
Abstract
Background
Canine circovirus is reported in dogs in many countries, including the USA, China and Thailand. It has been detected in healthy dogs and dogs with diarrhea, hemorrhagic gastroenteritis, and vasculitis. It comprises five genotypes and is frequently found as a coinfection with canine parvovirus-2 (CPV-2).
Aim
To characterize canine circovirus genotypes co-circulating with CPV-2 in Vietnam.
Method
PCR assessment of 81 CPV-2-positive fecal samples from Vietnamese diarrheic dogs up to seven months of age for other viral enteric pathogens, including canine bocavirus, canine adenovirus, paramyxovirus, canine coronavirus, porcine circovirus-3 and canine circovirus. In addition, eight selected full genome sequences of Vietnamese canine circovirus were analyzed and used for phylogeny.
Results
In total 19.8% of samples were found to be positive for canine circovirus. Phylogeny revealed that the Vietnamese canine circovirus strains were clustered in two different genotypes (genotype-1 and -3). The genetic diversity among Vietnamese canine circovirus was 86.0–87.2%. The nucleotide discrepancy among both genotypes altered the deduced amino acid sequence in 14 and ten residues of the replicase and capsid proteins, respectively. Genetic recombination analysis revealed that the Vietnamese canine circovirus-6 strain has the American and Chinese canine circovirus as its major and minor parents, respectively. Only a single dog revealed triple detections of CPV-2c, Canine circovirus and canine adenovirus (1.2%).
Conclusion
The co-circulation of two different genotypes of canine circovirus and CPV-2c in dogs in Vietnam has been illustrated.
Clinical relevance
The mortality rate with CPV-2 only (22%) doubled in dogs with canine circovirus and CPV-2 co-infection.
1. Introduction
Canine circovirus (CanineCV) is a member of the genus Circovirus, family Circoviridae, along with other mammal and avian circoviruses that have been found in humans, pigs, ducks, geese and pigeons (Walker et al. Citation2020). In non-human mammals, the porcine circovirus (PCV) infection in pigs causes an immunosuppressive condition, particularly PCV type 2 (PCV-2), and results in devastating syndromes for the swine industry (Segales et al. Citation2005). The CanineCV was first detected in serum samples collected from dogs in the USA (Kapoor et al. Citation2012). Subsequently, the virus has been reported in dogs in Italy, Germany, Thailand, Taiwan, Argentina, China, Brazil, and Colombia (Li et al. Citation2013; Decaro et al. Citation2014; Hsu et al. Citation2016; Weber et al. Citation2018; Piewbang et al. Citation2018b; Kotsias et al. Citation2019; Sun et al. Citation2019; Giraldo-Ramirez et al. Citation2020). CanineCV is considered to be the second, non-human mammalian circovirus. Recently, CanineCV has been divided into five genotypes (CanineCV-1, -2, -3, -4, and -5) (Urbani et al., Citation2021). The CanineCV-1 genotype has been identified in dogs, wolves and a badger from the USA, Europe and China, which corresponds to the Cosmopolitan genotype proposed by Giraldo-Ramirez et al. (Citation2020). The CanineCV-2, -3, and -4 genotypes are reported in Asia, including China and Thailand (Niu et al. Citation2020; Urbani et al., Citation2021), and this classification agrees with the China, Asia-I, and Asia-II genotypes reported by Giraldo-Ramirez et al. (Citation2020). Additionally, the CanineCV-5 genotype has been detected only in the arctic foxes and red foxes in Norway (Urbani et al., Citation2021).
CanineCV is an icosahedral, non-enveloped virus containing a small monomeric single circular strand DNA of 2,063 nucleotides in length. Its genome is comprised of two major putative open reading frames (ORFs) that encode for replicase (Rep) and capsid (Cap) protein. The Rep protein is comprised of 303 amino acids and is essential for viral replication (Cheung Citation2003), while the Cap protein is comprised of 270 amino acids and plays the role of a structural protein in the virus (Kapoor et al. Citation2012; Li et al. Citation2013). Among the two major ORFs, there is a non-coding region containing nine nucleotides (TAGTATTAC), which is a thermodynamically stable stem-loop that acts during the initiation of rolling-circle replication (Kapoor et al. Citation2012). Recently, an additional ORF, ORF-3, was identified in the antisense of ORF1 of a Thai strain of CanineCV, but its function has yet to be elucidated (Piewbang et al. Citation2018b).
CanineCV has been detected in dogs with various pathological conditions, including vasculitis, gastroenteritis, and infectious respiratory disease complex (Li et al. Citation2013; Decaro et al. Citation2014; Anderson et al. Citation2017; Piewbang et al. Citation2018b). Since viral isolation in vitro has not been successful, knowledge on the pathogenesis of CanineCV infection in dogs remains poorly understood. However, CanineCV has been considered as a cause of enteritis and is frequently identified in dual infections attributed to canine parvovirus-2 (CPV-2) (Hsu et al. Citation2016; Thaiwong et al. Citation2016; Giraldo-Ramirez et al. Citation2020), leading to an increased mortality rate (Anderson et al. Citation2017). Therefore, it has been suggested that CanineCV likely acts in synergy with other infectious agents in the development of disease. A few studies in dogs have reported coinfections of CanineCV with CPV-2 and that these coinfected dogs developed disease with severe clinical signs (Thaiwong et al. Citation2016; Kotsias et al. Citation2019).
In Vietnam, outbreaks of CPV-2 occur frequently and cause massive numbers of deaths in dogs, which may be caused by the humid subtropical climate and poor vaccination programs. However, the study of CanineCV in Vietnam is limited. This study, therefore, investigated the incidence of CanineCV in CPV-2-infected dogs and characterized the molecular genetics of Vietnamese CanineCV (CanineCV-VN) strains using phylogenetic analysis and recombination analysis.
2. Materials and methods
2.1. Samples, viral nucleic acid extraction and CPV-2 detection
Eighty-one diarrheic dogs were presented to veterinary hospitals for medical treatment. Fecal swabs were collected from all dogs exhibiting CPV-positive detection upon immunosorbent assay (Nguyen Manh et al. Citation2021). Tested dogs resided in Hanoi (n = 41), Da Nang (n = 16), and Ho Chi Minh (n = 24), Vietnam, during September-December 2017. The swabs were immersed in sterile phosphate buffer saline (pH 7.4) and preserved at −80 °C until assayed. All procedures were approved by the Chulalongkorn University Animal Care and Use Committee (No. 2031005). Samples were subjected to viral nucleic acid extraction using a commercial extraction kit (Geneaid Biotech, New Taipei city, Taiwan) and further assayed via polymerase chain reaction (PCR) to confirm the presence of CPV-2, as previously described (Nguyen Manh et al. Citation2021). These positive amplicons were then subjected to genomic sequencing to elucidate the original genotype of the obtained CPV-2. Essential data on the animals, including their sex, age, breed, vaccination history, and clinical outcome were also documented ().
Table 1. Detection of other enteric viruses as a coinfection with canine parvovirus-2 (CPV-2) in 81 Vietnamese dogs.
2.2. PCR amplification and full-length genome sequencing of CanineCV
The primer pairs, including CanineCV-605F/-1041R and CanineCV-1022F/-1538R, were retrieved from a previous study (Piewbang et al. Citation2018b), while CanineCV-1448F/-110R and CanineCV-2014F/-776R were designed based on the alignment of the available CanineCV sequences from the GenBank database. These were used to detect and amplify the complete genome of the CanineCV strains obtained in this study (). The PCR was performed using Gotaq Green Master mix (Promega, USA) and amplified the target amplicons according to the specific primer pairs. The thermal cycling was performed at an initial temperature of 94 °C for 7 min, followed by 35 cycles of 94 °C for 30 sec, 55 °C for 5 min and 72 °C for 1 min, and then a final 72 °C for 7 min. The PCR products were resolved on a 1.0% (w/v) agarose gel containing 0.5% (v/v) ethidium bromide in-gel staining and visualized under a UV transilluminator. All PCR products were purified using NucleoSpin Extract II (Macherey-Nagel, Düren, Germany) and submitted for bi-directional Sanger’s sequencing (Macrogen, Seoul, Korea), to confirm the specificity. Selected positive samples were further characterized and sequenced to obtain the full-length genome for subsequent analysis.
Table 2. Nucleotide composition of Vietnamese Canine circovirus (CanineCV-VN) strains.
Other canine viral enteric pathogens, including canine bocavirus (CBoV), canine adenovirus (CAdV), paramyxovirus (PMX), canine coronavirus (CCoV) and porcine circovirus-3 (PCV-3), previously reported circovirus in dogs, were also identified using specific primers and PCR programs, as previously reported (Posuwan et al. Citation2010; Piewbang et al. Citation2017; Zhang et al. Citation2018; Piewbang et al., Citation2018a).
2.3. Genetic characterization and phylogenetic and amino acid analyses
The derived full-length genome sequences were aligned using the Multiple Alignment algorithm in the Fast Fourier Transform (MAFFT) program version 7 (https://mafft.cbrc.jp). They were then compared to the reference CanineCV sequence (NC_020904) and those CanineCV sequences available in the GenBank database. These alignments were then further used for nucleotide and deduced amino acid sequence analysis using the BioEdit software package version 7.2 (http://www.mbio.ncsu.edu). The full-length genome sequences and individual Rep and Cap gene sequences of the CanineCV-VN strains of this study were then subjected to further genetic analyses in order to construct maximum likelihood phylogenetic trees. A dataset of 72 full-genome CanineCV sequences, originally derived from domestic dogs, wolves, foxes and a badger between 1996 and 2020 and available in the GenBank were used to accomplish this analysis. The best-fit model of substitution (TN93 + I + G) was determined using a program implemented in the MEGA 7.0 software (https://www.megasoftware.net/). Phylogenies were constructed via maximum likelihood (ML) methods with the bootstrapping (BS) of 1,000 replicates and accepting significance at BS values > 70%. Prediction of potential N-linked glycosylation was performed using the NetNglyc 1.0 (http://www.cbs.dtu.dk/services/NetNGlyc/) (Gupta and Brunak, Citation2002).
2.4. Recombination analysis
Recombination analysis was implemented using two independent programs, The Recombination Detection Program (RDP) and Simplot analysis, to define the potential natural genetic recombination events in the evolution of the CanineCV-VNs. Initially, the possibility of genetic recombination in CanineCV-VN was statistically analyzed using GENECONV, BootScan, MaxChi, Chimaera, SiScan and 3Seq with the default settings for all parameters, which were embedded in RDP package version 4.0 (http://web.cbio.uct.ac.za/). A potential positive recombination was recorded when at least four of the six methods showed a breakpoint signal with p-values of < 0.01. The recombinant breakpoint was then further confirmed using bootscanning and a similarity plot, implanted in the Simplot software package v. Beta 4.94 (Piewbang et al. Citation2018b).
3. Results
3.1. Detection of CanineCV in CPV-2-positive Vietnamese dogs
Among 81 tested samples, 80 (98.8%) samples were found to be positive for CPV-2c, while a single sample (1.2%) was positive for CPV-2a (). All 81 CPV-2-infected samples were then examined for CanineCV genomic detection, revealing 16 positive samples (19.8%) via the presence of the target PCR amplicons of CanineCV (). Of note, CanineCV was identified in the fecal samples of all dogs positive for the CPV-2c genotype. The rate of CanineCV detection was highest in the samples from Hanoi (24.4%, 10/41), followed by Ho Chi Minh (16.7%, 4/24), and Da Nang (12.5%, 2/16). Other canine pathogens were simultaneously detected, including CBoV (6.2%, 5/81), CAdV (3.7%, 3/81), and PMX (2.5%, 2/81). However, there were no detected cases of CCoV and PCV-3 coinfection in all the tested dogs ().
Dogs in this study that showed coinfection with CanineCV and CPV-2c were purebred (five Poodles, three Pomeranians, two Pugs, two Chihuahuas, two Bulldogs, one Malinois, and one Shih Tzu). The dogs’ ages ranged from 2 to 7 months old, with the highest prevalence at 3 months (37.5%, 6/16). Of note, only one dog (No.2 from Hanoi) revealed a triple detection of CPV-2c, CanineCV and CAdV (1.2%). The clinical signs of all dogs varied from mild to severe, including fever, depression, anorexia, vomiting, and diarrhea (data not shown). Sixty-one (75.3%) dogs were vaccinated for CPV-2, while other 41 (19.8%) and four (4.9%) dogs were unvaccinated or had no data available, respectively. The clinical outcome revealed whether dogs were dead or alive. The mortality rate was shown to be 22.2% (12/54) when dogs were infected with only CPV-2, while this percentage was double in dogs with CanineCV and CPV-2 co-infection (43.8%, 7/16) ( and ).
3.2. Genome organization and phylogenetic analysis of Vietnamese CanineCV
Eight out of 16 CanineCV-positive samples were further PCR amplified and sequenced using the respective primers () to obtain the full-length genome sequence of CanineCV. The obtained genome sequences of the CanineCV-VN strains were submitted to GenBank (MT740194-MT740201). A sequence analysis of these CanineCV-VN strains revealed a mean nucleotide composition of 52.14 ± 0.26% GC and 47.86 ± 0.25% AT, which was similar to the nucleotide composition of the reference CanineCV UCD1-1698 strain (NC_020904) from the USA (). The Rep gene located at nt 1-912 and encoded for 304 amino acids, while the Cap gene was placed in the antisense nucleotide sequences from nt 1,116–1,928 and encoded for 271 amino acids. The thermodynamical stem-loop sequence (TAGTATTAC) was located at nt 2,012–2,020.
The ML phylogenetic trees were constructed based on the eight full-genome CanineCV-VN sequences derived from this study and 72 sequences published from 1996 to 2020 and retrieved from GenBank. A total of 80 CanineCV strains were segregated into five clades, corresponding with five genotypes (Urbani et al., Citation2021). The CanineCV-VN-3,- 4, -5, and -8 strains were clustered together with the CanineCV-1 genotype strain, which was comprised of strains from Italy, Germany, Argentina, Colombia, Brazil, the USA and China. In contrast, the CanineCV-VN-1, -2, -6, and -7 strains were grouped in the CanineCV-3 genotype, along with other strains from China (). The phylogenetic analyses based on the individual Rep and Cap genes also showed similar ML phylogenetic trees results as compared to the phylogeny based on the full genomes ().
Figure 1. Phylogenetic tree (ML) with time, based on 80 full-length genomes of Canine circovirus (CanineCV) collected during 1996 to 2020 (▼ CanineCV-VN strains in this study).
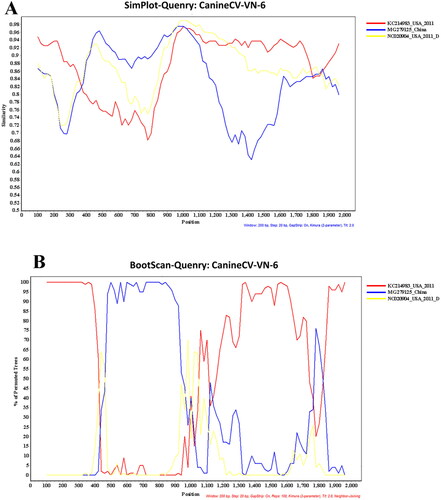
Figure 2. Potential recombination events of Vietnamese Canine circovirus (CanineCV-VN-6 as query) strains compared to published CanineCV strains; (A) Similarity plot, and (B) Bootscan analysis.
3.3. Genetic and amino acid characterizations of different CanineCV genotypes in Vietnam
A genetic analysis of eight full-length genomes revealed that CanineCV-VN strains shared 86.0–99.8% similarity. Meanwhile, the nucleotide similarities among strains of the Vietnamese CanineCV-1 genotype (CanineCV-VN-3, -4, -5, and -8) and Vietnamese CanineCV-3 genotype (CanineCV-VN-1, -2, -6, and -7) were 95.2–98.0% and 98.9–99.8%, respectively. However, the nucleotide comparison between both Vietnamese genotypes was 86.0–87.2% ( and ). In addition, the nucleotide similarity analysis among genotypes revealed 83.8–85.9% and 85.3–86.8% similarity for the Rep and Cap genes, respectively. The deduced amino acid similarities between the Vietnamese CanineCV-1 and CanineCV-3 genotypes were 92.7–97.3% for the Rep protein and 92.9–95.1% for the Cap protein ( and ).
Table 3. Similarity percentage of nucleotides and deduced amino acids of Vietnamese Canine circovirus (CanineCV-VN) and reference strains.
Between the Vietnamese CanineCV-1 and CanineCV-3 genotypes, amino acid variations were clearly observed in 14 amino acid positions of the Rep protein and ten amino acid positions of the Cap protein (). Among these positions, almost all the amino acid sequences of the Rep and Cap proteins of the Vietnamese CanineCV-1 genotype were identical to the reference UCD1-1698 strain (NC_020904), except for amino acids at 269 (Rep) and 74 (Cap). A further analysis illustrated the unique amino acid variations in both the Rep and Cap proteins of each CanineCV genotype, as shown in and . In addition, a single potential N-glycosylation site at position N134AT was found in the Cap protein of seven of the CanineCV-VN strains, but only one strain (CanineCV-VN-7) presented H134AT. However, no N-glycosylation site was found in the Rep protein.
Table 4. Amino acidTable Footnotea variations in the Replicase (Rep) and Capsid (Cap) proteins of Vietnamese canine circovirus (CanineCV-VN) strains.
Table 5. Relevant amino acidTable Footnotea changes in the Replicase (Rep) and Capsid (Cap) protein sequence in each Canine circovirus (CanineCV) genotypes.
3.4. Recombination analysis
Recombination analysis was applied to all eight CanineCV-VN strains obtained in this study using RDP4 software, including RDP, GENECONV, Bootscan, Maxchi, Chimaera, SiScan, 3Seg and LARD. A potential recombination breakpoint was found in the Vietnamese CanineCV-1 genotype (CanineCV-VN-3, -4, -5, and -8). However, the potential recombination signal was only confirmed in the CanineCV-VN-6 strain (Vietnamese CanineCV-3 genotype), located at nucleotide position 420–1020. The recombinant CanineCV-VN-6 strain had an American CanineCV (KC241983, as an outgroup strain) and a Chinese CanineCV (MG279125, CanineCV-2 genotype) as its major and minor parents, respectively ().
4. Discussion
This study reports the first identification of CanineCV in Vietnam and provides evidence of CanineCV circulation in Vietnamese dogs. The CanineCV genome was detected in CPV-2-positive fecal samples at an incidence of 19.8%, which is higher than those reported in Germany (13.0%) and South America (16.6%) (Anderson et al. Citation2017; Giraldo-Ramirez et al. Citation2020). The incidence of CanineCV detected from this study varied in the Northern, Central and Southern areas of Vietnam. The fecal samples collected in Hanoi (Northern Vietnam) showed the highest rate of CanineCV at about 24.4%, while those samples from Da Nang (Central Vietnam) and Ho Chi Minh (Southern Vietnam) were about 12.5% and 16.7%, respectively. Since this study did not investigate the presence of CanineCV in either healthy or CPV-2-negative dogs and the number of samples has been limited, the actual prevalence of CanineCV in Vietnam could not be ascertained. Further investigation into these groups is required to understand the epidemiology of CanineCV in Vietnam.
The pathogenesis of CanineCV-associated disease and the potential synergistic effects of CanineCV and other pathogens have not been clarified. However, several studies have indicated that CanineCV may act in synergy with other infectious agents in the development of a disease (Thaiwong et al. Citation2016; Zaccaria et al. Citation2016; Kotsias et al. Citation2019). In this study, CanineCV was co-detected with CPV-2c in enteritis-suffering dogs, which is in agreement with the previous findings (Kotsias et al. Citation2019; Giraldo-Ramirez et al. Citation2020). Since CPV-2c has been reported as a predominant genotype detected in Vietnam (Hoang et al. Citation2019), which is in agreement with our study, co-infection with CanineCV may play a significant role as a negative co-factor in disease outcomes in dogs with CPV-2 infection (Anderson et al. Citation2017). This is shown in our study as a higher mortality rate for dogs co-infected with CanineCV and CPV-2 (43.8%) than for dogs having a single CPV-2 infection (22.2%). However, a recent report indicated that the CanineCV may play a role as a primary pathogen that causes acute hemorrhagic diarrheic disease in dogs (Anderson et al. Citation2017). Thus, the definitive role of CanineCV-associated diseases should be explored in further studies.
Here, we described the characteristics of the full-length genomes and deduced amino acids of the eight CanineCV-VN strains in order to add information on the geographical extent of the virus. The viral genome encompasses two ORFs encoding for the Rep and Cap proteins, similar to a previous report (Kapoor et al. Citation2012). However, genetic diversity among the CanineCV-VN genotypes was detected. The complete genome sequences of the Vietnamese CanineCV-1 genotype showed a close similarity (94.4%) to the reference strain, but the other genotype had a lower similarity (88.3%). The further surveillance of CanineCV genotypes is vital in understanding viral molecular genetics.
Previous studies on PCV-2 have suggested that amino acid mutations in the Rep protein may affect viral replication ability, while amino acid mutations in the Cap protein may alter viral antigenicity (Lekcharoensuk et al. Citation2004; Cheung Citation2012). Changes in several amino acids in PCV-3 are related to viral evolutionary dynamics (Sun et al. Citation2018). However, knowledge of mutations in CanineCV is limited. Selective pressure analysis on the PCV-2 genome has been demonstrated to be the factor driving mutations in the Cap gene, resulting in the virus escaping the host’s immune response in pigs (Franzo et al. Citation2016). However, the factor driving the mutation in CanineCV has not been investigated, but evidence for positive selection in both the Cap and Rep proteins of CanineCV has been reported (Giraldo-Ramirez et al. Citation2020). Thus, further study is still necessary to understand the evolutionary pattern of this virus.
The ML phylogenetic analysis, based on the full-length genome and the individual Rep and Cap genes, divided the CanineCV strains into five clades corresponding to five genotypes (CanineCV-1 to -5) (Giraldo-Ramirez et al. Citation2020). Notably, the CanineCV-1 genotype was mainly detected in Europe (Italy and Germany) and the Americas (USA, Brazil, Colombia and Argentina), except for one Chinese sequence (MG266899) (Niu et al. Citation2020) and four Vietnamese sequences (MT740198-MT740201) from this study. The CanineCV-2 and -4 genotypes were distributed only in China, while the CanineCV-3 genotype was detected in both China and Vietnam, representing four sequences (MT740194-MT740197) from this report. Of note, the CanineCV-5 genotype is found only in arctic foxes (Vulpes lagopus) and red foxes (Vulpes vulpes) in Norway and the UK (Bexton et al. Citation2015; Urbani et al., Citation2021). Interestingly, each genotype presented the typical mutation in both the Rep and Cap proteins. Since the geographic distribution pattern has been well-described in canine distemper virus (CDV; a fatal Morbillivirus in dogs), this indicates that the origin and global evolution of CDV derives from specific mutations of the ancestral strain (Panzera et al. Citation2015). Therefore, further study is required to verify this observation in CanineCV, as to whether any mutations in the ancestral sequence (KC241983 as an outgroup in the phylogeny) play a role in the global evolution of CanineCV. In addition, this study confirms that the CanineCV-1 and -3 genotypes are now circulating in CPV-2 enteritis-infected dogs in Vietnam, and provides evidence for the distribution of the CanineCV-1 genotype in Asia.
Recombination events have previously been identified in CanineCV in the Rep gene and other parts of the genome (Piewbang et al. Citation2018b; Sun et al. Citation2019). In this study, a recombination event was found in the CanineCV-VN-6 strain at nucleotide position 420–1020 with the major parent sequence derived from an ancestral American strain (KC241983) and a minor parent sequence derived from the Chinese strain (MG279125; CanineCV-2 genotype). In the results of the phylogenetic analysis on the Cap gene, we found that the ancestral American strain KC214983 has been clustered within the most CanineCV sequences detected in China and Vietnam, suggesting that they shared evolutionary patterns among sequences. This result implies that the American strain may serve as the common ancestor of the strains detected in Vietnam. Further studies on evolutionary analysis and retrospective, larger-scaled investigations in the form of genetic analysis are, therefore, needed to elucidate the intermediate CanineCV strains that may have closer relationships to the American ancestor. This viral recombination is important because it has been associated with viral host range expansion, the emergence of novel progeny viruses that express new antigenic and serological characteristic, and increases in virulence and pathogenesis (Pérez-Losada et al. Citation2015). Our findings suggest that various genotypes of CanineCV have been prevalent in Vietnam and a novel recombinant CanineCV-CN-6, which belonged to the CanineCV-3 genotype, is apparent. This recombination event should be strictly monitored because the different genotypes of CanineCV have simultaneously circulated and been co-detected with other pathogens in the same host.
In conclusion, this is the first detection and characterization of the full-length genome of CanineCV in Vietnam. This study provides further evidence of CanineCV as a co-infectious agent for enteritis in dogs. Phylogenetic analysis revealed that CanineCV was classified into five genotypes marked by typical mutations of amino acid and specific geographic distribution. Further studies are vital in understanding the genotypes of CanineCV.
Supplemental Material
Download MS Word (5.8 MB)Supplemental Material
Download MS Word (588 KB)Acknowledgments
Nguyen Manh Tuong received grant funding via the Asean scholarship from Chulalongkorn University. Chutchai Piewbang received a grant from the Ratchadaphisek Somphot Fund for Postdoctoral Fellowship, Chulalongkorn University. This study was supported by the 90th Anniversary of Chulalongkorn University Fund (Ratchadaphiseksomphot Endowment Fund) and Chulalongkorn Academic Advancement Into Its 2nd Century Project, Faculty of Veterinary Science, Chulalongkorn University.
Disclosure statement
The authors declare no conflict of interests.
References
- Anderson A, Hartmann K, Leutenegger CM, Proksch AL, Mueller RS, Unterer S. 2017. Role of canine circovirus in dogs with acute haemorrhagic diarrhoea. Vet Rec. 180(22):542.
- Bexton S, Wiersma LC, Getu S, van Run PR, Verjans GMGM, Schipper D, Schapendonk CME, Bodewes R, Oldroyd L, Haagmans BL, et al. 2015. Detection of circovirus in foxes with meningoencephalitis, United Kingdom, 2009-2013. Emerg Infect Dis. 21(7):1205–1208.
- Cheung AK. 2003. Transcriptional analysis of porcine circovirus type 2. Virology. 305(1):168–180.
- Cheung AK. 2012. Porcine circovirus: transcription and DNA replication. Virus Res. 164(1–2):46–53.
- Decaro N, Martella V, Desario C, Lanave G, Circella E, Cavalli A, Elia G, Camero M, Buonavoglia C. 2014. Genomic characterization of a circovirus associated with fatal hemorrhagic enteritis in dog, Italy. PLoS One. 9(8):e105909.
- Franzo G, Tucciarone CM, Cecchinato M, Drigo M. 2016. Porcine circovirus type 2 (PCV2) evolution before and after the vaccination introduction: a large scale epidemiological study. Sci Rep. 6:39458.
- Giraldo-Ramirez S, Rendon-Marin S, Vargas-Bermudez DS, Jaime J, Ruiz-Saenz J. 2020. First detection and full genomic analysis of canine circovirus in CPV-2 infected dogs in Colombia, South America. Sci Rep. 10(1):17579.
- Gupta R, Brunak JE. 2004. Prediction of N-glycosylation sites in human proteins.
- Gupta R, Brunak S. 2002. Prediction of glycosylation across the human proteome and the correlation to protein function. Pac Symp Biocomput. 7:310–322.
- Hoang M, Lin WH, Le VP, Nga BTT, Chiou MT, Lin CN. 2019. Molecular epidemiology of canine parvovirus type 2 in Vietnam from November 2016 to February 2018. Virol J. 16(1):52.
- Hsu HS, Lin TH, Wu HY, Lin LS, Chung CS, Chiou MT, Lin CN. 2016. High detection rate of dog circovirus in diarrheal dogs. BMC Vet Res. 12(1).
- Kapoor A, Dubovi EJ, Henriquez-Rivera JA, Lipkin WI. 2012. Complete genome sequence of the first canine circovirus. J Virol. 86(12):7018.
- Kotsias F, Bucafusco D, Nunez DA, Lago Borisovsky LA, Rodriguez M, Bratanich AC. 2019. Genomic characterization of canine circovirus associated with fatal disease in dogs in South America. PLoS One. 14(6):e0218735.
- Lekcharoensuk P, Morozov I, Paul PS, Thangthumniyom N, Wajjawalku W, Meng XJ. 2004. Epitope mapping of the major capsid protein of type 2 porcine circovirus (PCV2) by using chimeric PCV1 and PCV2. J Virol. 78(15):8135–8145.
- Li L, McGraw S, Zhu K, Leutenegger CM, Marks SL, Kubiski S, Gaffney P, Dela Cruz Jr FN, Wang C, Delwart E, et al. 2013. Circovirus in tissues of dogs with vasculitis and hemorrhage. Emerg Infect Dis. 19(4):534–541.
- Nguyen Manh T, Piewbang C, Rungsipipat A, Techangamsuwan S. 2021. Molecular and phylogenetic analysis of Vietnamese canine parvovirus 2C originated from dogs reveals a new Asia-IV clade. Transbound Emerg Dis. 68(3):1445–1453.
- Niu L, Wang Z, Zhao L, Wang Y, Cui X, Shi Y, Chen H, Ge J. 2020. Detection and molecular characterization of canine circovirus circulating in northeastern China during 2014-2016. Arch Virol. 165(1):137–143.
- Panzera Y, Sarute N, Iraola G, Hernandez M, Perez R. 2015. Molecular phylogeography of canine distemper virus: geographic origin and global spreading. Mol Phylogenet Evol. 92:147–154.
- Pérez-Losada M, Arenas M, Galán JC, Palero F, González-Candelas F. 2015. Recombination in viruses: mechanisms, methods of study, and evolutionary consequences. Infect Genet Evol. 30:296–307.
- Piewbang C, Jo WK, Puff C, Ludlow M, van der Vries E, Banlunara W, Rungsipipat A, Kruppa J, Jung K, Techangamsuwan S, et al. 2018a. Canine Bocavirus type 2 infection associated with intestinal lesions. Vet Pathol. 55(3):434–441.
- Piewbang C, Jo WK, Puff C, van der Vries E, Kesdangsakonwut S, Rungsipipat A, Kruppa J, Jung K, Baumgärtner W, Techangamsuwan S, et al. 2018b. Novel canine circovirus strains from Thailand: Evidence for genetic recombination. Sci Rep. :8:7524.
- Piewbang C, Rungsipipat A, Poovorawan Y, Techangamsuwan S. 2017. Development and application of multiplex PCR assays for detection of virus-induced respiratory disease complex in dogs. J Vet Med Sci. 78(12):1847–1854.
- Posuwan N, Payungporn S, Thontiravong A, Kitikoon P, Amonsin A, Poovorawan Y. 2010. Prevalence of respiratory viruses isolated from dogs in Thailand during 2008-2009. Asian Biomed. 4(4):563–569.
- Segales J, Allan GM, Domingo M. 2005. Porcine circovirus diseases. Anim Health Res Rev. 6(2):119–142.
- Sun J, Wei L, Lu Z, Mi S, Bao F, Guo H, Tu C, Zhu Y, Gong W. 2018. Retrospective study of porcine circovirus 3 infection in China. Transbound Emerg Dis. 65(3):607–613.
- Sun W, Zhang H, Zheng M, Cao H, Lu H, Zhao G, Xie C, Cao L, Wei X, Bi J, et al. 2019. The detection of canine circovirus in Guangxi, China. Virus Res. 259:85–89.
- Thaiwong T, Wise AG, Maes RK, Mullaney T, Kiupel M. 2016. Canine circovirus 1 (CaCV-1) and canine parvovirus 2 (CPV-2): recurrent dual infections in a papillon breeding colony. Vet Pathol. 53(6):1204–1209.
- Urbani L, Tryland M, Ehrich D, Fuglei E, Battilani M, Balboni A. 2021. Ancient origin and genetic segregation of canine circovirus infecting arctic foxes (Vulpes lagopus) in Svalbard and red foxes (Vulpes vulpes) in Northern Norway. Transbound Emerg Dis. 68(3):1283–1293.
- Walker PJ, Siddell SG, Lefkowitz EJ, Mushegian AR, Adriaenssens EM, Dempsey DM, Dutilh BE, Harrach B, Harrison RL, Hendrickson RC, et al. 2020. Changes to virus taxonomy and the Statutes ratified by the International Committee on Taxonomy of Viruses (2020). Arch Virol. 165(11):2737–2748.
- Weber MN, Cibulski SP, Olegário JC, da Silva MS, Puhl DE, Mósena ACS, Alves CDBT, Paim WP, Baumbach LF, Mayer FQ, et al. 2018. Characterization of dog serum virome from Northeastern Brazil. Virology. 525:192–199.
- Zaccaria G, Malatesta D, Scipioni G, Di Felice E, Campolo M, Casaccia C, Savini G, Di Sabatino D, Lorusso A. 2016. Circovirus in domestic and wild carnivores: An important opportunistic agent? Virology. 490:69–74.
- Zhang J, Liu Z, Zou Y, Zhang N, Wang D, Tu D, Yang L, Deng Z, Yang Y, Jiang P, et al. 2018. First molecular detection of porcine circovirus type 3 in dogs in China. Virus Genes. 54(1):140–144.