemicals in human blood from the United Kingdom. Environ Pollut. 141:30–41.
- Trejo-Acevedo A, Diaz-Barriga F, Carrizales L, Dominguez G, Costilla R, Ize-Lema I, Yarto-Ramirez M, Gavilan-Garcia A, Jesus Mejia-Saavedra J, Perez-Maldonado IN. 2009. Exposure assessment of persistent organic pollutants and metals in Mexican children. Chemosphere. 74:974–980.
- Turski L, Bressler K, Rettig KJ, Loschmann PA, Wachtel H. 1991. Protection of substantia nigra from MPP +neurotoxicity by N-methyl-D-aspartate antagonists. Nature. 349:414–418.
- Vale C, Fonfra E, Bujons J, Messeguer A, Rodriguez-Farr E, Sunol C. 2003. The organochlorine pesticides gamma-hexachlorocyclohexane (lindane), alpha -endosulfan and dieldrin differentially interact with GABAA and glycine-gated chloride channels in primary cultures of cerebellar granule cells. Neuroscience. 117:397–403.
- van der Mark M, Brouwer M, Kromhout H, Nijssen P, Huss A, Vermeulen R. 2011. Is pesticide use related to parkinson disease? some clues to heterogeneity in study results. Environ Health Perspect. 120:340–347.
- van Noort H, Mandema E, Juul Christensen EK, Huizinga T. 1960. 4,6-Dinitro-o-cresol poisoning caused by sprays. Nederlands Tijdschrift voor Geneeskunde. 104:676–684.
- Venda LL, Cragg SJ, Buchman VL, Wade-Martins R. 2010. α-Synuclein and dopamine at the crossroads of Parkinson's disease. Trends Neurosci. 33:559–568.
- Walsh GM, Fink GB. 1972. Comparative toxicity and distribution of endrin and dieldrin after intravenous administration in mice. Toxicol Appl Pharmacol. 23:408–416.
- Weisskopf MG, Knekt P, O’Reilly EJ, Lyytinen J, Reunanen A, Laden F, Altshul L, Ascherio A. 2010. Persistent organochlorine pesticides in serum and risk of Parkinson disease. Neurology. 74:1055–1061.
- Westerink RH. 2013. Do we really want to REACH out to in vitro? Neurotoxicology. 39:169–172.
- Westerink RH, Hondebrink L. 2010. Are high-throughput measurements of intracellular calcium using plate-readers sufficiently accurate and reliable? Toxicol Appl Pharmacol. 249:247–248; author reply 249-250.
- Westerink RHS. 2006. Targeting Exocytosis: ins and outs of the modulation of quantal dopamine release. CNS Neurol Disord - Drug Targets. 5:57–77.
- Whitacre DM, Ware GW, Whitacre DM, Ware GW. 2004. The pesticide book. 6th ed. Willoughby (OH): Meister-Pro Information Resources (Meister Media Worldwide).
- Wilkaniec A, Strosznajder JB, Adamczyk A. 2013. Toxicity of extracellular secreted alpha-synuclein: Its role in nitrosative stress and neurodegeneration. Neurochem Int. 62:776–783.
- Wu J, Chan P, Schroeder KM, Ellsworth K, Partridge LDL. 2002. 1-Methyl-4-phenylpridinium (MPP+)-induced functional run-down of GABAA receptor-mediated currents in acutely dissociated dopaminergic neurons. J Neurochem. 83:87–99.
- Xu X, Dailey AB, Talbott EO, Ilacqua VA, Kearney G, Asal NR. 2010. Associations of serum concentrations of organochlorine pesticides with breast cancer and prostate cancer in U.S. adults. Environ Health Perspect. 118:60–66.
- Yasuda T, Nakata Y, Mochizuki H. 2013. α-Synuclein and neuronal cell death. Mol Neurobiol. 47:466–483.