
Abstract
Astroglia are a morphologically diverse and highly abundant cell type in the CNS. Despite these obvious observations, astroglia still remain largely uncharacterized at the cellular and molecular level. In disease contexts such as amyotrophic lateral sclerosis (ALS), it has been widely shown that astroglia downregulate crucial physiological functions, become hypertrophied, reactive, and toxic to motor neurons. However, little is known about the astroglia-specific transcriptomic changes that occur during ALS disease progression, especially early in disease. To address this, we FACS-isolated pure astroglia from early and mid-symptomatic superoxide dismutase 1 (SOD1) G93A spinal cord and performed microarray sequencing, in hopes to uncover markers and pathways driving astroglia dysfunction in ALS. After extensive analyses, we uncovered genes selectively enriched and downregulated in both control and SOD1 astroglia at both disease points. In addition, we were able to identify genes and pathways differentially expressed that may have relevance with other neurodegenerative diseases, such as Parkinson’s and Alzheimer’s disease, suggesting a common theme among astroglial dysfunction in neurodegenerative disease. In aggregate, this study sheds light on the common and unique themes of dysfunction that astroglia undergo during neurodegenerative disease progression and provides candidate targets for therapeutic approaches.
Introduction
Despite our understanding that astroglia are the major cell type in the central nervous system (CNS), they still remain largely misunderstood (Miller & Rothstein, Citation2016; Oberheim, Goldman, & Nedergaard, Citation2012; Zhang & Barres, Citation2010). The central dogma of astroglia exploits their ability to maintain homeostasis of the CNS and provide a supportive scaffolding environment for neurons and non-neuronal cells (Volterra & Meldolesi, Citation2005). Furthermore, studies are beginning to elucidated how astroglia influence neurogenic niches in the developing and adult brain to modulate the proper maturation of neuronal functions, particularly in the hippocampus (Sultan et al., Citation2015). Modulation of neurons by astrocytes typically involves the secretion of gliotransmitters and molecules such as thrombospondin, but also neurotransmitter recycling at perisynaptic terminals and astroglia wrapping of all neuronal synapses (Sofroniew & Vinters, Citation2010).
One major neurotransmitter that is dysregulated in amyotrophic lateral sclerosis (ALS) pathogenesis is glutamate (Rothstein, Van Kammen, Levey, Martin, & Kuncl, Citation1995). In ALS, glutamate-induced motor neuron excitotoxicity is due to excessive glutamate release, incompetent re-uptake and neurotoxic activation of motor neuron glutamate receptors leading to neuronal death. Human and rodent astrocytes are a key cell type that influences these aberrations (Rothstein et al., Citation1995). In particular, downregulation of the critical glutamate plasma membrane transporter, excitatory amino acid transporter 2 (EAAT2) or Glt1 in the mouse (Guo et al., Citation2010) occurs in human disease and rodent models. Downregulation of Glt1 exacerbates the glutamate toxicity by reducing the amount of glutamate being recycled from the extracellular space into astrocytes, rendering motor neurons susceptible to excitotoxicity (Rothstein et al., Citation1995).
The initiating steps that lead to Glt1 downregulation in astrocytes are not fully understood. Studies more than a decade later provided direct evidence, at least in vitro, that ALS astrocytes could contribute to neuronal degeneration (Re et al., Citation2014). Understanding this contributing cell type may provide new opportunities to treat ALS. To investigate this dysregulation and search for disease triggers, we performed an extensive transcriptomic array on a highly pure population of ALS-disease progressing adult astrocytes from the spinal cord of SOD1 G93A mouse model. We chose early-symptomatic and mid-symptomatic time points, as a novel way to evaluate the disease burden before it was too late (i.e. end stage). We identified genetic markers in the Cathepsin family such as in Ctss, which have previously been shown to be associated with neurodegeneration such Alzheimer’s disease and in ALS (Chiu et al., Citation2013). However, we also identified markers and pathways unique to ALS disease burdened astrocytes at these early time points compared to littermate controls. In particular, we identified over 1000 markers differentially regulated between control and ALS astrocytes with highest differential expression in receptor tyrosine kinase inhibitor, Spry2. Finally, our pathway analyses revealed that the tight junction signaling pathway was the most differentially regulated pathway between control and mutant SOD1 astroglia. In aggregate, we believe these markers and pathways provide insight into early astrocyte dysregulation in ALS, provide the scientific community with an astroglia-specific ALS transcriptomic database, and should be further evaluated for potential therapeutic intervention.
Materials and methods
Animal models
BAC-ALDH1L1-eGFP and the G93A SOD1 (Jackson Labs, Bar Harbor, ME) mice were used for in vivo experiments. The care and treatment of animals is in accordance with the NIH Guide for the Care and Use of Laboratory Animals, the Guidelines for the Use of Animals in Neuroscience Research, and the Johns Hopkins University IACUC (protocol M14M089). Mice were housed at standard temperature (21 °C) and in light-controlled environment with ad libitum access to the food and water. BAC-ALDH1L1-eGFP mice were crossed with G93A SOD1 mice to generate double transgenic mice. Littermates were used as control and for comparison between different astrocyte populations. Not more than five mice were kept in a cage, in accordance with Johns Hopkins IACUC. Mice were then sacrificed at a designated time point post breeding and aging.
Fluorescence-activated cell sorting (FACS)
Spinal cord astrocytes from BAC-ALDH1L1-eGFP and BAC-ALDH1L1-eGFP/G93A SOD1 double transgenic mice were analyzed by FACS. Mice were anesthetized with an intraperitoneal injection of ketamine xylazine. The spinal cord was immediately dissected and dissociated as previously described (Foo, Citation2013). Single cells were then sorted by using MoFlo high-speed cell sorter and gated based on eGFP fluorescence. Two to three mice were used for each group.
Microarray
Microarray procedure and analyses were performed as previously published. Briefly, total RNA was isolated from FACS sorted populations using the RNA isolation kit (Qiagen, Hilden, Germany) and the concentration was determined with Nanodrop (Thermo Fisher Scientific, Waltham, MA) and Bioanalyzer (Agilent Technologies, Santa Clara, CA). Only samples with a RNA integrity (RIN) score greater than five were used. Total RNA was lineally amplified and labeled in a Nugene protocol. Sample labeling and hybridization with Mouse Exon 1.0 ST chips (Affymetrix, Santa Clara, CA) were performed in the Johns Hopkins University microarray facility following manufacturer’s protocol. After hybridization, hybridization signals were acquired and normalized with the use of Partek Genomics Suite Software (Partek, St. Louis, MO). Differential gene expression between the different conditions was assessed by statistical linear model analysis using the Partek, Tibco Spotfire, and Prism 6 software packages. The moderated t-statistic p values derived from the Partek analysis above were further adjusted for multiple testing by Benjamini and Hochberg’s method to control false discovery rate (FDR). The FDR cutoff of <10% was used to obtain the list of differentially expressed genes. In total, a minimum of two mice were used for these experiment. (Note: Data deposition will be submitted with final manuscript, Supplemental Table 1).
Pathway analysis
Gene ontology and pathway analysis were performed using data obtained from Partek and Spotfire post statistical analyses using the Ingenuity Pathway Analysis software package.
Results
SOD1 G93A astroglia undergo transcriptome changes during disease progression
To generate a model that we could FACS isolate pure astroglia in ALS disease context, we bred the SOD1 G93A, fast progressing ALS mouse model, with the ALDH1L1-eGFP mouse (Weydt, Hong, Kliot, & Moller, Citation2003; Yang et al., Citation2011). ALDH1L1-eGFP provides adult astroglia specific eGFP expression and compared to the Glt1-eGFP model, we expected less variation between eGFP signal from early to mid-disease progression where Glt1 levels are continuously reduced (Olsen, Campbell, McFerrin, Floyd, & Sontheimer, Citation2010; Yang et al., Citation2011). After generating double transgenic, ALDH1L1-eGFP/SOD1 G93A mice, we subjected whole spinal cord from both pre-clinical and disease onset stages, P60 and P90, respectively, to tissue dissociation. To isolate single-cell astroglia from spinal cord, we performed the previously published methodology to ensure high viability and purity (Foo, Citation2013). Post tissue dissociation of both control and SOD1 G93A spinal cords, we FACS isolated ALDH1L1-eGFP positive astroglia and subjected the corresponding cell populations to microarray analyses (Supplemental Table 1) (Yang et al., Citation2011).
Next, to identify genes differentially regulated we subjected isolated astroglia to microarray analyses. Using stringent parameters (fold-change (FC) of ±1.5 and p values <.05) we uncovered 1063 and 1020 differentially regulated genes, including miRNA, at P60 and P90, respectively (Supplemental Tables 1 and 2; )). As expected, we noted a modest increase in markers associated with astrogliosis and reactivity such as GFAP at both time points in the mutant SOD1 mouse compared to the littermate controls (Supplemental Tables 2 and 3).
Figure 1. Heatmaps from early- and mid-symptomatic G93A SOD1 astroglia show dramatic gene dysregulation. (A) Early onset P60 control vs mutant SOD1 astroglia display differential gene expression, (B) Mid onset P90 control vs mutant SOD1 astroglia display differential gene expression. A total of four mice were used for each time point and all probes being illustrated.
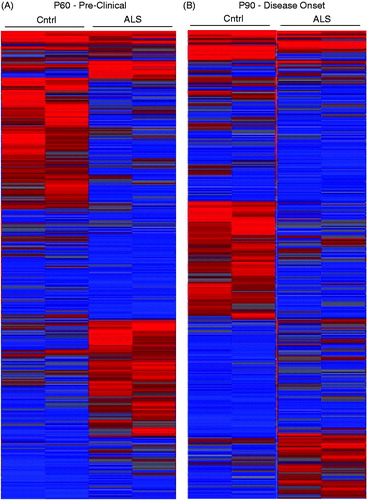
Furthermore, there was a notable change in intensity of the heatmap when comparing different ALS stage astroglia, consistent with our differential gene results (). Meanwhile when comparing between control and mutant SOD1 heatmaps, there is a dramatic difference supporting the notion that astroglia undergo widespread dysregulation in disease (). We next focused on which genes followed the strongest trend in disease progression from P60 to P90 (). Expectedly, we saw an upregulation in Connexin-43 (Gja1) and Glial fibrillary acidic protein (Gfap), both well-known to non-specifically upregulate with neuronal injury (Vargas & Johnson, Citation2010). Unexpectedly, we uncovered genes including protein kinase C inhibitor-1 (Ywhaz) and sodium bicarbonate cotransporter (Slc4a4) that showed strong trends of up- and downregulation in disease versus control astroglia, respectively. Lastly, a Venn diagram was generated displaying both upregulated and downregulated genes that followed the same trend between P60 and P90 and genes that were unique to each age point ().
Figure 2. Disease astroglia display a strong trend in genes differentially expressed during disease progression. (A) Correlation plot shows genes most correlated being upregulated and downregulated in control vs mutant SOD1 at both time points. (B) Venn diagram displays each time point of isolated astroglia and time points combined to illustrate genes unique to each and genes showing similar trend of differentiation. A fold change of 1.5 and p values of .05 was used to allow for stringent parameters.
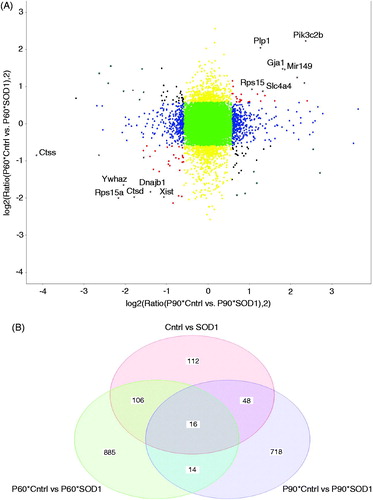
Of the 1000 differentially regulated genes, we saw the highest downregulation in the potassium channel KCNK1 in mutant at P60 and an increase in sodium cotransporter, Slc41a1 (). Meanwhile, at a later time point, P90, the strongest decrease in expression in mutant astroglia was calcium-binding protein, Sparcl1, and a dramatic increase in lysosomal protein, Ctss (). The results that ion proteins are the most dysregulated in astroglia of ALS disease is intriguing but quite consistent with former results that altered ion homeostasis exacerbates and is a hallmark of several different neurodegenerative disorders (Kaiser et al., Citation2006; LaFerla, Citation2002; Tong et al., Citation2014). Lastly, the myelin protein, PLP1 was shown to be downregulated in mutant SOD1 astroglia. The finding that PLP1 can be identified in astroglia, although at much lower levels than oligodendrocytes, is consistent with prior work (Cahoy et al., Citation2008).
Table 1. Top differentially enriched genes in pre-clinical P60 ALS spinal cord astroglia.
Table 2. Top differentially enriched genes in P90 disease onset ALS spinal cord astroglia.
Gene ontology of differentially regulated genes
Determining if differentially regulated genes are associated with a common biochemical pathway could provide us with useful insight into the molecular changes present in different stages of ALS. First, we subjected microarray data from P60 pre-clinical ALS astroglia to gene ontology analytics. We uncovered that even prior to obvious clinical disease, there already were a series of genes dysregulated. The majority of downregulated astroglial genes in ALS were associated with protein transport and localization. Second to protein transport, the downregulated gene products clustered to cell–cell signaling and generation of a signal being involved in cell–cell signaling (Supplemental Table 4). Meanwhile, gene products downregulated in the mutant vs control astroglia were annotated to pathways involved in biosynthesis and organelle organization (Supplemental Table 4).
When looking at a time point that typically is first associated with clinical disease onset, P90, gene ontology data revealed significant downregulation in GTPase activity and signal transduction in the ALS astroglia. Furthermore, there was a downregulation in mutant astroglia at this time point for DNA repair and replication, in addition to the positive regulation of cell differentiation (Supplemental Table 4).
Lastly, combination of both pre-clinical and disease-onset microarray data showed significant downregulation in protein localization and transport in mutant astroglia with a marked increase in transcriptional regulation and cytoplasmic membrane-bound vesicle (Supplemental Table 4).
Ingenuity pathway analysis (IPA) and STRING map uncovers dysregulated signaling pathways
Lastly, we decided to use bioinformatics tools to uncovered pathways that are dysregulated in the ALS disease progression. To address this, we chose to use Ingenuity Pathway Analysis (IPA) for its comprehensive analysis. We subjected the differentially regulated genes from time points P60 and P90 and found that several canonical signaling pathways were found to be effected. The top differentially regulated pathways consisted of glutamine biosynthesis and calcium transport at P60 (). Meanwhile, at P90 tight junction signaling and mTor signaling was noted to be highly differentiated between control and mutant SOD1 astroglia ().
Table 3. Top differentially regulated pathways in the P60 ALDH1L1-eGFP astroglia from mutant spinal cord compared to the control SOD1 G93A spinal cord.
Table 4. Top differentially regulated pathways in the P90 ALDH1L1-eGFP astroglia from mutant spinal cord compared to the control SOD1 G93A spinal cord.
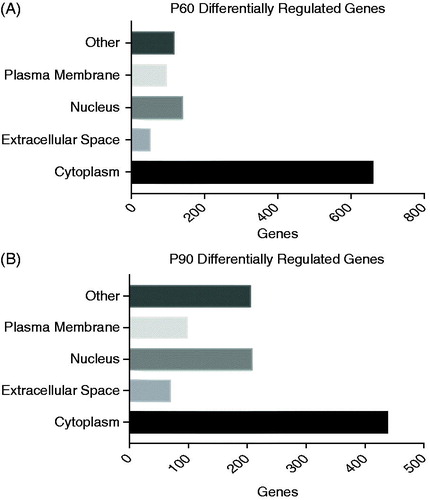
In order to understand if the differentially regulated genes and their protein products had a cellular compartment preference, we subjected the genes to additional ingenuity pathway analysis. We noted a preference for proteins localized to the cytoplasm in P60 and P90 mutant SOD1 astroglia (). Furthermore, we noted that differentially regulated genes were consistently enriched in the cytoplasm and nucleus compared to other compartments during disease progression ().
Figure 3. Cellular compartmentalization shows unique preference at early- and mid-disease onset. (A, B) P60 and P90 showed that the highest amount of hits to be differentially regulated compared to control resided in the cytoplasm and nucleus, respectively. Stringent parameters of fold change of 1.5 and p values of 0.05 was used for this analyses and ingenuity pathway analyses for identification of comparts of top hits.
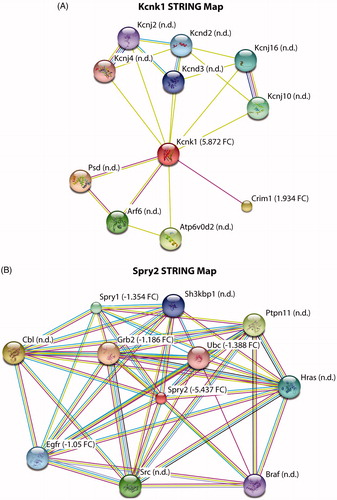
To supplement the IPA results, we also explored functional interactions of the protein products with the top candidate genes that were either downregulated or upregulated in diseased astroglia. In doing so, we focused heavily on Kcnk1, a potassium channel, that displayed significant downregulation and Spry2, a receptor tyrosine kinase inhibitor that was highly upregulated in disease contexts (Supplemental Table 1). In order to evaluate other interacting proteins with both Kcnk1 and Spry2, we utilized the software STRING. We found a Kcnk1-interacting protein, Crim1, to be significantly downregulated in disease astroglia compared to control (). We did not however detect from our microarray any other interacting proteins with Kcnk1. Lastly, when evaluating the upregulated Spry2 string map, we found significant enrichment in several Spry2-interacting proteins such as Ubc, Egfr, Grb2, and Spry1 (). These STRING map results consistently show either upregulation or downregulation in disease astroglia that is consistent with differential expressed markers in our microarray data (Supplemental Table 1, )).
Figure 4. STRING map analysis to determine interacting proteins for highly enriched markers. (A,B) Kcnk1 and Spry2 have several established interacting proteins with some not detected (n.d.) and others detected in our microarray results. Detected proteins are represented by fold-change value (FC ±), control vs mutant SOD1.
Discussion
Pathways dysregulated in the ALS SOD1 G93A astroglia
Our study is the first to analyze the full complement of endogenous in vivo adult astroglia from living ALS mice. Prior studies have examined laser capture astroglia and cultured astroglia from ALS mouse models. The ALDH1LI gene is the most reliable and most comprehensive reporter for adult spinal cord astroglia, especially in a disease context. Other reporters, such as GFAP- reflect only a subset of astroglia and GLT1/EAAT2, can be heavily dysregulated in cellular expression in disease states. Our prior studies have shown that ALDH1L1 is a suitable and constant marker of astroglia – even in disease states (Yang et al., Citation2011). As such this study serves as an additional comparator to the biology of astroglia in the course of ALS disease progression. In fact, our microarray results shed light on some common themes throughout neurodegenerative disorders. The glutamine biosynthesis pathway was remarkably differentially regulated in the ALS astroglia. Glutamate is a known component of ALS pathogenesis by promoting the excitotoxic effects seen in motor neurons (Robberecht & Philips, Citation2013; Rothstein & Cleveland, Citation2001). This accumulation of glutamate is likely a result of incompetent uptake by regional astrocytes. However, these findings support the notion that in addition to glutamate uptake that glutamine biosynthesis is also dysregulated in astroglia. Glutamine biosynthesis has been previously reported to be dysregulated in other neurodegenerative disorders such as Alzheimer’s disease and Huntington’s disease (Kulijewicz-Nawrot, Sykova, Chvatal, Verkhratsky, & Rodriguez, Citation2013). We, therefore, hypothesize a model that ALS astroglia have altered glutamate–glutamine cycle dysregulation. For example, there may be an increase in glutamine biosynthesis with subsequent release into the synaptic cleft for use in neurotransmitter glutamate resynthesis, ultimately altering excitotoxic pools of glutamate. However, this model needs to further explored and validated with functional data.
Meanwhile, numerous genes and pathways were involved in ion homeostasis and showed high dysregulation in mutant SOD1 astroglia compared to control. Astroglia play large roles in ion balance of the CNS and it is not surprising that prior art has shown that ion dyshomeostasis exacerbates neurodegeneration in many different disorders (Bataveljic, Nikolic, Milosevic, Todorovic, & Andjus, Citation2012; LaFerla, Citation2002; Nowacek, Kosloski, & Gendelman, Citation2009; Olsen et al., Citation2010; Sofroniew & Vinters, Citation2010; Tong et al., Citation2014). For example, in Huntington’s disease potassium dyshomeostasis in the striatum leads to hyperexcitable neurons but adeno-associated-viral delivery of potassium channel Kir4.1, which is downregulated in ALS, to astroglia restores this hyperexcitable deficit (Tong et al., Citation2014). In ALS, potassium imbalances occur in both the cerebral cortex and spinal cord (Bataveljic et al., Citation2012). These findings shed light on potential targets for restoring this homeostasis at early time points in the disease.
In comparison to prior observations, we found strong concordance in pathways being altered in ALS SOD1 G93A astroglia. Although our approach was different than prior work, with ours focusing on highly purified adult astroglia isolated from in vivo astroglia, we, like others, showed strong dysregulation in energy metabolism, signaling, cell cycle, immune responses, MAPK signaling, and apoptosis (Ferraiuolo et al., Citation2011; Phatnani et al., Citation2013). In Ferraiuolo et al. (Citation2011), authors showed that after laser capture of Ald1h1l1 astroglia that pathways involved in energy metabolism and cell cycle were the most differentially regulated, consistent with our results. Furthermore, Phatnani et al. (Citation2013) performed an elaborate and extensive array analyses on cultured ALS astrocytes along with other non ALS astroglia and showed strong dysregulation in apoptosis and MAPK signaling, processes we also found to be altered. Taken these findings, all three papers have consistently come to several pathways as being the most differentially regulated in the ALS SOD1 G93A disease context.
Early genetic dysregulation in ALS SOD1 G93A astroglia
Prior to clinical disease onset, at P60, we found levels of KCNK1 to be the highest downregulated in mutant SOD1 compared to control astroglia. KCNK1 (Potassium Two Pore Domain Channel Subfamily K Member 1, TWIK-1, or K2P1) is a member of the two-pore domain K+ (K2P) family of ion channels (Lesage et al., Citation1996). These channels consist of four membrane-spanning domains surrounding two pore-forming loops, and are considered ‘leak channels’ responsible for maintaining background K+ conductance and stabilizing resting membrane potential (Enyedi & Czirjak, Citation2010; Talley, Solorzano, Lei, Kim, & Bayliss, Citation2001). Interestingly, these channels are highly sensitive to modulation by a wide range of physiological factors such as neurotransmitters, pH, temperature, and membrane stretch as well as to pharmacological compounds such as anesthetics making them a possible target for selective modification in vivo (Talley et al., Citation2001). Initially, KCNK1 was thought to be inactive due to a lack of functional data, despite mRNA evidence showing its robust expression in the brain, kidney, and heart (Lesage et al., Citation1996; Talley et al., Citation2001). One early study suggested that this non-functionality was due to sumoylation, although other studies have failed to replicate this finding and instead propose that KCNK1 is regulated by rapid endocytosis from the cell surface and subsequent storage in the endosomal compartment (Feliciangeli et al., Citation2010; Rajan, Plant, Rabin, Butler, & Goldstein, Citation2005). Feliciangeli et al. (Citation2010) also provided evidence that stimulation of Gi-coupled serotonergic and adrenergic receptors lead to an increase of KCNK1 current, potentially through stabilizing its location and accumulation at the plasma membrane.
KCNK1 mRNA was found to be the most abundantly expressed in mouse astrocytes (Cahoy et al., Citation2008). It was recently reported that KCNK1 associates with another member of the K2P family, TREK-1 (TWIK-related K+ channel-1) to form a functional heterodimeric channel capable of passing K+ current (Hwang et al., Citation2014). Importantly, the KCNK1/TREK-1 heterodimer was found to be responsible for most of the background passive conductance of astrocytes (Hwang et al., Citation2014). While the specific contribution of TREK-1 to the electrophysiological properties of astrocytes is not yet defined, KNCK1 alone significantly contributes to passive conductance in mature hippocampal astrocytes (Du et al., Citation2016; Zhou et al., Citation2009). Surprisingly, neither individual knockout of TREK-1 or double knockout of the TWIK-1/TREK-1 heterodimer in rodents lead to remarkable alterations in the electrophysiological properties of astrocytes; however, knockout of KCNK1 in rodents does lead to noticeable alterations in astrocyte electrophysiology (Du et al., Citation2016; Wang et al., Citation2013). Intriguingly, the KCNK1/TREK-1 heterodimer has also been implicated fast glutamate release from astrocytes following stimulation of Gαi-coupled GPCRs (in particular CB1), indicating that the pore may change structure following Gβγ-binding (Hwang et al., Citation2014).
In our study, KCNK1 was found to be dysregulated in astrocytes prior to overt clinical disease (P60) in G93A SOD1 mice. Certainly this suggests a very dramatic alteration in spinal cord astroglia that well proceeds disease and neuronal death. It is not clear from the biology of KCNK1 alone what stimulus would cause its initial downregulation in pre-clinical astroglia, however, one potential consequence of this downregulation does emerge. Given that KCNK1 is regulated in part by endocytosis, along with the gene ontology results that the majority of downregulated genes in SOD1/G93A astrocytes are associated with protein transport and localization, it is tempting to speculate a scenario where very early KCNK1 dysfunction is further exacerbated by a disruption in its transport pathway from the cytoplasm to the plasma membrane. Lastly, using STRING map analysis we found that another Kcnk1 interacting protein, Crim1, was found to be downregulated in mutant SOD1 astroglia. Crim1 is a transmembrane protein and we speculate that translocation of these key plasma membrane proteins to the cell surface may be altered in mutant SOD1 astroglia consistent with their mRNA decline (Turtoi et al., Citation2012). Finally, Kcnj10, a potassium channel that interacts with Kcnk1 did not appear on our microarray results but has been widely shown to be downregulated in astroglia in mutant SOD1 animal models (Kaiser et al., Citation2006). However, the downregulation of these three interacting proteins remains unexplored and should be of focus for future studies.
The gene most upregulated in ALS astroglia prior to overt clinical disease was SPRY2 (Sprouty 2, Spry2), which belongs to the sprouty family of downstream negative feedback inhibitors of receptor tyrosine kinase (RTK) dependent-signaling pathways (Ras/Raf/ERK/MAPK signaling). The sprout family is involved in cell proliferation, differentiation, and survival (Casci, Vinos, & Freeman, Citation1999; Gross, Bassit, Benezra, & Licht, Citation2001; Mason, Morrison, Basson, & Licht, Citation2006). Depending on model organism and cellular context, Spry2 can act through different binding partners to regulate extracellular signal–receptor (ERK) signaling (Egan, Hall, Yatsula, & Bar-Sagi, Citation2002; Gross et al., Citation2001; Mason et al., Citation2006; Yusoff et al., Citation2002). Mammalian Spry2 protein contains a conserved cysteine-rich Spry domain that can inhibit EGF-induced MAPK activation via its binding to Raf1 (a member of the Ras family of GTPases), and a conserved region in the N-terminus containing an invariant tyrosine residue which may also mediate many of the inhibitory effects of Spry2 (Egan et al., Citation2002; Mason et al., Citation2006). Spry2 is localized to the plasma membrane in mammalian cells following RTK stimulation, and mutations that disrupt its localization also result in disruption of its ability to negatively regulate RTKs (Lim et al., Citation2002; Yusoff et al., Citation2002).
In human embryonic stem cells (hESCs), glioblastoma multiforme (GBM) cell lines, and in a GBM tumor xenograft in vivo model, Spry2 knockdown resulted in decreased cell proliferation, leading to significantly impaired tumor growth in the GBM xenograft (Felfly & Klein, Citation2013; Walsh et al., Citation2015). Not surprisingly, elevated Spry2 expression was associated with decreased GBM patient survival, making Spry2 a potential biomarker for GBM progression (Walsh et al., Citation2015). Taken these findings, it may be speculated that Spry2 elevation in mutant SOD1 astroglia may be a protective response for the cellular stress that astroglia undergo in neurodegeneration.
In addition to Spry2’s upregulation, other key Spry2-interacting proteins were also found to be upregulated in mutant SOD1 astroglia such as Egfr, Grb2, Ubc, and Spry1. Due to the number of interacting proteins showing consistent upregulation with Spry2, this pathway is of particular importance to study as it may provide very meaningful insight into astroglia dysfunction in disease. Of particular interest is Grb2, an interacting protein that selectively binds to Egfr (another protein upregulated in this pathway) at the cell surface and couples Egfr to intracellular signal transduction pathways. However, it has been shown that Spry2 can lead to the degradation of Egfr post-activation (Alice & Walsh, Citation2013; Yamazaki, Hailey, Presley, Lippincott-Schwartz, & Samelson, Citation2002). Therefore, we speculate that the Efgr pathway is upregulated in SOD1 disease astroglia and Spry2’s upregulation is part of the intrinsic feedback mechanism to maintain Egfr signaling and levels.
By the time of disease onset, P90, astroglial expression of SPARCL1 was found to have the most significant downregulation in ALS. SPARCL1 (synaptic cleft-1, SC1, MAST-9, RAGS1, ECM2, hevin) is a member of the SPARC (Secreted Protein Acidic and Rich in Cysteine) family of matricellular proteins that modulate cell–extracellular matrix interactions (Brekken, Citation2004; Johnston, Paladino, Gurd, & Brown, Citation1990). Members of this protein family contain an acidic domain l, a follistatin-like domain, and an extracellular calcium-binding (E-C) domain containing a pair of Ca2+ binding EF-hand motifs (Yan & Sage, Citation1999). SPARCL1 is a secreted glycoprotein that is strongly expressed in both neurodevelopment and adulthood, where it is localized to synapses in the mammalian CNS (Johnston et al., Citation1990; Lively, Ringuette, & Brown, Citation2007). SPARCL1 can additionally function as an anti-adhesive molecule, as well as a negative regulator of cellular growth and proliferation (Brekken & Sage, Citation2000; Claeskens et al., Citation2000). Interestingly, SPARCL1 is often found to be downregulated in neoplastic tissue, including several types of cancers such as gliomas and gastric cancers, and patients with higher levels of SPARCL1 are predicted to have a better survival outcome (Claeskens et al., Citation2000; Li et al., Citation2012; Turtoi et al., Citation2012).
SPARCL1 is highly expressed in early postnatal adult, and cultured astrocytes, with lower expression in neurons and oligodendrocytes (Cahoy et al., Citation2008; Eroglu, Citation2009). Several groups have demonstrated that hypertrophied/reactive astrocytes upregulate SPARCL1 in response to injury (Lively & Brown, Citation2007, Citation2008; Lively, Moxon-Emre, & Schlichter, Citation2011; McKinnon & Margolskee, Citation1996; Mendis, Ivy, & Brown, Citation1996). These groups also associated SPARCL1 with glial scarring, and showed SPARCL1 co-localization with markers of reactive gliosis such as GFAP. In the adult mammalian CNS, SPARCL1 is localized to excitatory synapses and perisynaptic glial processes (Lively et al., Citation2007). Lively et al. (Citation2007) hypothesized that this localization pattern, taken with the calcium-binding properties of SPARCL1 and its developmental expression, would implicate SPARCL1 in regulating synaptic plasticity. Later, SPARCL1 was demonstrated to be sufficient in inducing the formation of synapses between retinal ganglion cells (RGC) cultured in the presence of SPARCL1 alone or co-cultured with astrocytes (Kucukdereli et al., Citation2011). In vivo, astrocyte-secreted SPARCL1 can stimulate excitatory synapse formation between RGCs and the superior colliculus, as well as form thalamocortical synapses (Kucukdereli et al., Citation2011; Risher et al., Citation2014). Moreover, SPARCL1-null mice display lower synaptic density and synapse immaturity (Kucukdereli et al., Citation2011; Risher et al., Citation2014). Recently, astrocytes were shown to control synapse assembly via a trans-synaptic bridge between SPARCL1, NRX1alpha, and NL1, which in turn recruits’ NMDARs to the synapse (Singh et al., Citation2016). Synaptic disruption likely occurs early in ALS neurodegeneration and we speculate that astroglial SPARCL1 contributes to aberrant motorneuron and interneuron synaptic biology in the ALS spinal cord. We further speculate that as synaptic stripping and dendritic simplification occurs in ALS, even early in disease, a downregulation of SPARCL1 may result.
In this study, we also found the expression of Cathepsin S to be most upregulated in mutant SOD1 astroglia at age P90. Cathepsin S is a lysosomal cysteine protease with roles in MHC class II-mediated antigen presentation, extracellular proteolysis, and trafficking protein turnover in lysosomes (Driessen et al., Citation1999; Wilkinson, Williams, Scott, & Burden, Citation2015). Uniquely, Cathepsin S is able to maintain its enzymatic activity at neutral pH (Petanceska, Canoll, & Devi, Citation1996). Cathepsin S has restricted expression in mammalian tissue, but has strong expression in the brain and spinal cord (Berjaoui et al., Citation2015; Petanceska, Burke, Watson, & Devi, Citation1994; Shi et al., Citation1994). In the adult mammalian brain, Cathepsin S is expressed by all CNS cell types (Cahoy et al., Citation2008; Hickman et al., Citation2013; Petanceska et al., Citation1994; Wendt, Lubbert, & Stichel, Citation2008).
Cathepsin S dysregulation has been implicated in a broad range of diseases including cancer, neurodegenerative, and metabolic disorders (Wilkinson et al., Citation2015). In Multiple Sclerosis (MS), Cathepsin S is capable of cleaving myelin basic protein (MBP), a major component of the myelin sheath that undergoes degradation in MS (Beck et al., Citation2001). Additionally, Cathepsin S is also capable of generating toxic amyloidogenic fragments of amyloid-beta (Aβ) through its cleaving interaction with β-Amyloid Precursor Protein (βAPP), as demonstrated in vitro (Munger et al., Citation1995). In Alzheimer’s disease (AD) brain tissue, Cathepsin S mRNA was found to be upregulated in AD affected regions (Lemere et al., Citation1995). Together, these studies implicate a role for astroglial Cathepsin S in local inflammation and neurodegenerative events at the time of clinical disease onset. Importantly, Cathepsin S mRNA was significantly upregulated in anterior lumbar spinal cord samples from terminal-stage ALS patients when compared to controls, but other inflammatory markers were not significantly altered (Berjaoui et al., Citation2015). Cathepsin S was also previously shown to be significantly upregulated in spinal cord tissue, and degenerating neuronal regions from aged SOD1-G93A mice (Chiu et al., Citation2013; Wendt et al., Citation2008).
Our observation that CTSS is strongly upregulated in ALDH1L1-EGFP/SOD1 G93A mouse provides an intriguing new insight to ALS pathogenesis, given that many previous studies have focused on the role of Cathepsin S in microglial pathology, not astrocytic pathology. Given the evidence for the role of Cathepsin S in late-stage neurodegenerative disease and its role as a lysosomal protease, its increased expression could represent an upregulated cellular response to clear toxic or misfolded proteins from reactive astrocytes. Notably, this result is consistent with other studies (discussed above) that describe a transition in disease conditions whereby Cathepsin S is upregulated.
Therapeutic intervention and conclusions
Notably, analytics of large sets of genomic and proteomic data may be an important evolution in the understanding of normative and pathobiology in neuroscience. Recently, journals are pressing more than ever for authors to release large data files to allow transparency among scientists and the ability for them to pursue their own hypotheses in a data exposed manner (Ferguson, Nielson, Cragin, Bandrowski, & Martone, Citation2014). We believe the release of this true in vivo data on a CNS single cell type, rather than gross tissue homogenates, in the setting of disease progression will be beneficial to glia biologists and scientists who use the Glia Open Access Database (GOAD.education) system, but also to industrial partners looking to explore common themes in neurodegeneration (Holtman et al., Citation2015). We have successfully and delicately isolated pure adult astroglia from the whole spinal cord from ALS mice prior to disease onset and in the very earliest clinical phase of the disease. It is these time points that are most likely to provide clues as to the possible role for astroglia in disease pathobiology. We found several unique markers and pathways that undergo dramatic dysregulation in disease context and set the stage for future studies to exploit those findings here. Furthermore, we uncovered markers that are also involved in other neurodegenerative disorders, making it feasible that a common drug or target may be beneficial to several disorders.
Miller_et_al_supplemental_content.zip
Download Zip (1.3 MB)Acknowledgements
We would like to thank the Johns Hopkins Microarray and Bioinformatics core for their assistance and knowledge, in particular Connie Talbot for his guidance on the bioinformatic analyses, Zhuoxun Chen for assisting with sample preparation, and the Johns Hopkins University School of Public Health FACS Center.
Disclosure statement
The authors declare no competing interests.
Additional information
Funding
Related Research Data
References
- Alice, M., & Walsh, M.J.L. (2013). Regulation of EGFR trafficking and cell signaling by Sprouty2 and MIG6 in lung cancer cells. Journal of Cell Science, 126, 4339–4348. doi:10.1242/jcs.123208.
- Bataveljic, D., Nikolic, L., Milosevic, M., Todorovic, N., & Andjus, P.R. (2012). Changes in the astrocytic aquaporin-4 and inwardly rectifying potassium channel expression in the brain of the amyotrophic lateral sclerosis SOD1(G93A) rat model. Glia, 60, 1991–2003. doi:10.1002/glia.22414.
- Beck, H., Schwarz, G., Schroter, C.J., Deeg, M., Baier, D., Stevanovic, S., … Kalbacher, H. (2001). Cathepsin S and an asparagine-specific endoprotease dominate the proteolytic processing of human myelin basic protein in vitro. The European Journal of Immunology, 31, 3726–3736. doi:10.1002/1521-4141(200112)31:12<3726::AID-IMMU3726>3.0.CO;2-O.
- Berjaoui, S., Povedano, M., Garcia-Esparcia, P., Carmona, M., Aso, E., & Ferrer, I. (2015). complex inflammation mRNA-related response in ALS is region dependent. Neural Plasticity, 2015, 573784. doi: 10.1155/2015/573784.
- Brekken, R.A. (2004). Expression and characterization of murine hevin (SC1), a member of the SPARC family of matricellular proteins. Journal of Histochemistry and Cytochemistry, 52, 735–748. doi:10.1369/jhc.3A6245.2004.
- Brekken, R.A., & Sage, E.H. (2000). SPARC, a matricellular protein: at the crossroads of cell-matrix. Matrix Biology, 19, 569–580. doi:10.1016/S0945-053X(00)00105-0.
- Cahoy, J.D., Emery, B., Kaushal, A., Foo, L.C., Zamanian, J.L., Christopherson, K.S., … Barres, B.A. (2008). A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. The Journal of Neuroscience, 28, 264–278. doi:10.1523/jneurosci.4178-07.2008.
- Casci, T., Vinos, J., & Freeman, M. (1999). Sprouty, an intracellular inhibitor of Ras signaling. Cell, 96, 655–665. doi:10.1016/S0092-8674(00)80576-0.
- Chiu, I.M., Morimoto, E.T., Goodarzi, H., Liao, J.T., O'keeffe, S., Phatnani, H.P., … Maniatis, T. (2013). A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Reports, 4, 385–401. doi:10.1016/j.celrep.2013.06.018.
- Claeskens, A., Ongenae, N., Neefs, J.M., Cheyns, P., Kaijen, P., Cools, M., & Kutoh, E. (2000). Hevin is down-regulated in many cancers and is a negative regulator of cell growth and proliferation. The British Journal of Cancer, 82, 1123–1130. doi:10.1054/bjoc.1999.1051.
- Driessen, C., Bryant, R.A., Lennon-Dumenil, A.M., Villadangos, J.A., Bryant, P.W., Shi, G.P., … Ploegh, H.L. (1999). Cathepsin S controls the trafficking and maturation of MHC class II molecules in dendritic cells. Journal of Cell Biology, 147, 775–790.
- Du, Y., Kiyoshi, C.M., Wang, Q., Wang, W., Ma, B., Alford, C.C., … Zhou, M. (2016). Genetic deletion of TREK-1 or TWIK-1/TREK-1 potassium channels does not alter the basic electrophysiological properties of mature hippocampal astrocytes in situ. Frontiers in Cellular Neuroscience, 10, 13. doi:10.3389/fncel.2016.00013.
- Egan, J.E., Hall, A.B., Yatsula, B.A., & Bar-Sagi, D. (2002). The bimodal regulation of epidermal growth factor signaling by human Sprouty proteins. Proceedings of the National Academy of the Sciences in the United States of America, 99, 6041–6046. doi:10.1073/pnas.052090899.
- Enyedi, P., & Czirjak, G. (2010). Molecular background of leak K + currents: two-pore domain potassium channels. Physiological Reviews, 90, 559–605. doi:10.1152/physrev.00029.2009.
- Eroglu, C. (2009). The role of astrocyte-secreted matricellular proteins in central nervous system development and function. The Journal of Cell Communication and Signaling, 3, 167–176. doi:10.1007/s12079-009-0078-y.
- Felfly, H., & Klein, O.D. (2013). Sprouty genes regulate proliferation and survival of human embryonic stem cells. Science Reports, 3, 2277. doi:10.1038/srep02277.
- Feliciangeli, S., Tardy, M.P., Sandoz, G., Chatelain, F.C., Warth, R., Barhanin, J., … Lesage, F. (2010). Potassium channel silencing by constitutive endocytosis and intracellular sequestration. Journal of Biological Chemistry, 285, 4798–4805. doi:10.1074/jbc.M109.078535.
- Ferguson, A.R., Nielson, J.L., Cragin, M.H., Bandrowski, A.E., & Martone, M.E. (2014). Big data from small data: data-sharing in the ‘long tail’ of neuroscience. Nature Neuroscience, 17, 1442–1447. doi:10.1038/nn.3838.
- Ferraiuolo, L., Higginbottom, A., Heath, P.R., Barber, S., Greenald, D., Kirby, J., & Shaw, P.J. (2011). Dysregulation of astrocyte-motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain, 134, 2627–2641. doi:10.1093/brain/awr193.
- Foo, L.C. (2013). Purification of astrocytes from transgenic rodents by fluorescence-activated cell sorting. Cold Spring Harbor Protocol, 2013, 551–560. doi:10.1101/pdb.prot074229.
- Gross, I., Bassit, B., Benezra, M., & Licht, J.D. (2001). Mammalian sprouty proteins inhibit cell growth and differentiation by preventing ras activation. Journal of Biological Chemistry, 276, 46460–46468. doi:10.1074/jbc.M108234200.
- Guo, Y., Duan, W., Li, Z., Huang, J., Yin, Y., Zhang, K., … Li, C. (2010). Decreased GLT-1 and increased SOD1 and HO-1 expression in astrocytes contribute to lumbar spinal cord vulnerability of SOD1-G93A transgenic mice. FEBS Letters, 584, 1615–1622. doi:10.1016/j.febslet.2010.03.025.
- Hickman, S.E., Kingery, N.D., Ohsumi, T.K., Borowsky, M.L., Wang, L.C., Means, T.K., & El Khoury, J. (2013). The microglial sensome revealed by direct RNA sequencing. Nature Neuroscience, 16, 1896–1905. doi:10.1038/nn.3554.
- Holtman, I.R., Noback, M., Bijlsma, M., Duong, K.N., van der Geest, M.A., Ketelaars, P.T., … Boddeke, H.W. (2015). Glia Open Access Database (GOAD): a comprehensive gene expression encyclopedia of glia cells in health and disease. Glia, 63, 1495–1506. doi:10.1002/glia.22810.
- Hwang, E.M., Kim, E., Yarishkin, O., Woo, D.H., Han, K.S., Park, N., … Park, J.Y. (2014). A disulphide-linked heterodimer of TWIK-1 and TREK-1 mediates passive conductance in astrocytes. Nature Communications, 5, 3227. doi:10.1038/ncomms4227.
- Johnston, I.G., Paladino, T., Gurd, J.W., & Brown, I.R. (1990). Molecular cloning of SC1: a putative brain extracellular matrix glycoprotein showing partial similarity to osteonectin/BM40/SPARC. Neuron, 4, 165–176. doi:10.1016/0896-6273(90)90452-L.
- Kaiser, M., Maletzki, I., Hulsmann, S., Holtmann, B., Schulz-Schaeffer, W., Kirchhoff, F., … Neusch, C. (2006). Progressive loss of a glial potassium channel (KCNJ10) in the spinal cord of the SOD1 (G93A) transgenic mouse model of amyotrophic lateral sclerosis. Journal of Neurochemistry, 99, 900–912. doi:10.1111/j.1471-4159.2006.04131.x.
- Kucukdereli, H., Allen, N.J., Lee, A.T., Feng, A., Ozlu, M.I., Conatser, L.M., … Eroglu, C. (2011). Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proceedindgs of the National Academy of Sciences of the United States of America, 108, E440–E449. doi:10.1073/pnas.1104977108.
- Kulijewicz-Nawrot, M., Sykova, E., Chvatal, A., Verkhratsky, A., & Rodriguez, J.J. (2013). Astrocytes and glutamate homoeostasis in Alzheimer's disease: a decrease in glutamine synthetase, but not in glutamate transporter-1, in the prefrontal cortex. ASN Neuro, 5, 273–282. doi:10.1042/AN20130017.
- LaFerla, F.M. (2002). Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nature Reviews Neuroscience, 3, 862–872. doi:10.1038/nrn960.
- Lemere, C.A., Munger, J.S., Shi, G.P., Natkin, L., Haass, C., Chapman, H.A., & Selkoe, D.J. (1995). The lysosomal cysteine protease, cathepsin S, is increased in Alzheimer's disease and Down syndrome brain. An immunocytochemical study. American Journal of Pathology, 146, 848–860.
- Lesage, F., Guillemare, E., Fink, M., Duprat, F., Lazdunski, M., Romey, G., & Barhanin, J. (1996). TWIK-1, a ubiquitous human weakly inward rectifying K + channel with a novel structure. EMBO Journal, 15, 1004–1011.
- Li, P., Qian, J., Yu, G., Chen, Y., Liu, K., Li, J., & Wang, J. (2012). Down-regulated SPARCL1 is associated with clinical significance in human gastric cancer. Journal of Surgical Oncology, 105, 31–37. doi:10.1002/jso.22025.
- Lim, J., Yusoff, P., Wong, E.S.M., Chandramouli, S., Lao, D.H., Fong, C.W., & Guy, G.R. (2002). The cysteine-rich sprouty translocation domain targets mitogen-activated protein kinase inhibitory proteins to phosphatidylinositol 4,5-bisphosphate in plasma membranes. Molecular and Cellular Biology, 22, 7953–7966. doi:10.1128/mcb.22.22.7953-7966.2002.
- Lively, S., & Brown, I.R. (2007). Analysis of the extracellular matrix protein SC1 during reactive gliosis in the rat lithium-pilocarpine seizure model. Brain Research, 1163, 1–9. doi:10.1016/j.brainres.2007.05.052.
- Lively, S., & Brown, I.R. (2008). Extracellular matrix protein SC1/hevin in the hippocampus following pilocarpine-induced status epilepticus. Journal of Neurochemistry, 107, 1335–1346. doi:10.1111/j.1471-4159.2008.05696.x.
- Lively, S., Moxon-Emre, I., & Schlichter, L.C. (2011). SC1/hevin and reactive gliosis after transient ischemic stroke in young and aged rats. Journal of Neuropathology and Experimental Neurology, 70, 913–929. doi:10.1097/NEN.0b013e318231151e.
- Lively, S., Ringuette, M.J., & Brown, I.R. (2007). Localization of the extracellular matrix protein SC1 to synapses in the adult rat brain. Neurochemical Research, 32, 65–71. doi:10.1007/s11064-006-9226-4.
- Mason, J.M., Morrison, D.J., Basson, M.A., & Licht, J.D. (2006). Sprouty proteins: multifaceted negative-feedback regulators of receptor tyrosine kinase signaling. Trends in Cell Biology, 16, 45–54. doi:10.1016/j.tcb.2005.11.004.
- McKinnon, P.J., & Margolskee, R.F. (1996). SC1: a marker for astrocytes in the adult rodent brain is upregulated during reactive astrocytosis. Brain Research, 709, 27–36. doi:10.1016/0006-8993(95)01224-9.
- Mendis, D.B., Ivy, G.O., & Brown, I.R. (1996). SC1, a brain extracellular matrix glycoprotein related to SPARC and follistatin, is expressed by rat cerebellar astrocytes following injury and during development. Brain Research, 730, 95–106. doi:10.1016/0006-8993(96)00440-4.
- Miller, S.J., & Rothstein, J.D. (2016). Astroglia in thick tissue with super resolution and cellular reconstruction. PLoS One, 11, e0160391. doi:10.1371/journal.pone.0160391.
- Munger, J.S., Haass, C., Lemere, C.A., Shi, G.P., Wong, W.S., Teplow, D.B., … Chapman, H.A. (1995). Lysosomal processing of amyloid precursor protein to A beta peptides: a distinct role for cathepsin S. Biochemical Journal, 311, 299–305.
- Nowacek, A., Kosloski, L.M., & Gendelman, H.E. (2009). Neurodegenerative disorders and nanoformulated drug development. Nanomedicine (Lond), 4, 541–555. doi:10.2217/nnm.09.37.
- Oberheim, N.A., Goldman, S.A., & Nedergaard, M. (2012). Heterogeneity of astrocytic form and function. Methods in Molecular Biology, 814, 23–45. doi:10.1007/978-1-61779-452-0_3.
- Olsen, M.L., Campbell, S.C., McFerrin, M.B., Floyd, C.L., & Sontheimer, H. (2010). Spinal cord injury causes a wide-spread, persistent loss of Kir4.1 and glutamate transporter 1: benefit of 17 beta-oestradiol treatment. Brain, 133, 1013–1025. doi:10.1093/brain/awq049.
- Petanceska, S., Burke, S., Watson, S.J., & Devi, L. (1994). Differential distribution of messenger RNAs for cathepsins B, L and S in adult rat brain: an in situ hybridization study. Neuroscience, 59, 729–738. doi:10.1016/0306-4522(94)90190-2.
- Petanceska, S., Canoll, P., & Devi, L.A. (1996). Expression of rat cathepsin S in phagocytic cells. Journal of Biological Chemistry, 271, 4403–4409. doi:10.1074/jbc.271.8.4403.
- Phatnani, H.P., Guarnieri, P., Friedman, B.A., Carrasco, M.A., Muratet, M., O?Keeffe, S., … Maniatis, T. (2013). Intricate interplay between astrocytes and motor neurons in ALS. Proceedings of the National Academy of Sciences of the United States of America, 110, E756–E765. doi:10.1073/pnas.1222361110.
- Rajan, S., Plant, L.D., Rabin, M.L., Butler, M.H., & Goldstein, S.A. (2005). Sumoylation silences the plasma membrane leak K + channel K2P1. Cell, 121, 37–47. doi:10.1016/j.cell.2005.01.019.
- Re, D.B., Le Verche, V., Yu, C., Amoroso, M.W., Politi, K.A., Phani, S., … Przedborski, S. (2014). Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron, 81, 1001–1008. doi:10.1016/j.neuron.2014.01.011.
- Risher, W.C., Patel, S., Kim, I.H., Uezu, A., Bhagat, S., Wilton, D.K., … Eroglu, C. (2014). Astrocytes refine cortical connectivity at dendritic spines. Elife, 3, doi:10.7554/eLife.04047.
- Robberecht, W., & Philips, T. (2013). The changing scene of amyotrophic lateral sclerosis. Nature Reviews Neuroscience, 14, 248–264. doi:10.1038/nrn3430.
- Rothstein, D.W., & Cleveland, J.D. (2001). From charcot to lou gehrig: deciphering selective motor neuron death in als. Nature Reviews Neuroscience, 2, 806–819. doi:10.1038/35097565.
- Rothstein, J.D., Van Kammen, M., Levey, A.I., Martin, L.J., & Kuncl, R.W. (1995). Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Annals of Neurology, 38, 73–84. doi:10.1002/ana.410380114.
- Shi, G.P., Webb, A.C., Foster, K.E., Knoll, J.H., Lemere, C.A., Munger, J.S., & Chapman, H.A. (1994). Human cathepsin S: chromosomal localization, gene structure, and tissue distribution. Journal of Biological Chemistry, 269, 11530–11536.
- Singh, S.K., Stogsdill, J.A., Pulimood, N.S., Dingsdale, H., Kim, Y.H., Pilaz, L.J., … Eroglu, C. (2016). Astrocytes assemble thalamocortical synapses by bridging NRX1α and NL1 via Hevin. Cell, 164, 183–196. doi:10.1016/j.cell.2015.11.034.
- Sofroniew, M.V., & Vinters, H.V. (2010). Astrocytes: biology and pathology. Acta Neuropathologica, 119, 7–35. doi:10.1007/s00401-009-0619-8.
- Sultan, S., Li, L., Moss, J., Petrelli, F., Casse, F., Gebara, E., … Toni, N. (2015). Synaptic integration of adult-born hippocampal neurons is locally controlled by astrocytes. Neuron, 88, 957–972. doi:10.1016/j.neuron.2015.10.037.
- Talley, E.M., Solorzano, G., Lei, Q., Kim, D., & Bayliss, D.A. (2001). Cns distribution of members of the two-pore-domain (KCNK) potassium channel family. Journal of Neuroscience, 21, 7491–7505.
- Tong, X., Ao, Y., Faas, G.C., Nwaobi, S.E., Xu, J., Haustein, M.D., … Khakh, B.S. (2014). Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nature Neuroscience, 17, 694–703. doi:10.1038/nn.3691.
- Turtoi, A., Musmeci, D., Naccarato, A.G., Scatena, C., Ortenzi, V., Kiss, R., … Castronovo, V. (2012). Sparc-like protein 1 is a new marker of human glioma progression. Journal of Proteome Research, 11, 5011–5021. doi:10.1021/pr3005698.
- Vargas, M.R., & Johnson, J.A. (2010). Astrogliosis in amyotrophic lateral sclerosis: role and therapeutic potential of astrocytes. Neurotherapeutics, 7, 471–481. doi:10.1016/j.nurt.2010.05.012.
- Volterra, A., & Meldolesi, J. (2005). Astrocytes, from brain glue to communication elements: the revolution continues. Nature Reviews Neuroscience, 6, 626–640. doi:10.1038/nrn1722.
- Walsh, A.M., Kapoor, G.S., Buonato, J.M., Mathew, L.K., Bi, Y., Davuluri, R.V., … Lazzara, M.J. (2015). Sprouty2 drives drug resistance and proliferation in glioblastoma. Molecular Cancer Research, 13, 1227–1237. doi:10.1158/1541-7786.MCR-14-0183-T.
- Wang, W., Putra, A., Schools, G.P., Ma, B., Chen, H., Kaczmarek, L.K., … Zhou, M. (2013). The contribution of TWIK-1 channels to astrocyte K(+) current is limited by retention in intracellular compartments. Frontiers in Cellular Neuroscience, 7, 246. doi:10.3389/fncel.2013.00246.
- Wendt, W., Lubbert, H., & Stichel, C.C. (2008). Upregulation of cathepsin S in the aging and pathological nervous system of mice. Brain Research, 1232, 7–20. doi:10.1016/j.brainres.2008.07.067.
- Weydt, P., Hong, S.Y., Kliot, M., & Moller, T. (2003). Assessing disease onset and progression in the SOD1 mouse model of ALS. Neuroreport, 14, 1051–1054. doi:10.1097/01.wnr.0000073685.00308.89.
- Wilkinson, R.D., Williams, R., Scott, C.J., & Burden, R.E. (2015). Cathepsin S: therapeutic, diagnostic, and prognostic potential. Biological Chemistry, 396, 867–882. doi:10.1515/hsz-2015-0114.
- Yamazaki, T., Z, K., Hailey, D., Presley, J., Lippincott-Schwartz, J., & Samelson, L.E. (2002). Role of Grb2 in EGF-stimulated EGFR internalization. Journal of Cell Science, 115, 1791–1802.
- Yan, Q., & Sage, E.H. (1999). SPARC, a matricellular glycoprotein with important biological functions. Journal of Histochemistry and Cytochemistry, 47, 1495–1506. doi:10.1177/002215549904701201.
- Yang, Y., Vidensky, S., Jin, L., Jie, C., Lorenzini, I., Frankl, M., & Rothstein, J.D. (2011). Molecular comparison of GLT1+ and ALDH1L1+ astrocytes in vivo in astroglial reporter mice. Glia, 59, 200–207. doi:10.1002/glia.21089.
- Yusoff, P., Lao, D.H., Ong, S.H., Wong, E.S., Lim, J., Lo, T.L., … Guy, G.R. (2002). Sprouty2 inhibits the Ras/MAP kinase pathway by inhibiting the activation of Raf. Journal of Biological Chemistry, 277, 3195–3201. doi:10.1074/jbc.M108368200.
- Zhang, Y., & Barres, B.A. (2010). Astrocyte heterogeneity: an underappreciated topic in neurobiology. Current Opinion in Neurobiology, 20, 588–594. doi:10.1016/j.conb.2010.06.005.
- Zhou, M., Xu, G., Xie, M., Zhang, X., Schools, G.P., Ma, L., … Chen, H. (2009). TWIK-1 and TREK-1 are potassium channels contributing significantly to astrocyte passive conductance in rat hippocampal slices. Journal of Neuroscience, 29, 8551–8564. doi:10.1523/JNEUROSCI.5784-08.2009.