Abstract
Widespread use of zebrafish (Danio rerio) in genetic analysis of embryonic development has led to rapid advances in the technology required to generate, map and clone mutated genes. To identify genes involved in the generation and regulation of vertebrate circadian rhythmicity, we screened for dominant mutations that affect the circadian periodicity of larval zebrafish locomotor behavior. In a screen of 6,500 genomes, we recovered 8 homozygous viable, semi-dominant mutants, and describe one of them here. The circadian period of the lager and lime (lagdg2) mutant is shortened by 0.7 h in heterozygotes, and 1.3 h in homozygotes. This mutation also shortens the period of the melatonin production rhythm measured from cultured pineal glands, indicating that the mutant gene product affects circadian rhythmicity at the tissue level, as well as at the behavioral level. This mutation also alters the sensitivity of pineal circadian period to temperature, but does not affect phase shifting responses to light. Linkage mapping with microsatellite markers indicates that the lag mutation is on chromosome 7. A zebrafish homolog of period1 (per1) is the only known clock gene homolog that maps near the lag locus. However, all sequence variants found in per1 cDNA from lagdg2 mutants are also present in wild type lines, and we were unable to detect any defect in per1 mRNA splicing, so this mutation may identify a novel clock gene.
INTRODUCTION
Circadian rhythms of physiology and behavior in most organisms are driven by an intracellular biological clock. Within the past decade, a combination of genetic, molecular and biochemical approaches has significantly advanced our understanding of the circadian molecular clockwork. Several genes have been identified as essential for proper circadian clock function in mammals: two Period (mPer1/2) genes, two Cryptochrome (mCry1/2) genes, Clock, Bmal1, Rev-Erb α, and Casein Kinase I epsilon (CKIε) (Allada et al., Citation2001; Lowrey & Takahashi, Citation2000; Okamura, Citation2003; Reppert & Weaver, Citation2002) Phenotypic and biochemical analyses of rodents carrying mutations in each of these genes have revealed that the central timekeeping mechanism is based on transcriptional/post-translational negative feedback loops.
Several lines of evidence suggest that additional players within the feedback loops remain unidentified. First, biochemical data from liver indicate that clock protein complexes are up to 4–6 times larger than expected if the complexes contained a single molecule of each known clock component (Lee et al., Citation2001). Although the stoichiometry of the complex components is not known, this suggests that there may be unidentified proteins within this complex. Second, genome-wide analysis for genetic determinants of circadian behavior has revealed more than 14 quantitative trait loci (QTL) linked to various aspects of circadian behavior (Hofstetter et al., Citation2003; Mayeda & Hofstetter, Citation1999; Shimomura et al., Citation2001; Suzuki et al., Citation2000; Suzuki et al., Citation2001). Importantly, almost all of these loci are distinct from loci harboring known mammalian clock genes, indicating that there are several unknown genes involved in determining circadian behavior. Finally, the clock in Drosophila involves many of the same clock gene homologs and operates in a similar manner (Shirasu et al., Citation2003). However, there are additional components of the fly clock that have yet to be shown to act in any vertebrate circadian clock.
In order to identify molecular components involved in the generation or regulation of the vertebrate circadian clock, we screened for zebrafish mutants with altered circadian periods. Zebrafish (Danio rerio) is an attractive vertebrate model organism for forward genetic approaches to identify the genes underlying complex biological processes. Random mutations within the zebrafish genome are efficiently induced by chemical mutagens and viral DNA insertion (Amsterdam, Citation2003; Solnica-Krezel et al., Citation1994). Large-scale mutagenesis screens for mutations that affect zebrafish development and behavior have been very successful (Brockerhoff et al., Citation1995; Darland & Dowling, Citation2001; Felsenfeld, Citation1996; Golling et al., Citation2002; Granato & Nusslein-Volhard, Citation1996; Neuhauss, Citation2003).
Zebrafish exhibit circadian rhythms of behavior as larvae and as adults, (Cahill et al., Citation1998; Delaunay et al., Citation2000; Hurd et al., Citation1998; Kazimi & Cahill, Citation1999) with peaks of behavioral activity during the daytime. Oscillators within the brain presumably drive these rhythms since pinealectomy and ocular enucleation do not affect behavioral rhythmicity of larvae (M.W.H. and G.M.C., unpublished observations). Zebrafish have peripheral oscillators in a variety of tissues. Pineal glands and retinas are photoreceptive organs that express light-entrainable circadian rhythms of melatonin production in culture (Cahill, Citation1996). Zebrafish also have oscillators that control rhythmic expression of several zebrafish clock gene homologs in liver, kidney, heart, and in two embryonically derived cell lines; all when in culture (Pando et al., Citation2001; Whitmore et al., Citation1998; Whitmore et al., Citation2000). Interestingly, rhythms of gene expression within cultured heart and kidney, as well as cell lines (all classically non-photoreceptive) are also directly entrainable by light in culture (Whitmore et al., Citation2000; Pando et al., Citation2001; Vallone et al., Citation2004). Thus, several zebrafish tissues contain endogenous, light-entrainable circadian oscillators that are independent of the brain. It is unclear at this point if the molecular mechanisms in peripheral oscillators and a central oscillator in the brain are similar, but in mammalian systems, the molecular clockwork appears to be very similar in the SCN (the site of the central oscillator) and peripheral tissues (Lee et al., Citation2001; Yagita et al., Citation2001).
We measured the locomotor activity rhythms of larval progeny of mutagenized zebrafish to identify dominant mutations that affect the period length of circadian rhythmicity. Period length is a property of a circadian oscillator that can be modulated by environmental and physiological inputs, so any mutant gene products are predicted to be involved in the generation, entrainment, or modulation of the underlying circadian oscillation.We chose a screen for dominant mutations because many clock mutants identified in other species are semidominant, and because this reduces the number of animals that must be tested. In this article, we present the results of our initial screen and the phenotypic and genetic characterization of the first zebrafish rhythm mutant (lager and lime, lag: cockney rhyming slang for “time”).
MATERIALS AND METHODS
Animals
All of the zebrafish used in this study were bred, raised and maintained in our colony located at the University of Houston. Juvenile (>15 days post fertilization) to adult zebrafish were maintained in 2-liter modular aquaria (Marine Biotech) with continuous water exchange at 28–29°C under a 14:10 light:dark (LD) cycle. Eggs and larvae were maintained in beakers, with daily water changes, under the same LD cycle and similar temperatures. All adult and juvenile fish were fed with live brine shrimp or flake food 1–3 times during the day. Larval fish were fed filtered Paramecium. The AB strain was obtained from Harvard, the *AB strain was obtained from the University of Oregon, and the Northwestern (NW) strain was a gift from J. Takahashi, Northwestern University. All protocols were reviewed and approved by the University of Houston Institutional Animal Care and Use Committee.
Mutagenesis and Screening
Our mutagenesis consisted of randomly mutating zebrafish genomes by submerging adult male zebrafish of the AB strain in 3 mM N-ethylnitrosourea (ENU) for 1 h followed by a one-week recovery period (Solnica-Krezel et al., Citation1994). ENU treatments were repeated 3–4 times. This mutagenesis regime results in the optimal balance between spermatogonial stem cell mutation and acute lethality of the treated males (Solnica-Krezel et al., Citation1994). These males were group-housed with AB wild type females (10–20 fish/2L tank) and bred weekly. The resulting G1 progeny were screened for altered circadian periods by measuring the activity rhythms of 9–17 days old larval fish. On the first day of recording, the water was changed twice, then each fish was transferred to a 750 µl well of either a 100- or 150- well plate. The plate was placed under a video camera in an incubator maintained at 24°C and illuminated with constant infrared light, and behavior was recorded as previously described (Cahill et al., Citation1998). Data analysis was performed with the Chrono II or Chrono 68/PPC software (Roenneberg & Taylor, Citation2000). Activity levels from 24-minute intervals were smoothed with a 4 h moving average, and long-term (>24 h) trends in activity (high or low) were removed with a 25 h moving average. To quantify relative phase at the end of each record, it was cut modulo the period for the population average (25.3 h in wild type, 25.0 h in F1 families, and 24.7 in F2 families); the fourth, fifth and sixth cycles were averaged, and the phase of the resulting peak was measured. Individuals with shorter periods will result in earlier average peak phases than individuals with longer periods after prolonged exposures to constant conditions. Individuals with average phases more than 2.5 h (3 standard deviations) away from the group mean were raised as putative mutants. Putative mutants were backcrossed to wild type and tested to determine if the period difference was heritable. Heterozygous F1 mutants were intercrossed to determine if the mutation was semi-dominant.
Pineal Melatonin Rhythms
Adult fish were decapitated within 3-hours of dark-onset, and pineal glands were dissected in L-15 (Gibco BRL) culture medium supplemented with 2,000 units/mL penicillin, 2 mg/mL streptomycin, and 2.5 µg/mL amphotericin (Gibco BRL). Immediately after dissection, pineal glands were transferred to individual wells of a 96-well plate and maintained in L-15 medium supplemented with 1,000 units/mL penicillin and 1 mg/mL streptomycin until start of flow through culture. At dark onset, the pineal glands were placed in flow-through culture in constant dark and temperature. Flow through cultures were performed essentially as described (Cahill, Citation1996). Culture media consisting of a mixture of balanced salts and amino acids (Cahill, Citation1996) was continuously infused at 1.0 ml/h, collected at 125-minute intervals on a fraction collector, and then stored at 4°C or − 20°C until assayed for melatonin.
Melatonin content of the superfusate samples was determined by RIA performed (Cahill, Citation1996) with minor modifications. Briefly, 20 µl culture media samples were diluted 10 fold with phosphate-buffered saline containing 0.1% gelatin, and incubated for 36–72 hours at 4°C with a 1:256,000 dilution of melatonin antiserum (R1055, a generous gift from Dr. Mark Rollag) and 8,000–20,000 cpm of 125I-labeled melatonin analog in a final volume of 400 µl. After the incubation, each sample was precipitated with 3.0 ml of ice-cold 95% ethanol, and bound 125I-labeled melatonin analog was measured with a Micromedic Apex gamma counter. Melatonin content of samples was determined by comparing the inhibition of 125I-labeled melatonin binding to the antibody in the samples with a standard curve ranging from 0.5 to 500 pg/tube.
The circadian period of melatonin production for each pineal was determined from a cosine fit to the raw integer data by a Fast Fourier Transform coupled to a nonlinear least squares routine (FFT-NLLS; Hurd et al., Citation1998; Plautz et al., Citation1997). One-way and two-way analyses of variance (ANOVA), as well as post-hoc comparisons, were performed using Statistica ‘98 software (StatSoft Inc., Tulsa, OK). Q10 values were calculated using the following formula: [period(T2)/period(T1)](1/T1–T2) (Tosini & Menaker, Citation1998).
To measure phase response curves, pineal glands were cultured at 29°C (±0.2°C), and pulses of white light were delivered by reflecting light from a 150-watt tungsten halogen source (Dolan-Jenner Ind., Model 170-D) through a Plexiglas window located on the underside of the water jacket for 1-h during the second day of the experiment. Light intensities were reduced to ∼ 130 lux using a neutral density filter, and measured using a LX-101 luxmeter (Lutron) just outside the Plexiglass window. We found that this light regime produces sub-maximal phase advances and delays of wild type pineal gland oscillators (data not shown).Light pulses were administered at 3-h intervals around the clock. Mutant and wild-type pineal glands were pulsed at the same circadian time (CT) within the same experiments. Since the periods of lag and wild-type pineal glands differ by two hours when cultured at 29°C, circadian times were predicted based on each genotype's period (see ), using the timing of the entry into DD (the start of the experiment) as a reference. The timing of the light pulses was confirmed by measuring the timing of the half-maximal point between the first peak and trough (designated CT0). Phase shifts were measured relative to non-pulsed controls within the same experiment. For phase shift determination, melatonin levels in each sample were normalized to the average melatonin produced by a pineal during the duration of the experiment. The data were imported into the Chrono 68K/PPC software where they were smoothed with a 4-h moving average and long-term trends were subtracted by a 24-h moving average. The phase shift for each light-pulsed pineal is reported as the difference in average phase of the third and fourth days between the pulse pineal and average phase of the non-pulsed control pineal glands within the same experiment. The circadian time reported for each pulse is the difference between CT 0 and the starting time of the light pulse, converted to circadian hours by dividing by the average pineal period for each genotype (26.7 h for wild type, 24.6 h for homozygous mutants; see Table I).
TABLE I Mutant and Wild Type Pineal Rhythm Characteristics
Linkage Mapping
Bulked Segregant Analysis (Michelmore et al., Citation1991) was performed by testing the segregation of Simple Sequence Length Polymorphism (SSLP) markers with the mutation through three generations (G1, F1, and F2) of the following mapping crosses: The original male lagdg2 mutant individual (G1; AB strain) was crossed with a wild type female from the unrelated NW strain for linkage mapping to produce F1s. The F1 animals with the shortest periods (heterozygotes) were raised and crossed to generate F2 families. The behavioral rhythms of the F2 larvae from five pairs of F1 parents were then monitored as described above to assign phenotype. At the end of the behavioral monitoring, genomic DNA was extracted from whole larval fish as described previously (Zhang et al., Citation1998). Genomic DNA from 25–30 homozygous mutant and homozygous wild type F2 individuals were pooled. Genomic DNA samples from the original G1 mutant and its mate, as well as F1 individuals were obtained from caudal fin clips. For bulked segregant analysis, all genomic DNA samples were purified and resuspended in water to equal concentrations. Genotyping with SSLP markers was performed using 25 µL polymerase chain reactions (PCR) consisting of (per reaction) 0.5–1.0 µL of crude homogenate or 30–50 ng of purified genomic DNA (G1s, F1s, or pools), 1.5 mM MgCl2, 100 µM dNTPs, 240 nM of each primer (Research Genetics), 10 µCi 32P-dCTP (3000Ci/mmol; 10mCi/ml; NEN), 10 ng gelatin, 100 ng bovine serum albumin (Promega), and 2–2.5 units Taq polymerase (Promega). Each reaction was subjected to a 5-min incubation at 94°C, then 27 cycles of 94°C, 57°C, and 72°C (1 min each), and then kept at 4°C. PCR products were separated on 6.0% denaturing sequencing gels, and then analyzed by autoradiography. For linkage mapping of per1, we PCR amplified from genomic DNA a fragment containing a polymorphism within an SspI site (amino acid residue 763; see , below). The PCR conditions were as follows: 0.5–1.0 µl crude genomic DNA, 1.5 mM MgCl2, 100 µM dNTPs, 1.0 µM of each primer (forward: GAGCTCCGTCTCAGCTCATC; reverse: ATCCTGCGCAATTTCAAAAC), and 2 units Taq polymerase (Promega) in 25 µl reactions using the following cycling conditions: 95°C 2 mins, 20 cycles of 95°C, 60–50°C ( − 0.5°C/cycle), 72°C (1 min each), and 20 cycles of 95°C, 50°C, and 72°C (1 min each). A ∼ 400 base pair genomic DNA fragment containing this polymorphism and then digested it with 5–10 Units of SspI (Promega) added directly to the completed PCR reactions. Digestions were incubated at 37°C for 4-hours to overnight. The AB specific allele yields two digested fragments (∼300 and ∼ 100 bases), whereas the NW allele is undigested (∼400 bases). Map distances, LOD scores, and 95% confidence intervals were calculated using the Map Manager QTX software with the Kosambi map function (Manly et al., Citation2001).
TABLE II per1 Polymorphisms in Zebrafish Strains
Radiation Hybrid (RH) mapping of zebrafish clock gene homlogs was performed on the Goodfellow T51 (Geisler et al., Citation1999) or on the LN54 (or both) zebrafish RH panels (Hukriede et al., Citation2001; Hukriede et al., Citation1999) as described. PCR reactions are identical to those described above, and all reactions were performed at least twice. PCR products were separated on 1–3% agarose gels, and DNA was visualized with ethidium bromide staining. The radiation hybrid vector was submitted to http://wwwmap.tuebingen.mpg.de:8082/rh/ for the Goodfellow T51 panel or http://mgchd1.nichd.nih.gov:8000/zfrh/beta.cgi for the LN54 panel, where map positions were determined by the SAMapper or RHMapper programs, respectively.
Sequence Analysis of per1
Total RNA was isolated (using Trizol, Invitrogen) from both retinas of individual homozygous mutant and wild type siblings (2 individuals each) and reverse-transcribed using the SuperScript II kit (Invitrogen). PCR was carried out using the TaqPlus Precision system (Stratagene) and primers (a generous gift from R.K. Barrett and J.S. Takahashi, Northwestern University) designed to amplify overlapping ∼ 500 base pair fragments of the per1 cDNA and cloned into the TOPO-pCR4 vector (Invitrogen) for sequencing. Sequencing was done in both directions using an automated sequencer, and alignments performed using MacVector 6.5.3 and AssemblyLIGN software suites (Oxford Molecular). The sequence with variants has been submitted to Genbank (Accession number AY597250).
Northern Analysis
Larval zebrafish (4–6 days post fertilization) were collected 4 times a day at 6-hour intervals, and either homogenized in Trizol (Life Technologies) immediately or frozen on dry ice. All samples were stored at − 80°C until processed. Total RNA was extracted using Trizol reagent according to manufacturer's instructions, and resuspended in Diethylpyrocarbonate (DEPC) treated water. 10–15 µg, depending on experiment of total RNA was separated on 1% agarose/ 6% formaldehyde gels at 25–30 volts overnight. RNA quality was determined by ethidium bromide staining. Separated RNAs were transferred to neutral nylon membranes (Hybond-N, Pharmacia) using upward diffusion of 20X SSC (3 M NaCl, 0.3 M sodium citrate, pH 7) for 12–48 h. RNA was fixed to the membrane by ultraviolet crosslinking. Blots were incubated in prehybridization solution at 42°C for 1–3 days, and then radiolabeled probes were added directly to the prehybridization solution [prehybridization solution: 5X Denhardt's (Sigma), 50% deionized formamide, 50 mM Tris (pH 7.5), 0.8 M NaCl, 0.1% sodium pyrophosphate, 0.25% SDS, 75 µg/ml denatured sheared salmon sperm DNA (Stratagene), 10% Dextran sulfate (Pharmacia)]. Probes to the 3'untranslated regions of per1 were generated by random primed labeling [Gibco BRL; 10 µCi 32P-dCTP (3000Ci/mmol; 10mCi/ml, NEN)] of cDNA clones corresponding to bases 5000–6439 of accn no. AY597250 and incubated with the blot for 24 h longer than prehybridization (2–4 days). Washed blots were sealed, and exposed to X-ray film (Kodax BioMax MS), with two intensifying screens, at − 80°C for 5–7 days. Band quantification was done using MacBAS software (Fuji).
RESULTS
Behavioral Screening
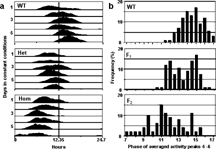
In a mutagenesis-based screen of 6,500 mutagenized genomes, we recovered 8 semi-dominant mutants, seven with short-period activity rhythms and one with a long period. The homozygous mutants’ average periods differ from wild type by 1.0 h to 2.5 h. The first mutant allele that we recovered is designated lagdg2. It is the only allele that we recovered at that locus so far (6 of our 8 mutants have been mapped to date). Activity rhythms have abnormally short periods in this mutant, but it has no obvious developmental or morphological defects. illustrates typical larval actograms for wild type (top), lagdg2 heterozygous (middle), and lagdg2 homozygous (bottom) individuals. The average period of activity rhythms of wild type, AB-strain zebrafish is 25.3 h under our standard recording conditions. The period in heterozygous lagdg2 is shortened to ∼ 24.7 h, and in homozygotes the period is ∼ 24.0 h. We have tested several approaches to quantification of zebrafish activity rhythm period length, and found that the least variable is to average the activity from the fourth, fifth and sixth full circadian cycles and measure the phase of the resulting activity peak; for rhythms that initially start out in the same phase, this measurement reflects period differences. In wild type families, these phase measurements are normally distributed (, top panel). In progeny of a lagdg2/ + X NWWT F1 cross, the variance is increased, and a second peak in the distribution is apparent about three hours earlier than the wild type peak, consistent with a 1:1 phenotypic ratio (, middle panel). In lagdg2 F2 families, the variance in peak phases is further increased, and the distribution is consistent with a phenotypic ratio expected of a semidominant, homozygous-viable, short period mutation.
Figure 1 The lagdg2 variant is a semi-Dominant, short-period mutant. A. Representative actograms of lagdg2 F2 siblings. The F2 individuals are the progeny of a cross between two heterozygous mutants. The circadian periods of most F2 individuals were categorized as wild type (WT, top), heterozygous (het, middle), or homozygous mutant (hom, bottom), based on the phase of the composite peak produced by averaging the last three cycles. A representative individual is shown for each phenotype. The data were smoothed with a 4 hr moving average and plotted modulo 24.7 h to emphasize period differences. B. Frequency distributions of circadian phases (days 4–6) for progeny of wild type and lagdg2 crosses. Each record was cut modulo 24.7 h; the fourth, fifth and sixth cycles were averaged, and the phase of the resulting peak was measured. Animals from a wild type population (WT), lagdg2 heterozygote outcross (F1), or lagdg2 heterozygote intercross (F2) from a typical experiment.
Effects of lagdg2 on Pineal Melatonin Rhythms
To determine whether the lagdg2 mutation affects the clock in the pineal gland, we measured melatonin release rhythms of cultured pineal glands from adult fish that had been previously identified behaviorally as homozygous mutant, heterozygous, or wild type (Cahill, Citation1996). Average rhythms of melatonin production measured from a F2 family in a typical experiment are shown in . There were no significant differences in rhythm amplitude or melatonin production among the groups. The average period of the wild type pineal clock was 24.6 hours (±0.1 h SEM) under these conditions. The average period of homozygous mutant pineal glands was significantly shorter (by 0.7 h) than either wild type or heterozygous periods (ANOVA, Sheffe's S, p < 0.01; Table I). The periods of the pineal clock in heterozygous lagdg2 pineal glands were not statistically different from the wild type periods (ANOVA, p > 0.05), so under these conditions, the mutation is recessive for this rhythm.
Figure 2 Pineal melatonin-production rhythms from lagdg2 F2 siblings. A. Average (n = 6 in each group) melatonin production from pineal glands isolated from lag homozygotes (lagdg2/lagdg2), lag heterozygotes (lagdg2/ + ), and wild type (+/+) siblings that were cultured at 21°C ± 0.1°C. B. Average periods (± SEM) of wild type (WT), heterozyotes (Het), and homozygous (Hom) rhythms shown in A. C. Average melatonin production from pineal glands isolated from lag homozygotes (lagdg2/lagdg;2 n = 13), lag heterozygotes (lagdg2/ + ; n = 10), and wild type ( + / + ; n = 14) siblings that were cultured at 32°C ± 0.1°C. D. Average periods (±SEM) of wild type (WT), heterozyotes (Het), and homozygous (Hom) rhythms shown in C. All individuals had been previously phenotyped by recording larval behavioral rhythms, as in . Furthermore, the genotypes of the individuals shown in C and D were confirmed using linkage to microsatellite z8604 (see below). White and black bars at the top of each A and C indicate light and dark, respectively. *=p < 0.05, ANOVA, Sheffe's S post-hoc test, from wild type. Periods were estimated as in Figure 3.
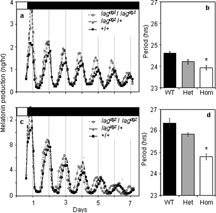
The period difference between homozygous lagdg2 and wild type pineal glands (0.7 h) is less than the period difference observed when larval behavioral rhythms were measured (1.3 h). Aside from the obvious differences in the recording systems (i.e., tissue culture versus moving animals), the temperatures of the monitoring systems were also different. Pineal rhythms were measured at 21°C, whereas larval behavioral rhythms were measured at 24°C. Therefore, we hypothesized that temperature compensation may also be altered in the lagdg2 mutants. To test this hypothesis, we measured melatonin production rhythms of pineal glands isolated from F2 family members and cultured at 32°C. illustrate that at this higher temperature, the periods of pineal gland oscillators of all three phenotypes lengthen with temperature, however, the period difference between wild type and homozygous mutant pineal glands was now 1.5 h (periods of 26.3 and 24.8 h, respectively), more than twice the difference observed at 21°C. These results indicate that the pineal clock slows as temperature increases, and that the lagdg2 alters this temperature sensitivity.
To further define the mutation's effects on the temperature compensation, we measured melatonin rhythms from homozygous lagdg2 and wild type pineal glands cultured in constant darkness at constant temperatures ranging from 15°C to 32°C for 7 days. In these experiments, we used lagdg2 fish from a line that had been backcrossed 3 generations to our wild type strain *AB, a lethal-free derivation of the AB strain (Johnson & Zon, Citation1999), to eliminate any possible effects of potential strain differences. The genotypes of the heterozygous and homozygous founders were confirmed by behavioral and pineal phenotype as well as segregation patterns for linked genetic markers (see below).
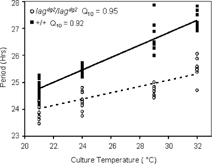
Pineal glands of wild type and lag lines cultured at 15°C and 18°C exhibit rhythms of melatonin production for 1 and 3 days in culture, respectively, after which melatonin was produced at trough levels for the remainder of the experiment (data not shown). Both mutant and wild type pineal glands exhibited circadian rhythms of melatonin production for 7 days in DD at temperatures tested above 18°C. The freerunning period of wild type pineal rhythms lengthened as the temperature increased (). The wild type periods ranged from 24.8 h at 21°C to 27.3 hours at 32°C (Table I). Freerunning periods of lagdg2 pineal rhythms lengthened, but to a lesser extent (), ranging from 24.1 hours at 21°C to 25.5 hours at 32°C (Table I). Two-way ANOVA indicates that the temperature effect on the period difference between lagdg2 and wild type is significant (p < 0.001). Q10 values of periods were 0.92 and 0.95 for wild type and mutant pineal glands, respectively.
Figure 3 Individual pineal periods at different temperatures. Pineal glands were dissected from our *AB wild type strain (+/+), or a backcrossed homozygous lag mutant line that had been generated by backcrossing to the *AB line for 3 generations (lagdg2/lagdg2). Pineal glands were cultured at the indicated temperatures (±0.1°C) for seven days in constant darkness. Periods were determined using FFT-NLLS for each pineal cultured at each temperature. The period data were fit with linear regressions. The R2 value and slope for lag pineal gland periods were 0.74 and 0.12, respectively. The R2 value and slope for the linear regression lines of wild type pineal periods were 0.89 and 0.22, respectively.
Light-Pulse PRCs of lagdg2 and Wild Type Pineal Clocks
Several period-altering mutations in other animals also alter the phase-dependent resetting responses of circadian clocks to light (Albrecht et al., Citation2001; Grosse et al., Citation1995; Shimomura & Menaker, Citation1994). To determine if this was the case in lagdg2 mutants, we compared light phase-response curves (PRCs) of mutant and wild type pineal glands. Pineal glands were cultured for five days in DD at 29°C, a temperature where there is a large difference in homozygous mutant and wild-type periods. Light pulses of white light at a subsaturating intensity were administered to mutant and wild-type pineal glands at the same circadian time (CT) within the same experiments, and measured relative to non-pulsed controls, also within the same experiments.
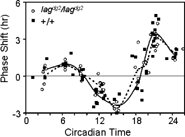
The PRCs for the lagdg2 and wild type pineal oscillators are qualitatively similar to light pulse PRCs in other systems (). Maximal phase delays of 2.3 hours were obtained at CT 15, and maximal phase advances of 3 h at CT 21. Light administered at CT 18 induced a wide variety of responses among the individual pineal glands of either line, resulting in an average phase shift of 0 for either group. Overall, the PRCs for both mutant and wild-type lines are very similar in phase, shape, magnitude of phase shifts. This finding suggests that that the pineal gland light-input pathway and/or the responsiveness of the pineal oscillator to light are not grossly altered by the mutation.
Figure 4 Phase response curves of wild Type and lag pineal glands. Pineal glands from either *AB wild type or our lag backcross line were cultured in constant darkness and 29°C temperature. One-hour white light pulses were administered at the circadian times indicated. PRCs for lag (dashed curve) and wild type (solid curve) pineal glands for average phase shifts at each CT time, with individual CT times and phase shifts are shown. Phase shifts are the difference in phase between light pulsed and control pineal glands (see Materials and Methods for a complete description).
Linkage Mapping of the lag Mutation
In order to map the mutation, we generated a mapping cross of lagdg2 (AB strain) with wild types of the NW strain. The distribution of F2 phenotypes ( was consistent with three populations with partially overlapping distributions at a 1:2:1 ratio. Therefore, we selected individuals from the extremes of the distribution as homozygotes, and individuals from the center of the distribution as heterozygotes. We first scanned the genome by Bulked Segregant Analysis (Michelmore et al., Citation1991; Postlethwait & Talbot, Citation1997) with microsatellite markers (Shimoda et al., Citation1999). We found two polymorphic markers that appeared to be linked to the lag mutation: z10487 and z11085. These are located at 53.4 and 33.2 cM respectively on linkage group 7 (http://zebrafish.mgh.harvard.edu/). We found no apparent linkage to 29 other markers on 20 of 24 other linkage groups/chromosomes. Polymorphic microsatellites were not tested for linkage groups 2, 4, 9 and 11.
Genotyping of individual F2s for 7 other LG 7 markers revealed that lag is most closely linked to z8604 at 59 cM on LG 7. We genotyped a panel of 500 F2s for the four closest polymorphic markers to produce a meiotic map (). The shortest calculated map agrees with the relative order of the microsatellites published previously (Mohideen et al., Citation2000; Shimoda et al., Citation1999). Lag mapped to the interval between z8604 and z20715, approximately 4.1 centiMorgans (34 recombinations/838 meioses) from z8604 (). Two-point LOD scores for linkage to z8604 and z20715 were 147.8 and 77.2, respectively (LOD scores > 3 are considered significant). However, in 6.4% of individuals, the predicted genotype for lag differed from both z8604 and z20715, while only 1.3% are expected to carry two recombinations in the 11.3 cM interval between these markers. This suggests that ∼ 5% of the lag genotypes were misassigned due to variability in the behavioral phenotype, so the maximum mapping resolution possible using behavioral rhythms as an assay is approximately 5 cM.
Figure 5 Genetic maps for Linkage Group Seven. Linkage mapping of the lag mutation was performed on a panel of 500 diploid animals. The thick bar indicates the approximate map position of lag. lag is within a 5.1 cM range, 0.6 cM away from z8604. Two point LOD scores between markers are all greater than greater than 70. The relative position of microsatellite markers within the TN54 radiation hybrid map is shown on the right for comparison.

Mapping of Candidate Genes for the lag Mutation
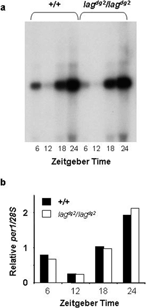
Over 30 zebrafish homologues of circadian clock or clock-related genes found in other animals have been mapped on Radiation Hybrid mapping panels by us or by others (Sprague et al., Citation2003). Mapping of genes, expressed sequence tags, large genomic DNA clones, and microsatellite markers is a major effort of the zebrafish community to facilitate the cloning of mutant genes via a “candidate gene” approach. This approach is much more amenable for identifying mutant genes that give rise to more subtle phenotypes, such as lag, than standard positional cloning/chromosome walking approaches that require high-resolution mapping. So far, this approach has been used to identify a number of mutant genes (Barrallo-Gimeno et al., Citation2004; Lele et al., Citation2002; Lyons et al., Citation2002). The circadian clock or clock-related genes mapped on these panels include nocturnin, two homologues each for gsk3, rev-erb. and slimb, three for clock/npas2, bmal1/2, and ck1 ϵ, four for per1/2/3, five for 4bp4, and six cryptochromes. Zebrafish period1 (per1) was mapped within 5 centiRays (cR) from microsatellite z8604, the marker closest to lag, on the T51 RH panel. All others that have been mapped so far are linked to SSLP markers that are > 20 cM from lag (data not shown). We identified a polymorphism that created an SspI restriction site within the mutant-linked per1 allele of our mapping cross (see below). We used this restriction-site polymorphism to map meiotically the per1 gene on the mapping panel used to map lag. We found, as illustrated in , that per1 mapped to a locus indistinguishable from microsatellite z8604 (no recombinations in 1000 meioses), thus closely linked to the lag locus. There were 34 apparent recombinations between per1 and lag. However, given the apparent frequency of misassigned genotypes described above, the possibility that per1 and lag map to the same position can't be ruled out. Sequencing of the per1 cDNA from homozygous mutant and wild-type siblings revealed 7 polymorphisms that result in predicted amino acid substitutions as well as 22 silent polymorphisms. However, sequencing cloned RT-PCR fragments amplified from several strains wild-type for lag indicated that none of the polymorphisms were limited to homozygous mutants (). Therefore, the zebrafish per1 gene does not appear to be mutated within lagdg2 mutants. Additionally, we performed Northern analysis of the per1 mRNA in homozygous mutant and wild type larvae to determine if there was a mutation within a non-coding region that might affect RNA splicing. Since we didn't know if, and what phase, per1cycled in, we chose timepoints every 6-hrs over the course a day in LD. This analysis revealed that per1 mRNA peaks during the night time, and that there is no obvious affect of the lagdg2 mutation on mRNA splicing in the mutant (). Furthermore, the mutation didn't seem to dramatically influence the per1 abundance rhythms in the mutant ().Taken together, these data indicate zebrafish per1 is tightly linked to the lag locus, but also indicate it is probably not responsible for the mutant phenotype. Thus, lagdg2 may identify a novel clock gene.
Figure 6 Northern-blot analysis of zebrafish per1. A.Total RNA was isolated from 5–6 day old zebrafish larvae at 6 hour intervals during a light:dark cycle (14:10). Lights on at Zeitgeber Time (ZT) 22, and off at ZT 12. Wild type larvae were of strain *AB, and the homozygous lag larvae were from our backcrossed line. B. The per1 signals were normalized for loading with the 28S, and plotted relative to the average signal for each phenotype, and the average of two independent blots is shown.
DISCUSSION
Screens for mutations that affect circadian period length have been performed in mice and extensively in Drosophila, and have identified many components involved in the generation and regulation of animal circadian clocks. We have used another vertebrate model, zebrafish, in a similar approach to identify components of vertebrate circadian clocks.
Zebrafish have become a very valuable model organism for genetic analyses of various aspects of vertebrate biology. Zebrafish have two main physical advantages over mammals for mutant screens in that they are very prolific breeders (>100 progeny/week from a single pair) and they are relatively small; both prerequisites for efficient high-throughput mutant screening approaches. High-throughput mutant screening allows for recovery of multiple mutant alleles for a single gene, and it is conceivable to screen until saturation, that is, until all the mutable clock genes are identified. Furthermore, using mutant screens is ideal for identifying novel clock genes since it doesn't make any assumptions about the clock except that there is a genetic component. The genetic tools one needs to identify most mutant genes recovered via mutagenesis screening have been developed, making this a complete system for this type of ‘forward genetic’ approach in vertebrates.
So far, we have recovered eight semidominant mutations that alter the freerunning period of locomotor rhythms, including the single allele of the lag locus described here. Period length is a property of a circadian oscillator that can be modulated by environmental and physiological inputs, so the mutated gene product is predicted to be involved in the generation, entrainment, or modulation of the underlying circadian oscillation.
We have further characterized the circadian phenotypes of the lagdg2 mutant by measuring the effect of the mutation on the clock within the pineal. Pineal glands isolated from homozygous mutants display short circadian periods compared to their wild type siblings when monitored in culture, indicating that the mutation affects this oscillator as well. This result indicates that the mutation affects circadian periodicity at the tissue level as well as behavior. Furthermore, because the mutation affects two independent circadian rhythms in similar ways (shortens period), it is possible that the gene product may be part of a mechanism common to many circadian oscillators. However, melatonin rhythms of heterozygous pineal glands in culture are indistinguishable from those of wild type pineal glands, regardless of temperature, suggesting that the period or temperature compensation phenotypes may be recessive in the pineal oscillator. This is different than the effect on behavioral rhythms, where the mutation is clearly semidominant.
Pineal glands of wild type and homozygous lag lines cultured at 15°C and 18°C exhibit rhythms of melatonin production for 1 and 3 days in culture, respectively; after which, melatonin was produced at trough levels for the remainder of the experiment. This result suggests that the pineal glands are still viable at these temperatures, since they still produce detectable amounts of melatonin. The arrhythmicity at these temperatures could be the result of a non-functioning clock or uncoupling of the clock from the output pathway to melatonin, but this has not been determined.
At permissive temperatures, pineal glands of both genotypes are temperature compensated, with Q10 values that are within the circadian range (0.8–1.2; Pittendrigh, Citation1957). However, pineal clocks from both wild types and mutants overcompensate for temperature since their Q10 values are less than 1. Temperature overcompensation is consistent with the notion that temperature compensation is not simply a lack of temperature sensitivity; rather, it is probably an evolved mechanism (Peixoto et al., Citation1998; Pittendrigh, Citation1957). Clocks within the chick pineal gland, Aplysia eye, mammalian cell lines, retina, and SCN all have temperature compensated periods when measured in culture with Q10s ranging from 0.8–1.1 (Barrett & Takahashi, Citation1995; Izumo et al., Citation2003; Ruby & Heller, Citation1996; Ruby et al., Citation1999; Tosini & Menaker, Citation1998; Tsuchiya et al., Citation2003; Jacklet, Citation1980). Circadian periods of lag mutant pineal glands are less sensitive to temperature relative to wild type. This phenotype has not been described previously for any other vertebrate circadian clock mutant, although there is limited data in this regard. The tau mutation adversely affects temperature compensation in the cultured hamster retina (Tosini & Menaker, Citation1998). The lagdg2 mutation seemingly improves compensation; however this phenotype may really reflect a modulatory effect of the mutation on a poorly understood mechanism. Identifying the mutant gene may shed light on this phenomenon.
We did not detect an effect of the mutation on the circadian light input pathway in the pineal. For these experiments, we used subsaturating white light intensities so that we might see an effect on light sensitivity. However, PRCs for subsaturating light intensities for both wild type and mutant pineal glands were indistinguishable; suggesting that transmission of light information to the clock is unaffected. However, since we only tested white light at a single intensity, we cannot rule out a subtle effect of the mutation on color light detection or light information flow to the clock.
The lag locus is strongly linked to per1, an obvious candidate gene on LG7. However, uncertainties in lag genotype assignment in our mapping cross prevent us from conclusively ruling it in or out as the affected gene. We did find seven predicted amino acid differences between lagdg2 and wild type per1, but all of the lagdg2 amino acid substitutions were also found in one or another wild type line, so it is unlikely that any was induced by ENU treatment. This level of polymorphism within the per1 coding region seems particularly high within the species, given that as of this writing, there have only been 2 silent coding region polymorphisms identified in either of mouse Clock, Bmal1, Per1, Per2, Cry1, or Cry2 according to the single nucleotide polymorphism database (SNP) maintained by NCBI (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD = search&DB = snp; as of July, 2004). It remains to be determined if other zebrafish clock gene homologs also have such high levels of polymorphism between strains.
Northern blot analysis of per1 mRNA did not reveal any obvious differences in abundance or splicing between homozygous mutant and wild type strains, so we found no evidence for a regulatory sequence mutation. Further experimentation is required to determine if there is a more subtle effect.One possibility that can't be excluded is that we selected a founder carrying a rare haplotype with the lagdg2 per1 sequence, and that this causes periods shorter than any that we have observed in wild type fish. We have not sequenced full-length per1 cDNAs to determine whether this is might be the case. An alternative is that lagdg2 is a mutation in another gene that is closely linked to per1. We haven't been able to identify any obvious candidates from existing radiation hybrid maps, nor from genome sequence information available as of this writing, so lag may be a novel clock gene.
Positional cloning of the gene mutated in lagdg2 will require linkage mapping with higher resolution than is possible using our current larval locomotor rhythm assay. Longer records or less noise in a rhythm assay would be expected to reduce the variability in period estimates and make this possible. Recording behavioral rhythms for longer than seven days would require more sophisticated apparatus to supply food ad lib and maintain water quality. Alternatively, pineal melatonin rhythms in vitro are less noisy and would provide more reliable period estimates, but would they would be difficult to record in large numbers for high resolution mapping. A third approach, used effectively in non-vertebrate screens (Liu et al., Citation1995; Millar et al., Citation1995; Kondo et al., Citation1994), would be to develop transgenic lines expressing a luciferase reporter under the control of a clock-driven promoter. We are currently pursuing this type of approach. Recently, Vallone et al. (Citation2004) showed that a period promoter drives strong and consistent rhythms of luciferase in zebrafish cell lines, suggesting that this will be feasible.
Ultimately, the work reported here illustrates the potential (recovery and mapping of mutations), and some of the limitations (subtle phenotypes), of zebrafish as a genetic system for understanding vertebrate circadian rhythmicity. However, as screening methods improve, as well as the completion of the zebrafish genome sequencing project, overcoming the difficulties in identifying such mutant-gene products should be feasible. Thus, we believe that zebrafish will be a remarkably useful system for genetic analyses of circadian rhythms in vertebrates.
*Present address: Department of Neurobiology, LRB 770A, University of Massachusetts Medical School, 364 Plantation Street, Worcester, MA 01605.
Acknowledgements
We thank Rhia Pascua and Ronit Spotts for technical assistance and Fanny S. Ng and Qin Zhao for help with the meiotic mapping. We also thank Keith Barrett (Northwestern University) and the Harvard and Oregon Zebrafish Facilities for zebrafish strains and Keith Barrett and Joseph Takahashi (Northwestern University) for the zebrafish per1 sequence, cDNA clone, and several sets of PCR primers. J.D. was supported in part by the Houston Area Vision Training Program institutional grant 5-T32-EY007024 (NIH). This research was supported by NIH grant MH60939 and Texas Advanced Research Program grant 3652–761 awarded to G.M.C.
REFERENCES
- Albrecht U., Zheng B., Larkin D., Sun Z.S, Lee C.C. 2001. mPer1 and mPer2 are essential for normal resetting of the circadian clock. J. Biol. Rhythms. 16: 100–104. [PUBMED], [INFOTRIEVE], [CSA]
- Allada R., Emery P., Takahashi J.S., Rosbash M.. 2001. Stopping time: the genetics of fly and mouse circadian clocks. Ann. Rev. Neurosci.. 24: 1091–1119. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Amsterdam A.. 2003. Insertional mutagenesis in zebrafish. Devel. Dyn.. 228: 523–534. [CSA], [CROSSREF]
- Barrallo-Gimeno A., Holzschuh J., Driever W., Knapik E.W.. 2004. Neural crest survival and differentiation in zebrafish depends on mont blanc/tfap2a gene function. Development. 131: 1463–1477. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Barrett R.K, Takahashi J.S. 1995. Temperature compensation and temperature entrainment of the chick pineal cell circadian clock. J. Neurosci.. 15: 5681–5692. [PUBMED], [INFOTRIEVE]
- Brockerhoff S.E., Hurley J.B., Janssen-Bienhold U., Neuhauss S.C., Driever W., Dowling J.E. 1995. A behavioral screen for isolating zebrafish mutants with visual system defects. Proc. Natl. Acad. Sci. US. 92: 10545–10549. [CSA]
- Cahill G.M. 1996. Circadian regulation of melatonin production in cultured zebrafish pineal and retina. Brain Res.. 708: 177–181. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Cahill G.M., Hurd M.W., Batchelor M.M.. 1998. Circadian rhythmicity in the locomotor activity of larval zebrafish. Neuroreport. 9: 3445–3449. [PUBMED], [INFOTRIEVE], [CSA]
- Darland T., Dowling J.E.. 2001. Behavioral screening for cocaine sensitivity in mutagenized zebrafish. Proc. Natl. Acad. Sci. US. 98: 11691–11696. [CSA], [CROSSREF]
- Delaunay F., Thisse C., Marchand O., Laudet V., Thisse B.. 2000. An inherited functional circadian clock in zebrafish embryos. Science. 289: 297–300. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Felsenfeld A.L.. 1996. Defining the boundaries of zebrafish developmental genetics. Nat. Genet.. 14: 258–263. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Geisler R., Rauch G.J., Baier H., van Bebber F., Bross L., Dekens M.P., Finger K., Fricke C., Gates M.A., Geiger H., Geiger-Rudolph S., Gilmour D., Glaser S., Gnugge L., Habeck H., Hingst K., Holley S., Keenan J., Kirn A., Knaut H., Lashkari D., Maderspacher F., Martyn U., Neuhauss S., Neumann C., Nicolson T., Pelegri F., Ray R., Rick J.M., Roehl H., Rowser T., Schauerte H.E., Schier A.F., Schonberger U., Schonthaler H.-B., Schulte-Merker S., Seydel C., Talbot W.S., Weiler C., Nüsslein-Volhard C., Haffter P.. 1999. A radiation hybrid map of the zebrafish genome. Nat. Genet.. 23: 86–89
- Golling G., Amsterdam A., Sun Z., Antonelli M., Maldonado E., Chen W., Burgess S., Haldi M., Artzt K., Farrington S., Lin S.Y, Nissen R.M., Hopkins N.. 2002. Insertional mutagenesis in zebrafish rapidly identifies genes essential for early vertebrate development. Nat. Genet.. 31: 135–140. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Granato M., Nusslein-Volhard C.. 1996. Fishing for genes controlling development. Curr. Opin. Genet. Devel.. 6: 461–468. [CSA], [CROSSREF]
- Grosse J., Loudon A.S., Hastings M.H.. 1995. Behavioural and cellular responses to light of the circadian system of tau mutant and wild-type Syrian hamsters. Neuroscience. 65: 587–597. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Hofstetter J.R, Trofatter J.A., Kernek K.L., Nurnberger J.I, Mayeda A.R.. 2003. New quantitative trait loci for the genetic variance in circadian period of locomotor activity between inbred strains of mice. J. Biol. Rhythms. 18: 450–462. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Hukriede N., Fisher D., Epstein J., Joly L., Tellis P., Zhou Y., Barbazuk B., Cox K., Fenton-Noriega L., Hersey C., Miles J., Sheng X., Song A., Waterman R., Johnson S.L., Dawid I.B., Chevrette M., Zon L.I, McPherson J., Ekker M.. 2001. The LN54 radiation hybrid map of zebrafish expressed sequences. Genome Res.. 11: 2127–2132. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Hukriede N.A., Joly L., Tsang M., Miles J., Tellis P., Epstein J.A, Barbazuk W.B., Li F.N., Paw B., Postlethwait J.H., Hudson T.J., Zon L.I., McPherson J.D., Chevrette M., Dawid I.B., Johnson S.L., Ekker M.. 1999. Radiation hybrid mapping of the zebrafish genome. Proc. Natl. Acad. Sci. US. 96: 9745–9750. [CSA], [CROSSREF]
- Hurd M.W., Debruyne J., Straume M., Cahill G.M.. 1998. Circadian rhythms of locomotor activity in zebrafish. Physiol Behav.. 65: 465–472. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Izumo M., Johnson C.H., Yamazaki S.. 2003. Circadian gene expression in mammalian fibroblasts revealed by real-time luminescence reporting: temperature compensation and damping. Proc. Natl. Acad. Sci. US. 100: 16089–16094. [CSA], [CROSSREF]
- Jacklet J.W.. 1980. Circadian rhythm from the eye of Aplysia: temperature compensation of the effects of protein synthesis inhibitors. J. Exp. Biol.. 84: 1–15. [PUBMED], [INFOTRIEVE]
- Johnson S.L., Zon L.I.. 1999. Genetic backgrounds and some standard stocks and strains used in zebrafish developmental biology and genetics. Meth. Cell Biol.. 60: 357–359
- Kazimi N., Cahill G.M.. 1999. Development of a circadian melatonin rhythm in embryonic zebrafish. Brain Res. Devel. Brain Res.. 117: 47–52. [CSA], [CROSSREF]
- Kondo T., Tsinoremas N.F., Golden S.S., Johnson C.H., Kutsuna S., Ishiura M.. 1994. Circadian clock mutants of cyanobacteria. Science. 266: 1233–1236. [PUBMED], [INFOTRIEVE]
- Lee C., Etchegaray J.P., Cagampang F.R., Loudon A.S., Reppert S.M.. 2001. Posttranslational mechanisms regulate the mammalian circadian clock. Cell. 107: 855–867. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Lele Z., Folchert A., Concha M., Rauch G.J, Geisler R., Rosa F., Wilson S.W., Hammerschmidt M., Bally-Cuif L.. 2002, parachute/n-cadherin is required for morphogenesis and maintained integrity of the zebrafish neural tube. Development 129. 3281–3294
- Liu Y., Golden S.S., Kondo T., Ishiura M., Johnson C.H.. 1995. Bacterial luciferase as a reporter of circadian gene expression in cyanobacteria. J. Bacteriol.. 177: 2080–2086. [PUBMED], [INFOTRIEVE]
- Lowrey P.L., Takahashi J.S.. 2000. Genetics of the mammalian circadian system: Photic entrainment, circadian pacemaker mechanisms, and posttranslational regulation. Ann. Rev. Genet.. 34: 533–562. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Lyons S.E., Lawson N.D., Lei L., Bennett P.E., Weinstein B.M., Liu P.P.. 2002. A nonsense mutation in zebrafish gata1 causes the bloodless phenotype in vlad tepes. Proc. Natl. Acad. Sci. US. 99: 5454–5459. [CSA], [CROSSREF]
- Manly K.F., Cudmore R.H., Jr., Meer J.M. 2001. Map Manager QTX, cross-platform software for genetic mapping. Mamm. Genome. 12: 930–932. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Mayeda A.R., Hofstetter J.R.. 1999. A QTL for the genetic variance in free-running period and level of locomotor activity between inbred strains of mice. Behav. Genet.. 29: 171–176. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Michelmore R.W., Paran I., Kesseli R.V.. 1991. Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl. Acad. Sci. US. 88: 9828–9832. [CSA]
- Millar A.J., Carre I.A., Strayer C.A., Chua N.H., Kay S.A.. 1995. Circadian clock mutants in Arabidopsis identified by luciferase imaging. Science. 267: 1161–1163. [PUBMED], [INFOTRIEVE]
- Mohideen M.A., Moore J.L., Cheng K.C.. 2000. Centromere-linked microsatellite markers for linkage groups 3, 4, 6, 7, 13, and 20 of zebrafish (Danio rerio). Genomics. 67: 102–106. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Neuhauss S.C.. 2003. Behavioral genetic approaches to visual system development and function in zebrafish. J. Neurobiol.. 54: 148–160. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Okamura H.. 2003. Integration of molecular rhythms in the mammalian circadian system. Novartis. Found. Symp.. 253: 161–170. [PUBMED], [INFOTRIEVE], [CSA]
- Pando M.P., Pinchak A.B., Cermakian N., Sassone-Corsi P.. 2001. A cell-based system that recapitulates the dynamic light-dependent regulation of the vertebrate clock. Proc. Natl. Acad. Sci. US. 98: 10178–10183. [CSA], [CROSSREF]
- Peixoto A.A., Hennessy J.M., Townson I., Hasan G., Rosbash M., Costa R., Kyriacou C.P. 1998. Molecular coevolution within a Drosophila clock gene. Proc. Natl. Acad. Sci. US. 95: 4475–4480. [CSA], [CROSSREF]
- Pittendrigh C.S. 1957. An oscillator model for biological clocks. Rhythmic processes and growth, D., Rudnick, Princeton, NJ, Princeton University Press. pp. 75–109
- Plautz J.D., Straume M., Stanewsky R., Jamison C.F., Brandes C., Dowse H.B., Hall J.C., Kay S.A.. 1997. Quantitative analysis of Drosophila period gene transcription in living animals. J. Biol. Rhythms 12. 20: 4–217
- Postlethwait J.H, Talbot W.S.. 1997. Zebrafish genomics: from mutants to genes. Trends Genet.. 13: 183–190. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Reppert S.M., Weaver D.R.. 2002. Coordination of circadian timing in mammals. Nature. 418: 935–941. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Roenneberg T., Taylor W.. 2000. Automated recordings of bioluminescence with special reference to the analysis of circadian rhythms. Meth. Enzymol.. 305: 104–119. [PUBMED], [INFOTRIEVE]
- Ruby N.F., Burns D.E., Heller H.C.. 1999. Circadian rhythms in the suprachiasmatic nucleus are temperature-compensated and phase-shifted by heat pulses in vitro. J. Neurosci.. 19: 8630–8636. [PUBMED], [INFOTRIEVE]
- Ruby N.F., Heller H.C.. 1996. Temperature sensitivity of the suprachiasmatic nucleus of ground squirrels and rats in vitro. J. Biol. Rhythms. 11: 126–136. [PUBMED], [INFOTRIEVE], [CSA]
- Shimoda N., Knapik E.W., Ziniti J., Sim C., Yamada E., Kaplan S., Jackson D., de Sauvage F., Jacob H., Fishman M.C.. 1999. Zebrafish genetic map with 2000 microsatellite markers. Genomics. 58: 219–232. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Shimomura K., Low-Zeddies S.S, King D.P., Steeves T.D., Whiteley A., Kushla J., Zemenides P.D., Lin A., Vitaterna M.H., Churchill G.A., Takahashi J.S.. 2001. Genome-wide epistatic interaction analysis reveals complex genetic determinants of circadian behavior in mice. Genome Res.. 11: 959–980. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Shimomura K., Menaker M.. 1994. Light-induced phase shifts in tau mutant hamsters. J. Biol. Rhythms. 9: 97–110. [PUBMED], [INFOTRIEVE], [CSA]
- Shirasu N., Shimohigashi Y., Tominaga Y., Shimohigashi M.. 2003. Molecular cogs of the insect circadian clock. Zoolog. Sci.. 20: 947–955. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Solnica-Kreze l.L., Schier A.F., Driever W.. 1994. Efficient recovery of ENU-induced mutations from the zebrafish germline. Genetics. 136: 1401–1420. [CSA]
- Sprague J., Clements D., Conlin T., Edwards P., Frazer K., Schaper K., Segerdell E., Song P., Sprunger B., Westerfield M.. 2003. The Zebrafish Information Network (ZFIN): the zebrafish model organism database. Nucleic Acids Res.. 31: 241–243. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Suzuki T., Ishikawa A., Nishimura M., Yoshimura T., Namikawa T., Ebihara S.. 2000. Mapping quantitative trait loci for circadian behavioral rhythms in SMXA recombinant inbred strains. Behav. Genet.. 30: 447–453. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Suzuki T., Ishikawa A., Yoshimura T., Namikawa T., Abe H., Honma S., Honma K., Ebihara S.. 2001. Quantitative trait locus analysis of abnormal circadian period in CS mice. Mamm. Genome 12. 27: 2–277. [CSA]
- Tosini G., Menaker M.. 1998. The tau mutation affects temperature compensation of hamster retinal circadian oscillators. Neuroreport. 9: 1001–1005. [PUBMED], [INFOTRIEVE], [CSA]
- Tsuchiya Y., Akashi M., Nishida E.. 2003. Temperature compensation and temperature resetting of circadian rhythms in mammalian cultured fibroblasts. Genes to Cells. 8: 713–720. [PUBMED], [INFOTRIEVE], [CSA]
- Vallone D., Gondi S.B., Whitmore D., Foulkes N.S.. 2004. E-box function in a period gene repressed by light. Proc. Natl. Acad. Sci. US. 101: 4106–4111. [CSA], [CROSSREF]
- Whitmore D., Foulkes N.S., Sassone-Corsi P.. 2000. Light acts directly on organs and cells in culture to set the vertebrate circadian clock. Nature. 404: 87–91. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Whitmore D., Foulkes N.S., Strahle U., Sassone-Corsi P.. 1998. Zebrafish Clock rhythmic expression reveals independent peripheral circadian oscillators. Nat. Neurosci.. 1: 701–707. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Yagita K., Tamanini F., Der Horst G.T., Okamura H.. 2001. Molecular mechanisms of the biological clock in cultured fibroblasts. Science. 292: 278–281. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Zhang J., Talbot W.S., Schier A.F. 1998. Positional cloning identifies zebrafish one-eyed pinhead as a permissive EGF-related ligand required during gastrulation. Cell. 92: 241–251. [PUBMED], [INFOTRIEVE], [CROSSREF]