
Abstract
Background
The transcriptional repressor B-cell lymphoma 6 (BCL6) has been reported to inhibit inflammation. So far, experimental evidence for the role of BCL6 in bronchopulmonary dysplasia (BPD) is lacking. Our study investigated the roles of BCL6 in the progression of BPD and its downstream mechanisms.
Methods
Hyperoxia or lipopolysaccharide (LPS) was used to mimic the BPD mouse model. To investigate the effects of BCL6 on BPD, recombination adeno-associated virus serotype 9 expressing BCL6 (rAAV9-BCL6) and BCL6 inhibitor FX1 were administered in mice. The pulmonary pathological changes, inflammatory chemokines and NLRP3-related protein were observed. Meanwhile, BCL6 overexpression plasmid was used in human pulmonary microvascular endothelial cells (HPMECs). Cell proliferation, apoptosis, and NLRP3-related protein were detected.
Results
Either hyperoxia or LPS suppressed pulmonary BCL6 mRNA expression. rAAV9-BCL6 administration significantly inhibited hyperoxia-induced NLRP3 upregulation and inflammation, attenuated alveolar simplification and dysregulated angiogenesis in BPD mice, which were characterized by decreased mean linear intercept, increased radical alveolar count and alveoli numbers, and the upregulated CD31 expression. Meanwhile, BCL6 overexpression promoted proliferation and angiogenesis, inhibited apoptosis and inflammation in hyperoxia-stimulated HPMECs. Moreover, administration of BCL6 inhibitor FX1 arrested growth and development. FX1-treated BPD mice exhibited exacerbation of alveolar pathological changes and pulmonary vessel permeability, with upregulated mRNA levels of pro-inflammatory cytokines and pro-fibrogenic factors. Furthermore, both rAAV9-BCL6 and FX1 administration exerted a long-lasting effect on hyperoxia-induced lung injury (≥4 wk).
Conclusions
BCL6 inhibits NLRP3-mediated inflammation, attenuates alveolar simplification and dysregulated pulmonary vessel development in hyperoxia-induced BPD mice. Hence, BCL6 may be a target in treating BPD and neonatal diseases.
Graphical Abstract
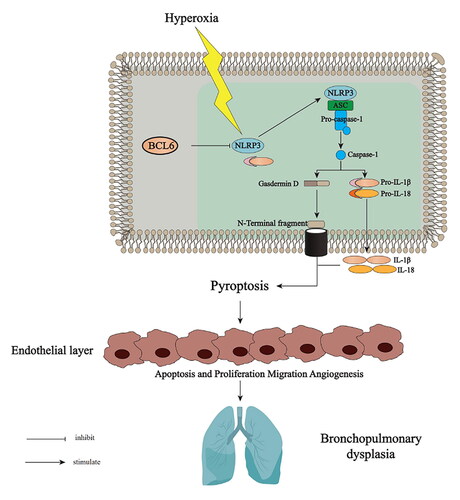
1. Introduction
Bronchopulmonary dysplasia (BPD) is a common chronic respiratory disease among preterm infants. It is characterized by abnormal pulmonary alveolarization and microvascular development,Citation1 as well as the presence of increased interstitial fibroproliferation.Citation2 In adulthood, BPD survivors often suffer from pulmonary diseases.Citation3 Clinically, hyperoxia is a major contributor to BPD. Long-term exposure to high levels of O2 induces an acute inflammation response, followed by a fibro-proliferative repair period, thereby leading to lung damage. Considering the sustained high prevalence of BPD and the associated high risk of pulmonary sequelae, further investigation on effective treatment options for this disease is warranted.
Chronic pulmonary inflammation exerts a major effect on the pathogenesis of BPD.Citation4 Pyroptosis is a type of programmed cell death, which leads to a sustained severe inflammatory response and contributes to inflammatory pathology in the host.Citation5 Thus far, activation of the nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3)-inflammasome activation and inflammatory response have been shown in lung tissues of BPD mice, inhibition of this activation has emerged as a novel approach for BPD therapy.Citation6,Citation7 Acetate attenuates lung injury in neonatal mice with BPD by downregulating the activation of NLRP3 inflammasomes.Citation8 NLRP3-knockout mice do not exhibit an inflammatory response and undergo normal alveolarization.Citation9 Mice expressing a gain-of-function NLRP3 exhibit abnormal pulmonary morphogenesis and expire shortly after birth.Citation10
The transcriptional repressor B-cell lymphoma 6 (BCL6) protein is regarded as an attractive therapeutic target for the treatment of inflammation autoimmunity disease and various types of cancer.Citation11 It has been reported that overexpression of BCL6 suppresses the generation of reactive oxygen species (ROS) and inhibits apoptosis in B-cell lymphoma cells treated with chemotherapeutic reagents.Citation12 In a previous study, we demonstrated that BCL6 could inhibit NLRP3 expression and inflammation.Citation13 Moreover, BCL6 overexpression alleviated ovalbumin-induced inflammation by inhibiting the activation of macrophages and NLRP3 inflammasomes.Citation14 Nevertheless, it remains unknown whether BCL6 participates in the regulation of the inflammatory response and lung damage caused by hyperoxic stimuli. The aim of this study was to explore the roles and possible underlying mechanisms of BCL6 in the progression of BPD.
2. Materials and methods
2.1. Experimental animals
Pregnancy C57BL/6J mice were obtained from Jiangnan University (Wuxi, China). All experiments were all approved by the Experimental Animal Care and Use Committee of Jiangnan University (JN.No20210915c0420408[342]) and carried out according to the Guide for the Care and Use of Laboratory Animal published by the US National Institutes of Health (8th edition, 2011). All mice were all housed in a temperature-controlled room under a 12-h light/dark cycle with ad libitum access to standard chow and water. At the end of the experiments, each mouse was euthanized by an overdose of pentobarbital sodium (50 mg/kg, intraperitoneally), and the lung tissues were collected for histological and molecular biological analyses.
2.2. Animal model and study design
To examine the effect of BCL6 on BPD mice, 90% O2 was utilized to mimic a hyperoxia challenged BPD model, as previously described.Citation15 Briefly, newborn mouse pups (within 12 h of birth) were exposed to hyperoxia (90% O2) from postnatal day 0 to 14 (PN0-14; alveolar stage) and subsequently returned to room air (RA), whereas the control mice remained in RA. For BCL6 overexpression, PN1 mice were intranasally incubated with recombination adeno-associated virus serotype 9 expressing BCL6 (rAAV9-BCL6) (GeneChem, Shanghai, China) at a total of 4 μl containing 2 × 1012 viral genome copies (GC) (2 μl per nostril).Citation16,Citation17 The control group received the same volume of rAAV9-Vector intranasally. Experiments were mainly carried out 2, 4 and 6 wk after the induction. Mothers nursing the pups take turns every 24 h to prevent oxygen poisoning. The timeline of this experimental study is presented schematically in Supplementary Figure 1.
To study the effect of BCL6 inhibitor FX1 (Supplementary Figure 2A) on BPD mice, 75% O2 was utilized to mimic a hyperoxia-challenged BPD model, as previously reported.Citation18 Briefly, newborn mice (within 12 h of birth) were exposed to 75% O2 for 14 days (PN14) and returned to RA. FX1 (25 mg/kg)Citation19 was intraperitoneally injected from PN14 to PN17. The control mice were housed under normoxic conditions throughout lactation. Short-term and long-term assessments were performed on PN25 and PN49, respectively. The timeline of this experimental study is schematically presented in Supplementary Figure 2B.
Lipopolysaccharide (LPS; 1 mg/kg) was also used to mimic a BPD murine model, as we previously reported.Citation20 Briefly, mice at day 5 of life were injected intraperitoneally with LPS (Sigma, St. Louis, MO) or sterile saline solution for 1 week, respectively.
2.3. Collection and analysis of bronchoalveolar lavage fluid (BALF)
The BALF was collected through lung lavage. The BCA reagent kit (Yeasen, Shanghai, China) was used to detect the total protein concentration in the cell-free supernatant, while the remaining cells were used to determine the cell count.
2.4. Analysis of the exudation of Evans blue
Evans blue dye was used to examine the pulmonary barrier permeability, as previously reported.Citation21,Citation22 The absorbance of the supernatant was measured by spectrophotometry at 620 nm wavelength. The total content of Evans blue is calculated based on the standard absorbance curve.
2.5. Histological staining analysis
Mice were anesthetized, and left lung lobes were collected, fixed overnight in 4% paraformaldehyde solution, and embedded in paraffin. Hematoxylin-and-eosin staining and Masson’s trichrome staining were performed to observe the histomorphology and fibrosis of the lung, respectively. The mean linear intercept (MLI), alveoli numbers, and radical alveolar count (RAC) were calculated as previously reported.Citation20
2.6. Immunohistochemistry
To detect the pulmonary vascular development, lung tissue sections (thickness: 5 μm) were immunostained with antibodies against CD31 and or α-smooth muscle actin (α-SMA; Cell Signaling Technology, Beverly, MA, USA) at a dilution of 1:100, respectively. Next, the avidin-biotin complex signal was detected using the 3,3′-diaminobenzidine solution.
2.7. Immunofluorescence
Lung tissue sections (thickness: 5 μm) were immunostained with the antibody against CD31 (Cell Signaling Technology, Beverly, MA, USA) at a dilution of 1:100. Images were captured using a Zeiss LSM880 confocal microscope (Carl Zeiss, Oberkochen, Germany).
2.8. Cell culture
Human primary pulmonary microvascular endothelial cells (HPMECs) were purchased from ScienCell Research Laboratories (Carlsbad, CA, USA) and cultured in endothelial-cell growth medium (ScienCell) supplemented with 5% fetal bovine serum (FBS) and antibiotics (100 units/mL penicillin and 100 mg/mL streptomycin) in a humidified atmosphere at 37 °C and 5% CO2.
2.9. Cell viability test
HPMECs cells were exposed to hyperoxia (95% O2) for 24, 36, and 48 h.Citation23,Citation24 Subsequently, the attached HPMECs cells were cultured with a mixture of Cell Counting Kit-8 solution (Beyotime Institute of Biotechnology, Shanghai, China) and culture medium for 2 h at 37 °C. The absorbance was measured at 450 nm wavelength with a microplate reader (ELX800; BioTek, Winooski, VT, USA).
2.10. BCL6 overexpression in HPMECs
The BCL6 overexpression plasmid was constructed as previously reported.Citation13 Briefly, after reaching 80% confluence, the cells were transfected with the pcDNA3.1 plasmid (control) or BCL6 overexpression plasmid (1 μg/mL) for 24 h. Thereafter, the transfected HPMECs were stimulated with hyperoxia (95% O2) for 48 h.
2.11 5-Ethynyl-2’-deoxyuridine (EdU) incorporation assay in HPMECs
The EdU assay kit (Guangzhou RiboBio, Guangzhou, China) was used to examine the effect of BCL6 on the proliferation of HPMECs, as previously reported.Citation25
2.12. Examination of HPMECs apoptosis
The Annexin V-FITC/PI Apoptosis Detection Kit (CoWin-Biotech, Beijing, China) was used to examine the effect of BCL6 on the apoptosis of HPMECs, as previously reported.Citation25
2.13. Tube formation assay in HPMECs
The tube formation assay was performed to examine the effect of BCL6 on angiogenesis in HPMECs, as previously reported.Citation20
2.14. Real-Time quantitative polymerase chain reaction (qPCR)
Real-time PCR was performed using the LightCycler® 480 PCR detection system (Roche, Foster City, CA, USA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control to normalize the data. The sequences of primers used in this analysis are listed in .
Table 1. Primer sequence of qPCR.
2.15. Western blotting analysis
Western blotting analysis was performed to examine the protein expression in lung tissues and HPMECs, as previously reported.Citation20 The antibodies used in this experiment are listed in . GAPDH was used as a loading control to normalize the data.
Table 2. Antibodies used for Western blotting.
2.16. Statistical analysis
All results were expressed as the mean ± standard error. Comparisons between the two groups were performed using the unpaired two-tailed Student’s t-test. Analysis of variance (ANOVA) techniques were used to compare the significance of differences between more than two groups followed by Bonferroni’s post hoc analysis. p-value <.05 denoted a statistically significant difference.
3. Results
3.1. Lung NLRP3 inflammasome activation and BCL6 downregulation in BPD mice
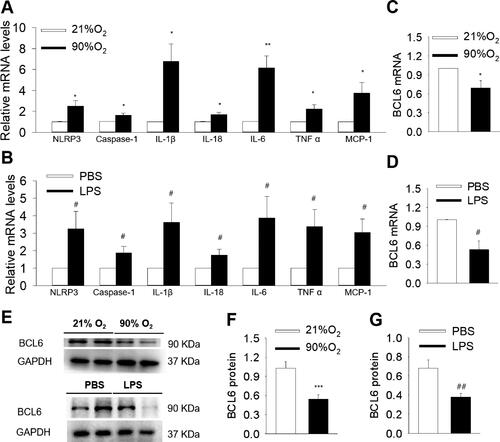
The mRNA levels of NLRP3, Caspase-1, IL-1β, IL-18, IL-6, TNF α and chemokine MCP-1 were significantly up-regulated in lungs both in hyperoxia- and LPS-induced BPD models (). Moreover, the mRNA and protein expression of BCL6 was down-regulated in the lungs compared with those of control (). These results imply that the downregulation of BCL6 aggravated the inflammatory response in BPD mice, and suggest that BCL6 plays a role in BPD.
Figure 1. NLRP3 activation and BCL6 expression in lung tissues of hyperoxia and LPS induced BPD mice. (A and B) Expression levels of NLRP3, Caspase-1, IL-1β, IL-18, IL-6, TNF α and MCP-1 mRNA. (C and D) mRNA levels of BCL6 in hyperoxia and LPS induced BPD model, respectively. (E) Representative images of Western blots. (F and G) Expression levels of BCL6 protein in hyperoxia and LPS induced BPD model, respectively. *p < .05 vs. 21% O2; **p < .01 vs. 21% O2; ***p < .001 vs. 21% O2. #p < .05 vs. PBS; ##p < .01 vs. PBS. Values are mean ± SE, n = 4–6 per group.
3.2. Effects of BCL6 overexpression on alveolar development in hyperoxia-induced BPD mice
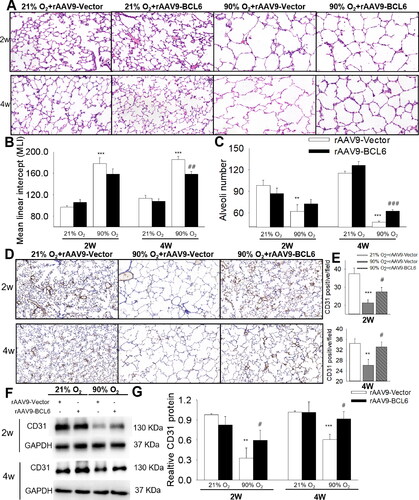
rAAV9-BCL6 was used to induce overexpression of BCL6 in control and BPD mice. At 2 wk after intranasal administration of rAAV9-BCL6, the BCL6 mRNA levels exhibited a two-fold elevation compared with the rAAV9-vector. This expression reached a peak at 4 wk, and was sustained for 6 wk (Supplementary Figure 3A). BCL6 protein expression was also measured, showing that the administration of rAAV9-BCL6 administration increased BCL6 expression in the lung, but not in the heart. These findings indicated the specificity and effectiveness of BCL6 overexpression in the lung (Supplementary Figure 3B,C). Subsequently, the effect of BCL6 overexpression on lung alveolar development was examined. As shown in , exposure to 90% O2 impaired alveolar growth, as evidenced by the increase of the MLI and decrease in alveoli numbers. At 2 wk after rAAV9-BCL6 administration, BPD mice exhibited reduced MLI and increased alveoli numbers, although the differences were not statistically significant. Surprisingly, 4 wk after treatment with rAAV9-BCL6, BPD mice showed significantly reduced MLI and increased alveoli numbers versus those treated with the rAAV9-Vector. These results indicate the beneficial role of BCL6 against hyperoxia-induced alveolar simplification.
Figure 2. Effects of BCL6 overexpression (OE) on alveoli and pulmonary vessels development in hyperoxia-induced BPD mice aged 2 and 4 wk old, respectively. (A) Representative H&E stained lung sections (magnification, ×400). (B) Quantification of mean linear intercept (MLI). (C) Quantification of alveoli number. (D) Representative Immunohistochemistry images of CD31 (magnification, ×200). (E) Quantification of CD31 immunoreactivity. (F) Representative images of Western blots. (G) The relative protein expression of CD31 in the lung. **p < .01 vs. 21% O2+rAAV9-Vector; ***p < .001 vs. 21% O2+rAAV9-Vector; #p < .05 vs. rAAV9-Vector; ##p < .01 vs. rAAV9-Vector; ###p < .001 vs. rAAV9-Vector. Values are mean ± SE, n = 4–6 per group.
3.3. Effects of BCL6 overexpression on angiogenesis during lung development in hyperoxia-treated mice
Abnormal angiogenesis is recognized as another major feature of BPD.Citation26 Thus, we examined the effect of BCL6 on angiogenesis in BPD mice. The presence of CD31, a vascular marker, was detected. As shown in , the levels of CD31 in lung tissues were reduced during exposure to hyperoxia (age: 2 wk), and during recovery to room air (RA) (age: 4 wk). In addition, And BCL6 overexpression significantly increased CD31 expression, indicating the beneficial effect of BCL6 on pulmonary vessel development under hyperoxic stimuli.
3.4. Effects of BCL6 overexpression on NLRP3-mediated inflammatory response in hyperoxia-treated mice
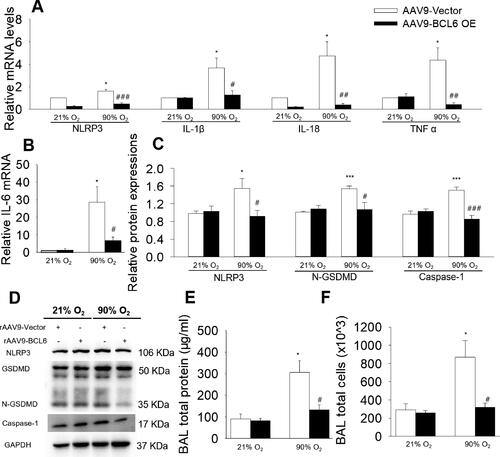
As shown in , exposure to hyperoxia followed by 2 wk of RA recovery increased the mRNA levels of pro-inflammatory cytokines (IL-1β, IL-18, TNF α and IL-6), which were all repressed by BCL6 overexpression. Our previous data demonstrated that BCL6 could inhibit NLRP3 expression.Citation13 Importantly, it was suggested that NLRP3 is a new promising therapeutic target in BPD.Citation6 Thus, we detected the expressions of NLRP3 and its downstream molecules Caspase-1 and N terminus-gasdermin-D (N-GSDMD) (key proteins in pyroptosis). As shown in , BCL6 overexpression significantly reduced the expression of NLRP3, N-GSDMD and Caspase-1 in BPD mice. Moreover, the protective roles of BCL6 were further confirmed by the fact that BCL6 overexpression significantly reduced the total protein concentration and the total cell numbers in BALF of BPD mice (), thereby confirming the protective role of BCL6. These results imply that BCL6 inhibits the hyperoxia-induced inflammatory response. Furthermore, the protective role of BCL6 against BPD may be attributed to its inhibitory effect against NLRP3 inflammasome activation.
Figure 3. Effects of BCL6 OE on NLRP3-mediated inflammation in hyperoxia-induced BPD mice aged 4 wk old. (A and B) Relative mRNA levels of NLRP3, IL-1β, IL-18, TNF α and IL-6 in the lung. (C and D) the protein expressions of NLRP3, N-GSDMD and Caspase-1 in the lung. (E) total protein concentration in bronchoalveolar lavage fluid (BALF). (F) Total cell count in BALF. *p < .05 vs. 21% O2+rAAV9-Vector; ***p < .001 vs. 21% O2+rAAV9-Vector; #p < .05 vs. rAAV9-Vector; ##p < .01 vs. rAAV9-Vector; ###p < .001 vs. rAAV9-Vector. Values are mean ± SE, n = 4–6 per group.
3.5. Effects of BCL6 overexpression on lung development in mice aged 6 wk
We also examined the effect of BCL6 overexpression on the lungs of BPD mice aged 6 wk old. Despite recovery from hyperoxia (90% O2) to RA for 4 wk, BPD mice also exhibited alveoli simplification as evidenced by the reduced alveolar numbers, increased MLI and RAC, and BCL6 overexpression significantly reversed these disadvantages (Supplementary Figure 4A–D). Considering the fact that the presence of increased interstitial fibroproliferation existed in BPD,Citation2 we also determined the effect of BCL6 on pulmonary fibrosis. As shown in Supplementary Figure 4E, the immunoreactivity of fibrosis marker α-SMA increased in BPD mice, and this effect was blocked by rAAV9-BCL6 administration. These results implied that BCL6 ameliorates hyperoxia-induced alveolar simplification and attenuates pulmonary epithelial cell alteration in BPD mice.
3.6. Effects of BCL6 overexpression on cell proliferation, apoptosis, angiogenesis, and inflammation in hyperoxia-induced HPMECs
Thereafter, we examined the effect of BCL6 on HPMECs under hyperoxia stimulation. We found that the mRNA and protein levels of BCL6 were remarkably downregulated after exposure to hyperoxia (95% O2) for 36 and 48 h (Supplementary Figure 5). Exposure to hyperoxia significantly reduced the proliferation of HPMECs, as evidenced by the reduced number of EdU-positive cells and decreased PCNA protein expression. Notably, overexpression of BCL6 significantly stimulated the proliferation of HPMECs (). Moreover, BCL6 overexpression decreased hyperoxia-induced apoptosis (). In addition, angiogenesis of HPMECs was greatly reduced following exposure to hyperoxia, whereas BCL6 overexpression exerted the opposite effect (). These results suggest that BCL6 overexpression promoted proliferation and angiogenesis, and inhibited apoptosis in hyperoxia-stimulated HPMECs.
Figure 4. Effects of BCL6 OE on cell proliferation, apoptosis, angiogenesis and NLRP3-mediated inflammation in hyperoxia-induced HPMECs. (A) Representative images of EdU incorporation assay. Blue fluorescence shows cell nuclei and green fluorescence stands for cells with DNA synthesis. (B) The protein expression of PCNA. (C) Apoptosis of HPMECs via flow cytometry. (D) Tube formation of HPMECs. (E) Representative images of Western blots. (F) The protein expressions of NLRP3, N-GSDMD and Caspase-1. *p < .05 vs. 21% O2; **p < .01 vs. 21% O2; #p < .05 vs. pcDNA3.1; ##p < .01 vs. pcDNA3.1. Values are mean ± SE, n = 4 per group.
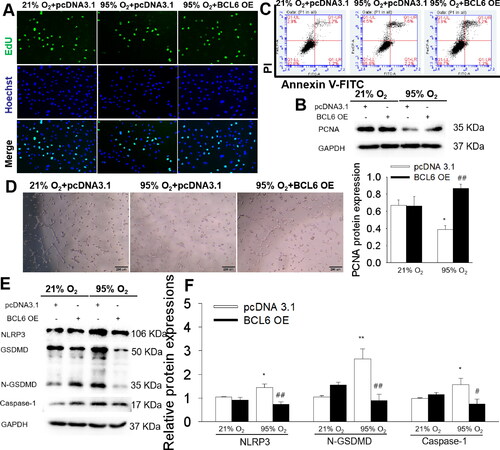
3.7. Effects of BCL6 overexpression on hyperoxia-induced inflammation in HPMECs
The anti-inflammatory effect of BCL6 was also determined in hyperoxia-treated HPMECs as presented in . Under hyperoxic conditions, HPMECs showed remarkably increased expressions of NLRP3 and its downstream molecule, i.e. Caspase-1 and N-GSDMD, while BCL6 overexpression significantly inhibited NLRP3-mediated inflammation, which strongly supporting the anti-inflammatory effect of BCL6 on HPMECs stimulated through hyperoxia.
3.8. Effect of BCL6 inhibitor FX1 on the development of alveoli and pulmonary vessels in mice with BPD
FX1, the specific BCL6 inhibitor, was also used in this study to examine the beneficial effect of BCL6. We found that in 90% O2-induced BPD mice, FX1 administration significantly inhibited the growth and development of mice. These effects were evidenced by the reduced body weight and the remarkably elevated brain-to-liver weight ratio (Supplementary Figure 6A–C). Meanwhile, FX1 treated BPD mice also exhibited more severe pulmonary injury, as manifested by the arrest of alveolarization and more obvious inflammatory cell infiltration (Supplementary Figures 6D,E). Hence, administration of FX1 in BPD mice exposed to 90% O2 resulted in marked growth arrest. Considering the welfare of animals, 75% O2 was selected to examine the effect of FX1 on BPD mice.
Following exposure to hyperoxia (75% O2) exposure for 14 days, BPD mice showed a continuous decrease in body weight by approximately 12% versus control mice until the age of 25 d. The administration of FX1 further reduced the body weight by approximately 33% and 25.8% at the age of 17 and 25 d, respectively when compared with BPD mice (Supplementary Figure 7A). Furthermore, the brain, liver, lungs, heart, and kidneys were collected and weighed. We found that hyperoxic (75% O2) exposure significantly increased brain weight, whereas it decreased liver weight, along with the increased brain-to-liver weight ratio (Supplementary Figure 7B,C). These results indicated that hyperoxia-induced BPD mice presented asymmetrical growth, while administration of FX1 aggravated this asymmetrical growth. In addition, treatment with FX1 increased the organ index of the lungs and heart, although the difference was not statistically significant. FX1 reduced the kidney index in BPD mice (Supplementary Figure 7D). These results imply a harmful effect of BCL6 inhibition on the growth and organ development of mice with BPD.
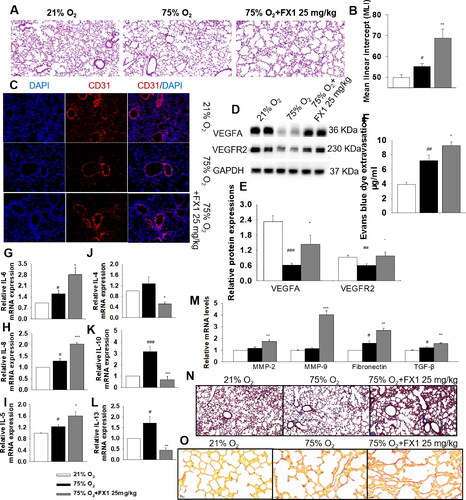
A short-term assessment was performed on PN25. Exposure of neonate mice to hyperoxia resulted in alveolar simplification while FX1 worsened the lung morphology and increased the alveolar size (). The immunofluorescence images revealed that hyperoxia decreased the immunoreactivity of CD31. Notably, treatment with FX1 did not have a significant effect on the immunoreactivity of CD31 (), there seems that FX1 does not affect angiogenesis. However, we found that both the mRNA and protein levels of VEGFA and VEGFR2 were significantly increased after the administration of FX1 (, Supplementary Figure 8A). As we know, VEGFA binds to VEGFR2, thereby mediating vascular sprouting, proliferation, and endothelial cell migration, as well as promoting angiogenesis.Citation27,Citation28 It has also been reported that exaggerated signaling by VEGFA and its receptor (i.e., VEGFR2) in pathologies results in poor vessel function.Citation29,Citation30 Thus, the Evans blue method was used to confirm the disruption of the alveolar-capillary barrier. We found that BPD mice treated with FX1 showed significantly more extravasation compared with both normal and BPD mice, indicating that treatment with FX1 exacerbated the disruption of the alveolar-capillary barrier (). According to these results, we think that inhibition of BCL6 exerts a deleterious effect on lung morphology and pulmonary vascular development.
Figure 5. Effect of FX1 on the lung in mice with BPD aged 4 wk. (A) Representative H&E stained lung sections (magnification, ×100). (B) Quantification of MLI. (C) Representative CD31 immunofluorescence staining. (D) Representative images of Western blots. (E) The protein expressions of VEGFA and VEGFR2. (F) The quantitative analysis of Evans blue dye leakage in lung tissues. (G–I) The relative mRNA levels of pro-inflammatory cytokines. (J–L) The relative mRNA levels of anti-inflammatory cytokines. (M) The mRNA levels of matrix metalloproteinases, including MMP-2, MMP-9, fibronectin and TGF-β. (N) Representative Masson’s staining of lung sections. (O) Representative Sirius red staining of lung sections. #p < .05 vs. 21% O2; ##p < .01 vs. 21% O2; ###p < .001 vs. 21% O2; *p < .05 vs. 75% O2; **p < .01 vs. 75% O2; ***p < .001 vs. 75% O2. Values are mean ± SE, n = 6–8 per group.
3.9. Effect of BCL6 inhibitor FX1 on inflammation in mice with BPD
As shown in and Supplementary Fig 8B, mice exposed to hyperoxia (75% O2) had increased mRNA levels of pro-inflammatory cytokines, including IL-1α, IL-6, TNF α, IL-8, IL-18, IL-5, and interferon-γ (IFN-γ) compared with normal mice. Administration of FX1 remarkably upregulated the mRNA levels of IL-6, IL-8, and IL-5. Moreover, FX1 significantly reduced the levels of anti-inflammatory factors, including IL-4, IL-10, and IL-13 (). These results suggest that the BCL6 inhibitor FX1 accelerates the hyperoxia-induced inflammatory response in lung tissues.
3.10. Effect of FX1 on pulmonary fibrosis in mice with BPD
We also examined the effect of BCL6 inhibitor FX1 on pulmonary fibrosis. As shown in , FX1 significantly increased the hyperoxia-induced upregulation of pro-fibrogenic factors, including MMP-2, MMP-9, fibronectin, and TGF-β. In addition, the pro-fibrogenic effect of FX1 was also revealed by Masson’s and Sirius red staining (). These results suggest that the BCL6 inhibitor FX1 exacerbates lung fibrosis in BPD.
3.11. Effects of FX1 on mice aged 7 wk
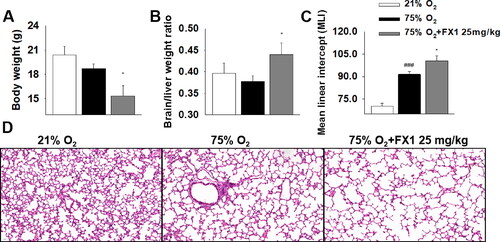
We also determined the long-term effects of FX1 on BPD mice. We found that FX1-treated BPD mice continued to exhibit a lower body weight and a higher brain-to-liver weight ratio versus BPD mice (). These data suggested that, despite the short duration of treatment (i.e., only 4 d), the effects of FX1 on the growth and development of BPD mice persisted into adulthood. Moreover, despite a recovery period of almost 5 wk, BPD mice continued to show impaired alveolar growth, as evidenced by the increase in the MLI. Furthermore, we investigated the effect of FX1 on alveolar simplification under exposure to hyperoxia. Expectedly, BPD mice treated with FX1 showed a significantly increased MLI as compared with those exposed to hyperoxia alone (). These results further support the aforementioned findings that inhibition BCL6 function led to long-lasting impairment of lung development in hyperoxia-induced BPD mice.
Figure 6. The effects of FX1 on mice aged 7 wk. (A) Effect of FX1 on body weight. (B) Effect of FX1 on brain/liver weight ratio. (C) Quantification of mean linear intercept (MLI). (D) Representative H&E stained lung sections (magnification, ×200). #p < .05 vs. 21% O2; ##p < .01 vs. 21% O2; ###p < .001 vs. 21% O2; *p < .05 vs. 75% O2; **p < .01 vs. 75% O2. Values are mean ± SE, n = 6–8 per group.
4. Discussion
The main findings of our study are that (1) BCL6 expression in the lung was reduced in hyperoxia or LPS induced BPD mice model. BCL6 overexpression reversed hyperoxia-induced NLRP3 upregulation and inflammation, then improved alveolar simplification and dysregulated pulmonary vessel development in BPD mice. (2) BCL6 overexpression promotes HPMECs proliferation and angiogenesis, inhibits apoptosis and inflammation in hyperoxia conditions. (3) BCL6 specific inhibitor FX1 exacerbated alveolar simplification and vascular damage, worsened inflammation and led to pulmonary fibrosis in BPD mice. These results reveal that BCL6 inhibits NLRP3-mediated inflammation, improves alveolar simplification and pulmonary vessel development in hyperoxia-induced BPD.
Long-term exposure to high concentrations of O2 causes inflammation, consequently resulting in BPD-like symptoms in offspring.Citation31 Thus far, several experimental mouse models induced by different concentrations of O2 have been established to mimic BPD-like disease.Citation15,Citation18,Citation32 In this investigation, 90% O2 and 75% O2 were used in our study, respectively. We found that exposure of neonatal mice to both 90% O2 and 75% O2 for 14 d resulted in alveolar simplification (with increased MLI, decreased RAC and reduced alveoli numbers) and abnormal pulmonary vascular development (with decreased CD31-stained pulmonary blood vessels), suggesting that exposure to hyperoxia during the alveolar phase of pulmonary development can lead to BPD in preterm infants. To examine the effects of BCL6 on lung alveolarization and abnormal angiogenesis in BPD mice were examined. Both rAAV9-BCL6 and BCL6 specific inhibitor FX1 were used. We found that in 90% O2-induced BPD mice, FX1 administration significantly inhibited the growth and development of mice. Meanwhile, FX1 treatment BPD mice also exhibited more severe pulmonary injury, as manifested by alveolarization arrest and more obvious inflammatory infiltration. In a word, FX1 administration in 90% O2-induced BPD mice showed severe growth arrest. Taking into account the welfare of animals, 75% O2 was selected to examine the effect of FX1 on BPD mice.
The increased pro-inflammatory cytokines such as IL-1β, IL-6 have been found in the BALF of infants with BPD. Pro-inflammatory cytokines antagonist, such as interleukin-1 receptor antagonist prevents hyperoxia-induced alveolar disruption and airway fibrosis in mice.Citation33 These results imply that inhibiting inflammation can improve alveolar and pulmonary microvascular development in BPD. Pyroptosis is a newly identified form of inflammatory programmed cell death that is mainly activated by the NLRP3/Caspase-1 axis.Citation34,Citation35 GSDMD, the executive protein of pyroptosis, once activated, it transferred to the membrane and cleaved the N-terminal sequence, then resulting in the release of more inflammatory mediators.Citation36,Citation37 The elevation of Caspase-1 activity and expression was found in neonatal mice induced by hyperoxia,Citation9 suggesting the possible involvement of pyroptosis in the pathological process of BPD. It was shown that Ac-YVAD-CMK, an irreversible caspase-1 inhibitor, decreases IL-1β and GSDMD expression, and improved alveolar and vascular development in hyperoxia-exposed lungs.Citation38 Actually, in our study, we also found the increased NLRP3, Caspase-1 and N-GSDMD expressions in hyperoxia-exposed murine lungs and HPMECs. BCL6 overexpression significantly inhibited NLRP3, Caspase-1 and N-GSDMD expressions both in vivo and in vitro. Meanwhile, BCL6 overexpression reduced the elevated mRNA levels of pro-inflammatory cytokines (IL-1β, IL-6, TNF α and IL-18). What’s more, BCL6 overexpression decreased the total cells and protein concentration in BALF. However, BCL6 inhibitor FX1 aggravated hyperoxia-induced inflammation, which was evidenced by the upregulation of pro-inflammatory cytokines (IL-6, IL-8, and IL-5) and the downregulation of anti-inflammatory cytokines (IL-4, IL-10, and IL-13). Considering the fact that inhibition of inflammation attenuates BPD.Citation6 Thus, it is reasonable to conclude that the beneficial role of BCL6 on hyperoxia-induced pulmonary injury may be attributed to the attenuation of NLRP3-mediated inflammation.
In addition to BPD, the chronic inflammatory microenvironment is also a key factor in the development of fibrosis.Citation39 Genes encoding extracellular proteins, such as the extracellular matrix (ECM) protein, fibronectin, and matrix metalloproteinases (MMPs), have been considered as the main hallmarks of fibrosis.Citation40,Citation41 Furthermore, TGF-β plays a critical role in the initiation and development of all types of tissue fibrosis, including pulmonary fibrosis. It can promote gene transcription to increase the production of collagen and fibronectin, thereby elevating the production and deposition of ECM.Citation42–44 In response to injury, epithelial cells within the lungs produce an array of pro-inflammatory and anti-inflammatory cytokines that stimulate fibroblast proliferation and matrix synthesis, leading to fibrosis.Citation45 Several profibrotic factors, such as TGF-β and fibronectin, are increased in patients with BPD.Citation46,Citation47 In the present study, we found that BPD mice aged 6-7 wk exhibited fibrotic phenotype in the lung, rAAV9-BCL6 administration reduced the fibrosis marker α-SMA, and BCL6 inhibitor FX1 aggravated pulmonary fibrosis as further confirmed by the increased expressions of pro-fibrogenic factors (MMP-2, MMP-9, fibronectin, and TGF β), Masson’s and Sirius red staining. These results suggest that BCL6 inhibits fibrosis. The pro-fibrogenic property of BCL6 inhibitor FX1 may be, at least partially, attribute to its enhancement of inflammation.
Angiogenesis is a key event in fundamental physiological processes, including growth and development, wound healing, organ regeneration, inflammation, and reproduction. Pulmonary vascular dysangiogenesis is one of the characteristics of BPD. We found that hyperoxic exposure reduced the expression of vessel marker CD31 in the lung. And BCL6 overexpression significantly increased CD31 expression, suggesting the beneficial effect of BCL6 on vasculogenesis. Abnormal angiogenesis is influenced by VEGF and its receptor; VEGF is essential for the normal development of blood vessels, and stimulates the generation of new, immature, and leaky blood vessels.Citation48 It has been previously reported that, in hyperoxia-induced lung injury, the pulmonary capillary endothelium is damaged, thereby exposing the capillary basement membrane and increasing vascular endothelial permeability.Citation49,Citation50 Prolonged exposure of neonates to hyperoxia can cause endothelial and epithelial injury, as well as enhance the leakage of alveolar capillary proteins, consequently increasing pulmonary permeability and fluid accumulation in the airways.Citation51 VEGFA, a predominant isoform of VEGF, binds to VEGFR2 and mediates vessel formation and vascular leakage.Citation28,Citation52 Therapeutic agents, such as bevacizumab, aflibercept, and ranibizumab, were designed to inhibit VEGFA through the prevention of binding to VEGFR2. It has been confirmed that treatment with these drugs decreases neovascular formation and edema.Citation53 Angiotensin-converting enzyme 2 (ACE2) antagonized the VEGFA-mediated increases in lung vessel permeability during acute lung injury and improved lung function.Citation54 We found that BCL6 inhibitor FX1 significantly activated the VEGFA/VEGFR2 signaling pathway in the lungs in hyperoxia-induced BPD mice. Meanwhile, FX1 exacerbated the disruption of the alveolar-capillary barrier, as shown by Evans blue extravasation. In addition, the excessive release of pro-inflammatory cytokines induced by FX1 may accelerate the damage in the alveolar-capillary barrier. Collectively, these results suggest that inhibition BCL6 function aggravated pulmonary vessel permeability via excessive activation of the VEGF pathway.
Honestly, there are still limitations in this study. Firstly, we didn’t examine the effect of BCL6 OE on proliferation, apoptosis and angiogenesis in HPMECs in room air. In addition, we found that BCL6 inhibits NLRP3-mediated inflammation and attenuates hyperoxia-induced BPD, it’s better to explore which part of the BCL6 protein works. The related experiments will continue to performed in the future.
5. Conclusions
This is the first study demonstrating that BCL6 inhibits NLRP3-mediated inflammation, and attenuates alveolar simplification and dysregulated pulmonary vessel development in BPD mice exposed to hyperoxia. These findings highlight the beneficial effects of BCL6 and imply that BCL6 may be a new target in the treatment of BPD.
Authors contributions
DC, RQY and QFP contributed to the conception and design of the research. DC, HMZ, XHD and SPL performed the experiments. DC, MHZ and SPL analyzed the data. DC, MHZ, YXW, and YT interpreted the results of the experiments. DC prepared the figures and drafted the manuscript. DC, RQY and QFP edited and revised the manuscript. All authors read and approved the final manuscript.
Ethical approval
All the in vivo experiments were approved by the Experimental Animal Care and Use Committee of Jiangnan University (JN.No20210915c0420408[342]) and carried out according to the Guide for the Care and Use of Laboratory Animal published by the US National Institutes of Health (Eighth edition, 2011).
| Abbreviations | ||
| ACE2 | = | angiotensin converting enzyme 2 |
| BALF | = | bronchoalveolar lavage fluid |
| BCL6 | = | B-cell lymphoma 6 |
| BPD | = | bronchopulmonary dysplasia |
| ECM | = | extracellular matrix |
| GSDMD | = | Gasdermin D |
| IL-1α | = | interleukin-1α |
| IFN γ | = | interferon-γ |
| MLI | = | mean linear intercept |
| MMP | = | matrix metalloproteinases |
| NLRP3 | = | nucleotide-binding oligomerization domain-like receptor protein 3 |
| RAC | = | radical alveolar count |
| ROS | = | reactive oxygen species |
| TNF α | = | tumor necrosis factor α |
| VEGFA | = | vascular endothelial growth factor A |
| VEGFR2 | = | vascular endothelial growth factor receptor 2 |
Supplemental Material
Download MS Word (1.5 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All data generated or analyzed during this study are available from the corresponding author upon reasonable request.
Additional information
Funding
References
- Thebaud B, Goss KN, Laughon M, et al. Bronchopulmonary dysplasia. Nat Rev Dis Primers. 2019;5(1):78. doi:10.1038/s41572-019-0127-7.
- Coalson JJ. Pathology of new bronchopulmonary dysplasia. Semin Neonatol. 2003;8(1):73–81. doi:10.1016/s1084-2756(02)00193-8.
- Higano NS, Spielberg DR, Fleck RJ, et al. Neonatal pulmonary magnetic resonance imaging of bronchopulmonary dysplasia predicts short-term clinical outcomes. Am J Respir Crit Care Med. 2018;198(10):1302–1311. doi:10.1164/rccm.201711-2287OC.
- Shahzad T, Radajewski S, Chao CM, Bellusci S, Ehrhardt H. Pathogenesis of bronchopulmonary dysplasia: when inflammation meets organ development. Mol Cell Pediatr. 2016;3(1):23. doi:10.1186/s40348-016-0051-9.
- Broz P, Pelegrin P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol. 2020;20(3):143–157. doi:10.1038/s41577-019-0228-2.
- Omer M, Melo AM, Kelly L, et al. Emerging role of the NLRP3 inflammasome and interleukin-1beta in neonates. Neonatology. 2020;117(5):545–554. doi:10.1159/000507584.
- Coll RC, Schroder K, Pelegrin P. NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol Sci. 2022;43(8):653–668. doi:10.1016/j.tips.2022.04.003.
- Zhang Q, Ran X, He Y, Ai Q, Shi Y. Acetate down regulates the activation of NLRP3 inflammasomes and attenuates lung injury in neonatal mice with bronchopulmonary dysplasia. Front Pediatr. 2020;8:595157. doi:10.3389/fped.2020.595157.
- Liao J, Kapadia VS, Brown LS, et al. The NLRP3 inflammasome is critically involved in the development of bronchopulmonary dysplasia. Nat Commun. 2015;6(1):8977. doi:10.1038/ncomms9977.
- Stouch AN, McCoy AM, Greer RM, et al. IL-1beta and inflammasome activity link inflammation to abnormal fetal airway development. J Immunol. 2016;196(8):3411–3420. doi:10.4049/jimmunol.1500906.
- Dong L, He Y, Cao Y, et al. Functional differentiation and regulation of follicular T helper cells in inflammation and autoimmunity. Immunology. 2021;163(1):19–32. doi:10.1111/imm.13282.
- Kurosu T, Fukuda T, Miki T, Miura O. BCL6 overexpression prevents increase in reactive oxygen species and inhibits apoptosis induced by chemotherapeutic reagents in B-cell lymphoma cells. Oncogene. 2003;22(29):4459–4468. doi:10.1038/sj.onc.1206755.
- Chen D, Xiong XQ, Zang YH, et al. BCL6 attenuates renal inflammation via negative regulation of NLRP3 transcription. Cell Death Dis. 2017;8(10):e3156–e3156. doi:10.1038/cddis.2017.567.
- Zhang S, Lin S, Tang Q, Yan Z. Knockdown of miR2055p alleviates the inflammatory response in allergic rhinitis by targeting B cell lymphoma 6. Mol Med Rep. 2021;24(5):1–11 doi:10.3892/mmr.2021.12458.
- Sun Y, Chen C, Di T, et al. Human beta-defensin-2 improves hyperoxia-induced lung structural and functional injury in neonatal rats. Med Sci Monit. 2019;25:6074–6084. doi:10.12659/MSM.915814.
- Durrani-Kolarik S, Pool CA, Gray A, et al. miR-29b supplementation decreases expression of matrix proteins and improves alveolarization in mice exposed to maternal inflammation and neonatal hyperoxia. Am J Physiol Lung Cell Mol Physiol. 2017;313(2):L339–L349. doi:10.1152/ajplung.00273.2016.
- Sureshbabu A, Syed M, Das P, et al. Inhibition of regulatory-associated protein of mechanistic target of rapamycin prevents hyperoxia-induced lung injury by enhancing autophagy and reducing apoptosis in neonatal mice. Am J Respir Cell Mol Biol. 2016;55(5):722–735. doi:10.1165/rcmb.2015-0349OC.
- Willis GR, Fernandez-Gonzalez A, Reis M, et al. Mesenchymal stromal cell-derived small extracellular vesicles restore lung architecture and improve exercise capacity in a model of neonatal hyperoxia-induced lung injury. J Extracell Vesicles. 2020;9(1):1790874. doi:10.1080/20013078.2020.1790874.
- Cardenas MG, Yu WB, Beguelin W, et al. Rationally designed BCL6 inhibitors target activated B cell diffuse large B cell lymphoma. J Clin Invest. 2016;126(9):3351–3362. doi:10.1172/Jci85795.
- Chen D, Gao ZQ, Wang YY, et al. Sodium propionate enhances Nrf2-mediated protective defense against oxidative stress and inflammation in lipopolysaccharide-induced neonatal Mice. J Inflamm Res. 2021;14:803–816. doi:10.2147/JIR.S303105.
- Hu Y, Sun X, Wang S, et al. Toll-like receptor-2 gene knockout results in neurobehavioral dysfunctions and multiple brain structural and functional abnormalities in mice. Brain Behav Immun. 2021;91:257–266. doi:10.1016/j.bbi.2020.10.004.
- Wang C, Xu K, Wang Y, et al. Spinal cannabinoid receptor 2 activation reduces hypersensitivity associated with bone cancer pain and improves the integrity of the blood-spinal cord barrier. Reg Anesth Pain Med. 2020;45(10):783–791. doi:10.1136/rapm-2019-101262.
- Zhang Y, Coarfa C, Dong X, et al. MicroRNA-30a as a candidate underlying sex-specific differences in neonatal hyperoxic lung injury: implications for BPD. Am J Physiol Lung Cell Mol Physiol. 2019;316(1):L144–L156. doi:10.1152/ajplung.00372.2018.
- Zhang Y, Dong X, Shirazi J, Gleghorn JP, Lingappan K. Pulmonary endothelial cells exhibit sexual dimorphism in their response to hyperoxia. Am J Physiol Heart Circ Physiol. 2018;315(5):H1287–H1292. doi:10.1152/ajpheart.00416.2018.
- Chen D, Wu YX, Qiu YB, et al. Hyperoside suppresses hypoxia-induced A549 survival and proliferation through ferrous accumulation via AMPK/HO-1 axis. Phytomedicine. 2020;67:153138. doi:10.1016/j.phymed.2019.153138.
- Thebaud B, Ladha F, Michelakis ED, et al. Vascular endothelial growth factor gene therapy increases survival, promotes lung angiogenesis, and prevents alveolar damage in hyperoxia-induced lung injury: evidence that angiogenesis participates in alveolarization. Circulation. 2005;112(16):2477–2486. doi:10.1161/CIRCULATIONAHA.105.541524.
- Wu JB, Tang YL, Liang XH. Targeting VEGF pathway to normalize the vasculature: an emerging insight in cancer therapy. Onco Targets Ther. 2018;11:6901–6909. doi:10.2147/OTT.S172042.
- Pitulescu ME, Schmidt I, Giaimo BD, et al. Dll4 and Notch signalling couples sprouting angiogenesis and artery formation. Nat Cell Biol. 2017;19(8):915–927. doi:10.1038/ncb3555.
- Testini C, Smith RO, Jin Y, et al. Myc-dependent endothelial proliferation is controlled by phosphotyrosine 1212 in VEGF receptor-2. EMBO Rep. 2019;20(11):e47845. doi:10.15252/embr.201947845.
- Li X, Redfors B, Sainz-Jaspeado M, et al. Suppressed vascular leakage and myocardial edema improve outcome from myocardial infarction. Front Physiol. 2020;11:763. doi:10.3389/fphys.2020.00763.
- Giusto K, Wanczyk H, Jensen T, Finck C. Hyperoxia-induced bronchopulmonary dysplasia: better models for better therapies. Dis Model Mech. 2021;14(2):dmm047753. doi:10.1242/dmm.047753.
- Nardiello C, Mizikova I, Silva DM, et al. Standardisation of oxygen exposure in the development of mouse models for bronchopulmonary dysplasia. Dis Model Mech. 2017;10(2):185–196. doi:10.1242/dmm.027086.
- Royce SG, Nold MF, Bui C, et al. Airway remodeling and hyperreactivity in a model of bronchopulmonary dysplasia and their modulation by IL-1 receptor antagonist. Am J Respir Cell Mol Biol. 2016;55(6):858–868. doi:10.1165/rcmb.2016-0031OC.
- Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481(7381):278–286. doi:10.1038/nature10759.
- Chen X, He WT, Hu L, et al. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 2016;26(9):1007–1020. doi:10.1038/cr.2016.100.
- Shi J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–665. doi:10.1038/nature15514.
- Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42(4):245–254. doi:10.1016/j.tibs.2016.10.004.
- Dapaah-Siakwan F, Zambrano R, Luo S, et al. Caspase-1 inhibition attenuates hyperoxia-induced lung and brain injury in neonatal mice. Am J Respir Cell Mol Biol. 2019;61(3):341–354. doi:10.1165/rcmb.2018-0192OC.
- Rout-Pitt N, Farrow N, Parsons D, Donnelley M. Epithelial mesenchymal transition (EMT): a universal process in lung diseases with implications for cystic fibrosis pathophysiology. Respir Res. 2018;19(1):136. doi:10.1186/s12931-018-0834-8.
- Scanlon CS, Van Tubergen EA, Inglehart RC, D’Silva NJ. Biomarkers of epithelial-mesenchymal transition in squamous cell carcinoma. J Dent Res. 2013;92(2):114–121. doi:10.1177/0022034512467352.
- Pardo A, Selman M. Matrix metalloproteases in aberrant fibrotic tissue remodeling. Proc Am Thorac Soc. 2006;3(4):383–388. doi:10.1513/pats.200601-012TK.
- Rockey DC, Bell PD, Hill JA. Fibrosis–a common pathway to organ injury and failure. N Engl J Med. 2015;372(12):1138–1149. doi:10.1056/NEJMra1300575.
- Wuyts WA, Agostini C, Antoniou KM, et al. The pathogenesis of pulmonary fibrosis: a moving target. Eur Respir J. 2013;41(5):1207–1218. doi:10.1183/09031936.00073012.
- Hewlett JC, Kropski JA, Blackwell TS. Idiopathic pulmonary fibrosis: epithelial-mesenchymal interactions and emerging therapeutic targets. Matrix Biol. 2018;71-72:112–127. doi:10.1016/j.matbio.2018.03.021.
- Brody AR. Occupational lung disease and the role of peptide growth factors. Curr Opin Pulm Med. 1997;3(3):203–208. doi:10.1097/00063198-199705000-00004.
- Toti P, Buonocore G, Tanganelli P, et al. Bronchopulmonary dysplasia of the premature baby: an immunohistochemical study. Pediatr Pulmonol. 1997;24(1):22–28. doi:10.1002/(sici)1099-0496(199707)24:1<22::aid-ppul4>3.0.co;2-l.
- Sinkin RA, Roberts M, LoMonaco MB, Sanders RJ, Metlay LA. Fibronectin expression in bronchopulmonary dysplasia. Pediatr Dev Pathol. 1998;1(6):494–502. doi:10.1007/s100249900068.
- McDonald DM. Angiogenesis and remodeling of airway vasculature in chronic inflammation. Am J Respir Crit Care Med. 2001;164(10 Pt 2):S39–S45. doi:10.1164/ajrccm.164.supplement_2.2106065.
- Crapo JD, Barry BE, Foscue HA, Shelburne J. Structural and biochemical changes in rat lungs occurring during exposures to lethal and adaptive doses of oxygen. Am Rev Respir Dis. 1980;122(1):123–143. doi:10.1164/arrd.1980.122.1.123.
- Perkett EA, Klekamp JG. Vascular endothelial growth factor expression is decreased in rat lung following exposure to 24 or 48 hours of hyperoxia: implications for endothelial cell survival. Chest. 1998;114(1 Suppl):52S–53S. doi:10.1378/chest.114.1_supplement.52s.
- You K, Xu X, Fu J, et al. Hyperoxia disrupts pulmonary epithelial barrier in newborn rats via the deterioration of occludin and ZO-1. Respir Res. 2012;13(1):36. doi:10.1186/1465-9921-13-36.
- Campochiaro PA. Molecular pathogenesis of retinal and choroidal vascular diseases. Prog Retin Eye Res. 2015;49:67–81. doi:10.1016/j.preteyeres.2015.06.002.
- Mintz-Hittner HA, Kennedy KA, Chuang AZ. Efficacy of intravitreal bevacizumab for stage 3+ retinopathy of prematurity. N Engl J Med. 2011;364(7):603–615. doi:10.1056/NEJMoa1007374.
- Yu X, Lin Q, Qin X, et al. ACE2 antagonizes VEGFa to reduce vascular permeability during acute lung injury. Cell Physiol Biochem. 2016;38(3):1055–1062. doi:10.1159/000443056.