
Abstract
Background
Bovine ephemeral fever (BEF) is a re-emerging disease caused by bovine ephemeral fever virus (BEFV). Although it poses a huge economic threat to the livestock sector, complete viral genome information from any South Asian country, including India, lacks.
Aim
Genome characterization of the first Indian BEFV isolate and to evaluate its genetic diversity by characterizing genomic mutations and their associated protein dynamics.
Materials and Methods
Of the nineteen positive blood samples collected from BEF symptomatic animals during the 2018-19 outbreaks in India, one random sample was used to amplify the entire viral genome by RT-PCR. Utilizing Sanger sequencing and NGS technology, a complete genome was determined. Genome characterization, genetic diversity and phylogenetic analyses were explored by comparing the results with available global isolates. Additionally, unique genomic mutations within the Indian isolate were investigated, followed by in-silico assessment of non-synonymous (NS) mutations impacts on corresponding proteins’ secondary structure, solvent accessibility and dynamics.
Results
The complete genome of Indian BEFV has 14,903 nucleotides with 33% GC with considerable genetic diversity. Its sequence comparison and phylogenetic analysis revealed a close relatedness to the Middle Eastern lineage. Genome-wide scanning elucidated 30 unique mutations, including 10 NS mutations in the P, L and GNS proteins. The mutational impact evaluation confirmed alterations in protein structure and dynamics, with minimal effect on solvent accessibility. Additionally, alteration in the interatomic interactions was compared against the wild type.
Conclusion
These findings extend our understanding of the BEFV epidemiological and pathogenic potential, aiding in developing better therapeutic and preventive interventions.
1. Introduction
Bovine ephemeral fever virus (BEFV) is a widespread and economically significant bovine pathogen associated with bovine ephemeral fever (BEF) disease. The first report on the emergence of BEFV outbreaks dates back to the mid-nineteenth century in Africa, which became endemic to Australia, Africa, the Middle East, and major parts of the Asiatic continent, including East Asia and South-East Asia (Niwa et al. Citation2015). The outbreaks persist during monsoon onset, coinciding with vectors propagation (culicoid midges and mosquitoes) that spreads the disease (Venter et al. Citation2003). The clinical manifestations of BEF include polyphasic fever, synovitis, somnolence, muscle stiffness, shifting lameness, reluctance to move, recumbency, and inappetence (St. George bCitation1986). Presently enzootic, the disease is characterized by high morbidity and occasional high case fatality (2-20%). The disease causes enormous economic loss arising from a reduction in milk production, impaired reproduction, traction decline, loss of body weight, and livestock-associated trade restrictions (Hsieh et al. Citation2005; Tonbak et al. Citation2013).
The BEFV belongs to the genus Ephemerovirus of the Rhabdoviridae family. It is an enveloped, bullet-shaped virion that enwraps an ∼14.9 kb, non-segmented (-) RNA genome encoding ten transcription units in the order 3′-l-N-P-M-G-[GNS-α1-α2-β-γ]-L-t-5′ (McWilliam et al. Citation1997). The virion contains five structural proteins, namely, nucleoprotein (N, 52kD), phosphoprotein (P, 43 kDa), matrix protein (M, 29 kDa), glycoprotein (G, 81 kDa), large polymerase protein (L, 180 kDa). Additionally, genome codes for a non-structural glycoprotein (GNS, 90 kDa) and few accessory proteins α, β and γ (each of <15kDa) (Walker and Klement Citation2015). Each gene junctions inhabit intergenic regions (IGRs), flanked by regulatory motifs termed transcription initiation (TI), transcription termination/polyadenylation (TTP) signals (Rose Citation1980; Barr et al. Citation1997). This arrangement likely regulates the sequential transcription by a “stop-start” process resulting in a 3′→5′ polar gradient of mRNA synthesis (Dietzgen et al. Citation2017). The phylogenetic analyses based on the glycoprotein (G) gene delineates circulating BEFV into geographically distinct Australian, Middle Eastern, Southeast Asian, and newly emerging South African lineages (Walker and Klement Citation2015; Omar et al. Citation2020). Although BEFV is serologically monotypic (Walker and Klement Citation2015), its diverse distribution and differential pathogenicity indicate its genetic variability. The mutation accumulation probability among all RNA viruses is driven by multiple factors, such as poor proofreading by polymerase protein, replicative repairing, selective pressure, etc. (Sardar et al. Citation2020; Wang et al. Citation2020). Notably, mutations could lead to protein structural alterations that affect the viral immunological properties, transmission, and pathogenicity (Woźniakowski et al. Citation2009; Sardar et al. Citation2020). Previous studies have suggested BEFV to possess high substitution rates (Trinidad et al. Citation2014). However, few recent reports on the generation of novel genogroups with higher viral virulence (Yanase et al. Citation2020) and limited vaccine effectiveness suggest BEFV evolution is enhancing variant's fitness landscape to help overcome the host immune system (Aziz-Boaron et al. Citation2014).
The new geographical expansions with enhanced case fatality rate and high production losses alarm BEFV as an evolving arbovirus that warrants closer scrutiny in surveillance, epidemiology, and ecological impact (Lee Citation2019; Yanase et al. Citation2020). Despite the multiple decades of episodic reporting on seasonal incursions from many South Asian countries, including India (Nandi and Negi Citation1999; Walker and Klement Citation2015), no complete sequence information was available until recently (Pyasi et al. Citation2020), which hinders progress for its detection and prevention. Field detection solely relies on the observed clinical signs accompanied by haemato-biochemical analysis in the lack of any serological diagnostics kits. However, molecular diagnosis affirms the viral suspicion. Thus, the current study aimed at genome sequencing and characterizing the first complete Indian BEFV genome. It further analysed its genetic diversity by investigating genomic mutations and their associated protein dynamics that could provide better insight into the viral pathobiology.
2. Materials and methods
2.1. Field sample collection
Blood samples (n = 25) were collected from symptomatic animals during the 2018–19 outbreaks in Madhya Pradesh, India. Bovine aged between 8 months to 8 years of age showed prominent clinical signs in the study. Veterinary professionals collected the blood sample. Haematological and biochemical analysis was performed with/without anticoagulants as required. Subsequently, serum and peripheral blood mononuclear cells (PBMCs) were separated by centrifugation at 3,000 rpm for 15 minutes and kept at −80 °C until future use.
2.2. RNA extraction and genome sequencing
Viral RNA was extracted from serum utilising TRIzol method (Invitrogen, Waltham, MA, USA) and converted to cDNA using Prime Script cDNA synthesis kit (Takara Bio Inc, Kusatsu, Japan). The viral presence was confirmed by reverse transcriptase PCR (RT-PCR) using published primers (Zheng and Qiu Citation2012) with previously described PCR conditions (Hsieh et al. Citation2005). Subsequently, overlapping RT-PCR using eleven primer pairs, designed as per the conserved regions of reference strain (accession no: NC_002526), was performed with optimized annealing between 55 °C and 62 °C as per the primer set (Table S1). Purified amplicons were directly sequenced using the ABI3100 platform (Applied Biosystems, USA). All sequences were subjected to Blastn search and assembled using BioEdit (Tom et al. Citation2011).
Alternatively, sequence accuracy was determined by performing Illumina sequencing, a commonly used next-generation sequencing (NGS) technology. Libraries preparation was done using total viral RNA with NEBNext mRNA Library Prep kit (NEB, Ipswich, MA, USA) and sequenced in a MiSeq instrument (150 cycles, paired-end sequencing). The sequenced raw data followed trimming and adapter removal using Trim Galore (Krueger 2015). Further, it reads classification by Kraken-2 utilizing a standard database (Wood et al. Citation2019). The only confirmed viral reads were snipped against other non-viral reads by BBMap (Bushnell Citation2014) and assembled into contigs using MEGAHIT v.1.1.3 (Li et al. Citation2015) to generate a de novo sequence.
2.3. Phylogenetic and genetic analysis
Phylogenetic analyses were inferred by the maximum likelihood method employing the Tamura-Nei model of nt substitution model using the MEGA X (1000 bootstrap replicates) (Kumar et al. Citation2018). Furthermore, the new genome sequence was aligned with all available BEFV complete genomes archived from GenBank using the ClustalW (Larkin et al. Citation2007) module of BioEdit to determine terminal sequence conservancy, molecular characterization, and relative similarity evaluation. Eventually, genetic variations among the consensus sequences in addition to the novel nucleotide (nt) and amino acid (aa) substitutions were inspected as synonymous (SN) and non-synonymous (NS). Lastly, the effect of NS mutations on structural alteration was evaluated.
2.4. Mutational assessment
The detected genomic mutations, emphasizing unique NS mutations, were examined to better understand the mutational consequences on protein structure and dynamics. P, L and GNS proteins of Indian BEFV isolate that possessed such mutations were extensively investigated utilizing several computational approach-based tools. We considered the first globally reported Australian isolate as wild type sequence (AF234533) for all comparisons.
2.4.1. Secondary structures prediction
The secondary structure of investigated proteins was predicted by CFSSP: Chou and Fasman secondary structure prediction server (Kumar Citation2013). The server predicts the probability of occurrence of regions such as α-helix, β-sheet and turns in secondary structure prediction. Eventually, NS mutation-driven relative alterations in structures and solvent accessibility were compared against the reference isolate.
2.4.2. Protein dynamics analysis
Analysing protein structure-based alterations due to mutation determines the energy change value, significant to drive functional alterations. To achieve this, structure elucidation was done relying on the Modeller 9v8 (http://salilab.org/modeller/download_installation.html) (Sali and Blundell Citation1993), as no crystal structure of the studied (P, L and GNS) proteins were available. The FASTA sequences of proteins of Indian and Australian isolates were retrieved from the NCBI database and modelled. Further validation to predict various stereochemical properties was done by PROCHECK (Laskowski et al. Citation1993) and verify 3 D (Colovos and Yeates Citation1993). All models were visualized using Chimera tool (Pettersen et al. Citation2004).
For investigating the mutational consequences on the protein structural conformations, DynaMut server was employed (Rodrigues et al. Citation2018). The server is based on graph-based signatures in a consensus predictor algorithm and outperforms predicting the effect of mutations on protein flexibility and stability (p-value < 0.001). It works by uploading the modelled proteins to evaluate the impact of mutation in native protein structures based on dynamicity and protein stability resulting from vibrational entropy changes (ΔΔSVibENCoM) and differences in the free energy (ΔΔG). It uses a well-established normal mode analysis (NMA) to interpret and visualize the practicality of protein structural dynamics.
Additionally, analysis on altered conformations, evaluating their interatomic interaction due to investigated mutations was done. By utilizing the DynaMut server, results of the interacting molecules demonstrate how often mutation disrupts the interacting bonds, using a pairwise alignment algorithm (Bauer et al. Citation2019) by comparing against the wild type.
2.5. Virus isolation
Virus isolation has also been attempted in Vero and Baby hamster kidney (BHK-21), cell lines, respectively. The cell monolayer has been inoculated by the extracted PBMCs and serum of BEFV positive samples as previously performed (Chen et al. Citation2010; Zaher and Ahmed Citation2011). These were eventually passaged every eighth day and observed routinely for visible cytopathic effect (CPE). The supernatant was further investigated by RT-PCR amplification for virus presence.
3. Results
3.1. Clinical and haemato-biochemical evaluations
The outbreaks were episodically epizootic from July-October during 2019. The studied group comprised animals suspected of BEF based on the typical clinical signs and hemato-biochemical data. Most of the affected animals showed significant neutrophilia and lymphocytopenia in the hematological analyses. Similarly, blood biochemical analysis revealed decreased calcium concentrations and elevated creatinine kinase activities compared to normal, attributing to recumbency and lameness in these animals. Details of the hemato-biochemical analyses are depicted in Table S2. Although the infected animals almost exhibited full recovery within 3-10 days, they suffered significant milk losses as evidenced by lactation cessation in 22 out of 25 diseased animals extending up to 8-10 months.
3.2. Virus confirmation and sequencing
The PCR analysis showed expected-sized amplicons, confirming BEFV suspicion in 19 samples. Of these, a randomly picked sample was further utilized for complete genome sequencing. The PCR purified amplicons were sequenced and assembled, generating a near-complete genome of IND/IDR/BEFV/2019. Analogously, Illumina sequencing yielded similar outcomes with a de novo assembled 14,875 nt long contigs. Blastn search returned ∼95% identity to other published BEFV sequences. However, the sequence lacked a stretch of nt (15 at 5′ and 13 at 3′) as determined by consensus sequences of aligned genomes. Importantly, a perfect conservancy was displayed at 5’end, while 3′ revealed an average of 96%, with a maximum 99% to Israeli isolate (with the insertion of two nt in the missing region) and hence adopted to the Indian isolate (Identity matrix as shown in Table S3). Altogether, the first complete genome of IND/IDR/BEFV/2019 revealed a length of 14,903 nt with 33.37% GC content and deposited in GenBank under accession number MN905763.
3.3. Sequence analysis
The aligned consensus sequences revealed a conserved genome pattern with each open reading frame (ORF) flanked by TI and TTP sequences and were separated by intergenic regions (). A partial complementarity at 3′ leader (l) of 52 nt (1–52) and 5′ trailer (t) of 72 nt (14832–14903 nt) ends were observed, a characteristic property of all Rhabdoviruses (Dhillon et al. Citation2000). While few variations in the intergenic regions with nt insertions at GNS/α1 and β/γ junctions in south-eastern isolates were observed, their significant difference in genome regulations requires experimental exploration. The details of each region are summarised in and their sequences are depicted in Table S3.
Figure 1. Schematic diagram illustrating the genome organization of Indian BEFV isolate. The complete length of single-stranded BEFV antigenome of 14,903 nucleotides (nt) along with the position of each terminal and gene, in the order from 3’ to 5’ polarity as 3’UTR (52 nt), nucleocapsid (N, 1328 nt), phosphoprotein (P, 858 nt), matrix (M, 691 nt), glycoprotein (G, 1897 nt), non-structural glycoprotein (GNS, 1785 nt), bicistronic alpha1(α1), alpha2 (α2) (630 nt), beta (β, 460 nt), gamma (γ, 400 nt) and large subunit of polymerase (L, 6470 nt) gene and 5’UTR (72 nt) are represented.
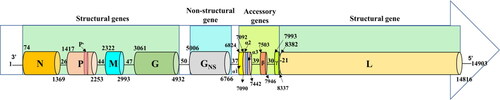
Table 1. Represents the characterisation of complete Indian BEFV genome sequences depicting transcription initiation (TI) and transcription termination (TTP), coding sequences of genes with their respective positions.
The Indian sequence revealed the highest (∼96.53%) identity to isolates of the Middle East, followed by East Asia (∼91%) and lastly, Australia (89.84%). Further comparison of each terminus and the concatenated non-coding regions also followed a similar pattern. However, aa similarity ranged from 85.65% to 92.93%. All comparisons are represented in .
Table 2. Depicting the comparison of nucleotide (nt) and amino acid (aa) similarity between Indian BEFV isolate with all globally available BEFV complete genome sequences isolates, along with each gene, 3’UTR, 5’UTR with concatenated non coding region. Information on country from where it is isolated, accession number, complete sequence length is denoted.
3.4. Phylogenetic analysis and genetic diversity
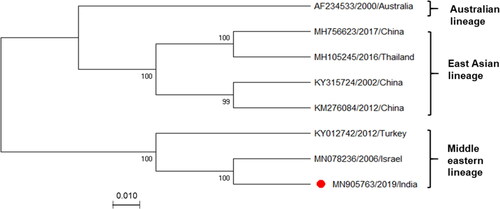
Phylogenetic analysis clustered IND/IDR/BEFV/2019 with the Israeli and Turkish isolates of Middle Eastern lineage (). The pairwise genetic distance of all corresponding sequences, estimated to be 0.03% to 0.10%, revealed high-level identity suggesting the constant evolutionary rate of BEFV. Additionally, the genetic distance had no clear correlation with geographical distance.
Figure 2. Phylogenetic analysis of Indian BEFV isolate. Phylogenetic analysis of complete Indian BEFV genome (marked with a red circle, n = 8) compared with corresponding globally available sequences. The maximum likelihood (ML) method and the Tamura-Nei model of nucleotide substitution were employed with bootstrap values (1,000 replicates) for tree construction. Bootstrap values are shown at the nodes representing the maximum likelihood bootstrap values and their subclades. There is a total of 14,959 positions in the final dataset. The GenBank accession numbers, along with the country name and year of the collection, are indicated for each virus.
Next, we comprehensively analysed the genomic mutational accumulation specific to the Indian isolate, in context to unique nt and aa variations, revealing 76 substitutions (34 in non-coding and 42 in coding regions) and 30 substitutions (20 in structural proteins and 10 in non-structural proteins), respectively (). Except for N, M, β, and γ proteins with 100% identity, SN mutations were prominent in P, G, GNS, α1, α2, and L proteins, with few NS mutations observed in P, L and GNS proteins as well (). Interestingly, the third nt substitution of codon seemed most frequent for identified NS mutations. The results indicated the L, P, and GNS proteins to show the highest variation with 10, 9, and 8 mutations, respectively, that specifically included 4, 3, and 3 as NS mutations.
Table 3. Represents the number and type of unique variations/substitutions in the nucleotide and amino acid of all the genes with respect to their position. Additionally, Synonymous mutation (SN) and Non-Synonymous mutation (NS) that were observed within the Indian BEFV isolate in comparison to globally available sequences are also summarized.
3.5. Assessment of genomic NS mutations on protein dynamics
The NS mutations stand unique to the particular isolate that profoundly impacts the overall structural properties and biological function (Lewin et al. Citation2018). Our study utilized the in-silico approach to analyse their impact on various parameters affecting protein secondary and tertiary structures. Additionally, the overall impact due to these NS mutations were evaluated to seek their biological impact.
3.5.1. Secondary structure and its mutational impacts
The impact of these mutations on the secondary structure of mutant proteins was investigated. Our findings demarcated the changes observed in the mutant protein. It shows that except for two mutations, including S152L and K173N in P protein, all the remaining eight sites, E45G, S115P, in P; K1635E, K1937N, E2072R in L and G490E, E515K, K520I in GNS proteins induced changes in secondary structure. The detailed analysis revealed that mutants attained considerable alterations specifically at mutation sites (E45G, S115P in P and E2072R in L) or in the neighbouring residues (remaining all other mutants), indicating alterations in the local protein conformations in the secondary structure as compared to the reference isolate ().
Figure 3. Representation of the effects of non-synonymous (NS) mutations on protein secondary structure. Depicts the prediction of the secondary structure due to change in mutation comparing Indian BEFV isolate to the Australian isolate (reference sequence) in P, L and GNS proteins. The rectangular box shows the mutant residue against the wild type. The position of the dashed box in the corresponding panels highlights the difference in secondary structure between Indian and Australian isolates.
The significant alterations in P protein were depicted as few charged (E) and polar residues (S) were substituted by uncharged, non-polar, and hydrophobic residues (GNS, P, and L), though with little altered solvent accessibility. Likewise, in L protein, mutant replaced few positively charged residue with a negative or uncharged residue (K→E/N), with nonsignificant changes observed. In contrast, the substitution of negative to positively charged residue (E→R) may lead to the breakage of hydrogen bonds between the bases, thus altering their structures. All residues showed their solvent accessibility conversion from exposed to slightly buried (K→E/N) and converse (E→R). Similarly, the GNS protein displayed transitions only in the neighbouring residues resulting in altering the secondary structure. For solvent accessibility, a transformation from strongly exposed charged lysine to most buried hydrophobic isoleucine and negatively charged slightly exposed (K→I/E) residues was observed.
3.5.2. Stability dynamics comparison of tertiary structure
By analyzing the 3 D protein models in DynaMut server, we correlated the interpretation of secondary structure alterations to tertiary structure protein dynamics. All the built models were refined and validated, assuring their high quality for dynamics analysis. Our data inferred, most of the identified NS mutations resulted in destabilization with negative ΔΔG value and enhanced molecular flexibility with a positive ΔΔSVibENCoM value. Except for K520I and E514K mutants of GNS protein with significantly higher positive ΔΔG (each as, −0.282 and −0.504 kcal/mol) and negative ΔΔSVibENCoM values (each as, 1.003 and 0.856 kcal.mol-1. K-1) demarcating enhanced stability and rigidity, respectively () (). The ΔΔSVibENCoM value represents an average of the protein configurational entropies within a single minimum energy landscape (Goethe et al. Citation2015). Whereas ΔΔG is the difference in ΔG of native and mutant type (Eriksson et al. Citation1992). Generally, ΔΔG positive value induces stabilization, and ΔΔSVibENCoM with negative value results in a decrease in flexibility. Additionally, other structure-based prediction tools such as mCSM, SDM, and DUET included as a module of the DynaMut server, further supported similar stabilization behavior, thus confirming our findings (Rodrigues et al. Citation2018).
Figure 4. Depicting the effects of non-synonymous (NS) mutations on protein structural dynamics. Analysis of proteins stability and flexibility due to NS mutations. Visual representation of change in vibrational entropy energy between wild-type and mutant proteins. The colour change in amino acids is according to the ΔΔSVibENCoM value as a result of mutation. Red represents a gain in flexibility and blue indicates rigidification of the structure.
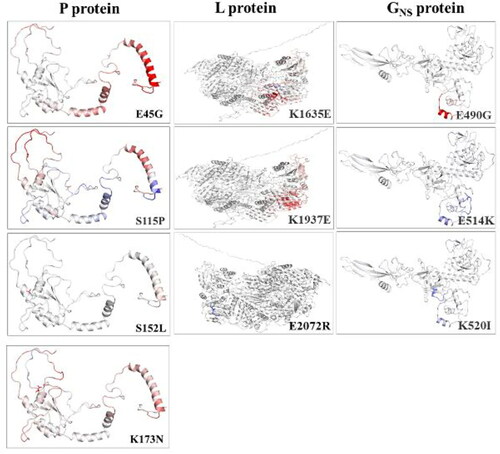
Table 4. Representing the analysis based on the differences in ΔΔSVibENCoM and ΔΔG value due to mutation affecting the stability and flexibility of the mutated protein of Indian BEFFV isolate (MN905763) as compared to wild type reference sequence (AF234533).
Furthermore, the significant relative variations in the mutants' stability and flexibility can investigate changes in protein confirmations. The comparison revealed significant alteration in the interatomic interaction among the variant and the wild-type. Major changes were observed in E490G and K514I mutants in GNS protein and S115P in P protein, while the rest showed slight alterations. The findings may contribute to explore the mutational influence on interatomic interactions of the proteins with their surrounding environment. All the mutated residues influenced the side chains altering the bond types, including H-bonds, halogen bonds, hydrophobic bonds, etc. That determines the interactions among residues existing in the surrounding area. The difference in structures representing variations is depicted in .
Figure 5. Visualization of comparative prediction of interatomic interactions due to NS mutations. The possible interatomic interaction rearrangements mediated by NS mutations in each of the proteins is represented. Wild-type and mutant residues are represented as light-green coloured sticks and all the surrounding residues involved in any type of interactions are denoted in different colours as mentioned in the box.
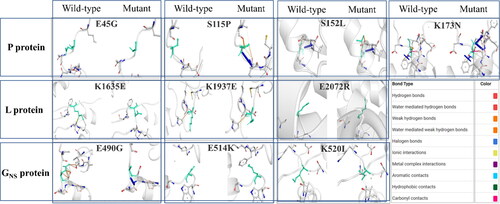
Subsequently, utilising the PROVEAN tool, we predicted their corresponding biological impacts as determined by the pairwise sequence alignment scores. Two mutations in P protein (S152L, K173N) showed deleterious effect, while the rest had a neutral effect ().
Table 5. Represents the pairwise sequence alignment scores based on the comparative score of wild and mutated protein sequence and the predicted effect of such identified NS mutations on the biological function of the mutated proteins.
3.6. Virus isolation
The cell monolayer evidenced profound CPE from the fourth to fifth passage after 48 hours post-inoculation. However, PCR confirmation produced unclear results. Thus, an assumption of an unknown virus that co-exists and co-circulates predominates its adaptability in cell culture compared to BEFV. This inference requires future experimental research. Hence, virus isolation proved unsuccessful.
4. Discussion
The enhanced endemicity, combined with high morbidity and increasing case-fatality associated with BEFV infection, seriously attributes to enormous economic losses to the global dairy sector. As the world's leading dairy producer, India is at immense risk to BEFV. This necessitates a thorough understanding of BEFV isolates and genome to assess the pathogenicity. This study performed RT-PCR and NGS analysis of prevalent BEFV isolate, accompanied by a mutation-linked assessment of protein structures, crucial in understanding the viral pathogenesis, transmission, evolution, and antiviral therapy development.
The nearly complete genome obtained through molecular approach was completed by utilizing the terminal sequence conservancy pattern of global BEFV sequences, which determined the IND/IDR/BEFV/2019 isolate, the first Indian BEFV genome sequence. The 14,903 nt long full-genome was comparable to the Australian and Middle eastern isolates but recorded ∼ 40 nt shorter than East Asian isolates. All eight available complete genomes (including ours) maintain a conserved genome architecture pattern, length similarities, and deduced proteins suggesting essentiality for the propagation and dispersion of BEFV within the host populations.
This isolate has an expected conserved TI and TTP sequences pattern, vital to recruiting RNA-dependent RNA polymerase (RdRp) for independent transcription (Zeng et al. Citation1998). Moreover, the IGRs separating each gene are crucial determinants in transcription initiation and termination events (Das and Pattnaik Citation2004; Leyrat et al. Citation2011). The current isolate showed a certain variation in nucleotide length in IGRs at GNS/α1 and β/γ junctions compared to the South-Eastern isolates, which might influence viral gene expression and replication, which needs further exploration.
Intriguingly, sequence and phylogenetic analysis revealed a close relatedness to Israeli and Turkish isolates, implying a common origin. The branching patterns coincide with the previous P and G genes based phylogenetic analysis, projecting its relative high pathogenicity (Yeruham et al. Citation2010; Abayli et al. Citation2017; Pyasi et al. Citation2020). Interestingly, no significant correlation between genotypes and geographical distance was observed. Despite the geographical closeness to Eastern and South-Eastern countries, the Indian isolate revealed the highest similarity to Middle Eastern isolates. As this region shares a significant portion of the livestock trade with India, it indicates a probable reason for the close relatedness of this cattle pathogen.
As geographical traversion of viruses may prompt certain adaptive responses in the form of mutations generating new variants that are more potent and fit (Pfeiffer and Kirkegaard Citation2005). Henceforth, we comprehensively scanned the IND/IDR/BEFV/2019 genome for unique mutations. We observed a prevalence of unique nt substitutions, with high codon degeneration, majorly contributing to silent or SN mutations (n = 20) and a few NS mutations (n = 10). The sequence conservation pattern followed M > G > N > L > P for structural proteins and γ > β>α2 > α1 > GNS for the non-structural proteins. Our results correlate well to other arboviruses (Domingo and Holland Citation1997; Pomeroy et al. Citation2008) that sustain constant viral evolution despite high substitution rates, thus similarly BEFV retained a single serotype characteristic worldwide (Dietzgen et al. Citation2017).
The highest 10 mutations were observed in L, followed by 9 in P, and 8 in GNS proteins that included few NS mutations. The L protein is the sole enzymatically active protein and is essential for genome transcription and replication. Similarly, P protein showed the highest divergence degree (relative to its size), supporting its significant role in genome evolution (Tordo et al. Citation1988; Leyrat et al. Citation2011). Since the rhabdovirus N-P-L complex regulates viral biological processes in collaboration (Das and Pattnaik Citation2004), experiencing mutations in any of these may presumably interfere with overall virus biology. Likewise, mutations in the GNS protein with understudied functions indicate an uncertain outcome.
We also examined unique NS mutations in the context of alteration to protein confirmations, contributing to the emergence of new genotypes. For instance, China, Taiwan and Turkey isolates (KY012742) demonstrated an elevated case fatality rate due to such mutations (Zheng and Qiu Citation2012; Abayli et al. Citation2017; Yanase et al. Citation2020). In total, 10 unique NS mutations comprising 4 in P, 3 in L, and 3 in GNS regions were structurally explored to infer any functional consequences (Cao et al. Citation2020)(Das and Pattnaik Citation2004). Studies suggest L protein to be highly conserved (Poch et al. Citation1989), while P protein shows the highest divergence degree (Leyrat et al. Citation2011). The computational analysis predicted two mutations in P (S152L and K173N) to be considerably deleterious and impact L protein's polymerase activities by interfering with L-P, protein-protein interaction functions (Cao et al. Citation2020) (). However, further in vitro analysis is needed to corroborate this observation. Similarly, mutations in the methyltransferase region of L could alter mRNA capping function.
Interestingly, our data on NS mutations in the corresponding protein revealed alteration to the structural stability. Most mutants' secondary structure analysis demonstrated the transition from charged to uncharged residues and hydrophilic to hydrophobic residue substitutions. Also, solvent accessibility decreased as substituted residues were buried deep in the protein structures compared to the earlier exposed ones. Eventually, we were able to link the aforesaid data to extrapolating the transition in the extremely vibrant protein tertiary structures by comparing it to the native type. The data revealed that most of the investigated NS mutations presumably resulted in the loss of rigidity and stability as is seen with positive ΔΔSVibENCoM and negative ΔΔG values, respectively. In contrast, GNS protein with E514K and K520I mutations are of concern as they stabilize conformations due to local helix backbone distortions (Cornish et al. Citation1994), leading to gain in rigidity. Further, most mutants showed significant alteration in the interatomic interaction compared to the wild-type.
However, our finding has limitations due to the unavailability of any crystal structure and limited knowledge of the functions of three proteins. Nonetheless, the Indian BEFV genome has acquired mutations that could potentially influence the protein structure. However, their influence on protein functions and viral virulency or transmission remains to be elucidated by performing wet-lab experiments.
5. Conclusion
The study presents the first complete genome sequence of Indian BEFV isolate and confirms its relatedness to Middle Eastern countries. Genome characterization revealed considerable genetic diversity with several unique mutations. Most of these mutations in P, L and GNS proteins were predicted to potentially impact the protein conformation and dynamics. Except for E514K and K520I mutants of GNS protein that achieved stabilized conformations and increased rigidity, all other mutants exhibited increased flexibility and destabilized effect on the structure. Mutations in P protein (S152L and K173N) are predicted to be deleterious for structural stability. As our study focused primarily on in-silico analysis, the inference from this could pave the way for prospective studies centered on experimental validation. Integrated knowledge of all these aspects with a larger sampling size would enhance diagnostics and vaccine development efforts of this understudied pathogen.
Data availability
All the data supporting this research study's findings are openly accessible in the GenBank repository (https://www.ncbi.nlm.nih.gov/genbank) with Accession numbers: AF234533, KM276084, KY315724, MH756623, MH105245, MN078236, KY012742 and MN905763. The protein id’s of all the sequences investigated (P, GNS and L proteins): QOU09201, QOU09205, QOU09210, NP_065399, NP_065403, NP_065409.
Supplemental Material
Download Zip (58.7 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Abayli H, Tonbak S, Azkur AK, Bulut H. 2017. Complete genome analysis of highly pathogenic bovine ephemeral fever virus isolated in Turkey in 2012. Arch Virol. 162(10):3233–3238.
- Aziz-Boaron O, Gleser D, Yadin H, Gelman B, Kedmi M, Galon N, Klement E. 2014. The protective effectiveness of an inactivated bovine ephemeral fever virus vaccine. Vet Microbiol. 173(1–2):1–8.
- Barr JN, Whelan SP, Wertz GW. 1997. cis-Acting signals involved in termination of vesicular stomatitis virus mRNA synthesis include the conserved AUAC and the U7 signal for polyadenylation. J Virol. 71(11):8718–8725. ]. https://www.ncbi.nlm.nih.gov/pubmed/9343230.
- Bauer JA, Pavlović J, Bauerová-Hlinková V. 2019. Normal mode analysis as a routine part of a structural investigation. Molecules. 24(18):3293.
- Bushnell B. 2014. BBMap: A fast, accurate, splice-aware aligner [Internet]. https://www.osti.gov/biblio/1241166.
- Cao D, Gao Y, Roesler C, Rice S, D’Cunha P, Zhuang L, Slack J, Domke M, Antonova A, Romanelli S, et al. 2020. Cryo-EM structure of the respiratory syncytial virus RNA polymerase. Nat Commun. 11(1):1–9. https://doi.org/http://dx.doi.org/10.1038/s41467-019-14246-3.
- Chen CY, Chang CY, Liu HJ, Liao MH, Chang CI, Hsu JL, Shih WL. 2010. Apoptosis induction in BEFV-infected Vero and MDBK cells through Src-dependent JNK activation regulates caspase-3 and mitochondria pathways. Vet Res. 41(2):15.
- Colovos C, Yeates TO. 1993. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 2(9):1511–1519.
- Cornish VW, Kaplan MI, Veenstra DL, Kollman PA, Schultz PG. 1994. Stabilizing and destabilizing effects of placing beta-branched amino acids in protein alpha-helices. Biochemistry. 33(40):12022–12031..
- Das SC, Pattnaik AK. 2004. Phosphorylation of vesicular stomatitis virus phosphoprotein P is indispensable for virus growth. J Virol. 78(12):6420–6430.
- Dhillon J, Cowley JA, Wang Y, Walker PJ. 2000. RNA polymerase (L) gene and genome terminal sequences of ephemeroviruses bovine ephemeral fever virus and Adelaide River virus indicate a close relationship to vesiculoviruses. Virus Res. 70(1–2):87–95.
- Dietzgen RG, Kondo H, Goodin MM, Kurath G, Vasilakis N. 2017. The family rhabdoviridae: mono- and bipartite negative-sense RNA viruses with diverse genome organization and common evolutionary origins. Virus Res. 227:158–170. https://pubmed.ncbi.nlm.nih.gov/27773769.
- Domingo E, Holland JJ. 1997. RNA virus mutations and fitness for survival. Annu Rev Microbiol. 51:151–178.
- Eriksson AE, Baase WA, Zhang XJ, Heinz DW, Blaber M, Baldwin EP, Matthews BW. 1992. Response of a protein structure to cavity-creating mutations and its relation to the hydrophobic effect. Science. 255(5041):178–183.
- Goethe M, Fita I, Rubi JM. 2015. Vibrational entropy of a protein: large differences between distinct conformations. J Chem Theory Comput. 11(1):351–359..
- Hsieh YC, Chen SH, Chou CC, Ting LJ, Itakura C, Wang FI. 2005. Bovine ephemeral fever in Taiwan (2001–2002). J Vet Med Sci. 67(4):411–416.
- Krueger F. 2015. Trim Galore!: a wrapper tool around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files. http://www.bioinformatics.babraham.ac.uk/projects/trim_. galore/
- Kumar TA. 2013. CFSSP: Chou and Fasman secondary structure prediction server. Wide Spectr. 1(9):15–19.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35(6):1547–1549.
- Larkin MA, Blackshields G, Brown NP, Chenna R, Mcgettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. 2007. Clustal W and Clustal X version 2.0. Bioinformatics. 23(21):2947–2948.
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 26(2):283–291. [Internet]..
- Lee F. 2019. Bovine ephemeral fever in Asia: recent status and research gaps. Viruses. 11(5):412.
- Lewin AC, Kolb AW, McLellan GJ, Bentley E, Bernard KA, Newbury SP, Brandt CR. 2018. Genomic, recombinational and phylogenetic characterization of global feline herpesvirus 1 isolates. Virology. 518:385–397.
- Leyrat C, Ribeiro EA, Gérard FCA, Ivanov I, Ruigrok RWH, Jamin M. 2011. Structure, interactions with host cell and functions of rhabdovirus phosphoprotein. Future Virol. 6(4):465–481.
- Li D, Liu C-M, Luo R, Sadakane K, Lam T-W. 2015. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 31(10):1674–1676.
- McWilliam SM, Kongsuwan K, Cowley JA, Byrne KA, Walker PJ. 1997. Genome organization and transcription strategy in the complex G(NS)-L intergenic region of bovine ephemeral fever rhabdovirus. J Gen Virol. 78(6):1309–1317.
- Nandi S, Negi BS. 1999. Bovine ephemeral fever: a review. Comp Immunol Microbiol Infect Dis. 22(2):81–91. http://www.sciencedirect.com/science/article/pii/S0147957198000277.
- Niwa T, Shirafuji H, Ikemiyagi K, Nitta Y, Suzuki M, Kato T, Yanase T. 2015. Occurrence of bovine ephemeral fever in Okinawa Prefecture, Japan, in 2012 and development of a reverse-transcription polymerase chain reaction assay to detect bovine ephemeral fever virus gene. J Vet Med Sci. 77(4):455–460.
- Omar R, Van Schalkwyk A, Carulei O, Heath L, Douglass N, Williamson AL. 2020. South African bovine ephemeral fever virus glycoprotein sequences are phylogenetically distinct from those from the rest of the world. Arch Virol. 165(5):1207–1210..
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera-a visualization system for exploratory research and analysis. J Comput Chem. 25(13):1605–1612.
- Pfeiffer JK, Kirkegaard K. 2005. Increased fidelity reduces poliovirus fitness and virulence under selective pressure in mice. PLoS Pathog. 1(2):e11.
- Poch O, Sauvaget I, Delarue M, Tordo N. 1989. Identification of four conserved motifs among the RNA-dependent polymerase encoding elements. Embo J. 8(12):3867–3874.
- Pomeroy LW, Bjørnstad ON, Holmes EC. 2008. The evolutionary and epidemiological dynamics of the paramyxoviridae. J Mol Evol. 66(2):98–106.
- Pyasi S, Sahu BP, Sahoo P, Dubey PK, Sahoo N, Byrareddy SN, Nayak D. 2020. Identification and phylogenetic characterization of bovine ephemeral fever virus (BEFV) of Middle Eastern lineage associated with 2018–2019 outbreaks in India. Transbound Emerg Dis. 67(5):2226–2232.
- Rodrigues CHM, Pires DV, Ascher DB. 2018. DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 46(W1):W350–W355. [Internet]..
- Rose JK. 1980. Complete intergenic and flanking gene sequences from the genome of vesicular stomatitis virus. Cell. 19(2):415–421.
- Sali A, Blundell TL. 1993. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 234(3):779–815.
- Sardar R, Satish D, Birla S, Gupta D. 2020. Integrative analyses of SARS-CoV-2 genomes from different geographical locations reveal unique features potentially consequential to host-virus interaction, pathogenesis and clues for novel therapies. Heliyon. 6(9):e04658.
- St. George TD. 1986. The epidemiology of bovine ephemeral fever in Australia and its economic effect. In: Arbovirus research in Australia. Proceedings Fourth Symposium May 6–9, Brisbane, Australia; p. 281–286.
- Tom H, Biosciences I, Carlsbad C. 2011. BioEdit: an important software for molecular biology. GERF Bull Biosci. 2(June):60–61.
- Tonbak S, Berber E, Deniz Yoruk M, Azkur AK, Pestil Z, Bulut H. 2013. A large-scale outbreak of bovine ephemeral fever in Turkey, 2012. J Vet Med Sci. 75(11):1511–1514.
- Tordo N, Poch O, Ermine A, Keith G, Rougeon F. 1988. Completion of the rabies virus genome sequence determination: Highly conserved domains among the L (polymerase) proteins of unsegmented negative-strand RNA viruses. Virology. 165(2):565–576. [accessed 2020 Apr 21]. https://www.sciencedirect.com/science/article/abs/pii/0042682288906009.
- Trinidad L, Blasdell KR, Joubert DA, Davis SS, Melville L, Kirkland PD, Coulibaly F, Holmes EC, Walker PJ. 2014. Evolution of bovine ephemeral fever virus in the Australian episystem. J Virol. 88(3):1525–1535. http://jvi.asm.org/content/88/3/1525.abstract.
- Venter GJ, Hamblin C, Paweska JT. 2003. Determination of the oral susceptibility of South African livestock-associated biting midges, Culicoides species, to bovine ephemeral fever virus. Med Vet Entomol. 17(2):133–137.
- Walker PJ, Klement E. 2015. Epidemiology and control of bovine ephemeral fever. Vet Res. 28:46:124.
- Wang C, Liu Z, Chen Z, Huang X, Xu M, He T, Zhang Z. 2020. The establishment of reference sequence for SARS-CoV-2 and variation analysis. J Med Virol. 92(6):667–674. [Internet].
- Wood DE, Lu J, Langmead B. 2019. Improved metagenomic analysis with Kraken 2. Genome Biol. 20(1):257.
- Woźniakowski G, Kozdruń W, Samorek-Salamonowicz E. 2009. Genetic variance of Derzsy’s disease strains isolated in Poland. J Mol Genet Med. 3(2):210–216. https://pubmed.ncbi.nlm.nih.gov/20076793.
- Yanase T, Murota K, Hayama Y. 2020. Endemic and emerging arboviruses in domestic ruminants in East Asia. Front Vet Sci. 7:168. https://pubmed.ncbi.nlm.nih.gov/32318588.
- Yeruham I, Van Ham M, Stram Y, Friedgut O, Yadin H, Mumcuoglu KY, Braverman Y. 2010. Epidemiological investigation of bovine ephemeral Fever outbreaks in Israel. Vet Med Int. 2010:1–5. https://pubmed.ncbi.nlm.nih.gov/20814543.
- Zaher KS, Ahmed WM. 2011. Investigations on Bovine Ephemeral fever virus in Egyptian cows and buffaloes with emphasis on isolation and identification of a field strain. Glob Vet. 6(5):447–452.
- Zeng L, Falgout B, Markoff L. 1998. Identification of specific nucleotide sequences within the conserved 3'-SL in the dengue type 2 virus genome required for replication . J Virol. 72(9):7510–7522. ]. https://pubmed.ncbi.nlm.nih.gov/9696848.
- Zheng F, Qiu C. 2012. Phylogenetic relationships of the glycoprotein gene of bovine ephemeral fever virus isolated from mainland China, Taiwan, Japan, Turkey, Israel and Australia. Virol J. 9(1):268.] http://virologyj.biomedcentral.com/articles/https://doi.org/10.1186/1743-422X-9-268.