
Abstract
Objective
The aim of this study is to assess the impact of Liver X receptors (LXRs) on airway inflammation, airway remodeling, and lipid deposition induced by cigarette smoke and lipopolysaccharide (LPS) exposure in the lung.
Methods
Wild mice and LXR-deficient mice were exposed to cigarette smoke and LPS to induce airway inflammation and remodeling. In addition, some wild mice received intraperitoneal treatment with the LXR agonist GW3965 before exposure to cigarette smoke and LPS. Lung tissue and bronchoalveolar lavage fluid were collected to evaluate airway inflammation, airway remodeling and lipid deposition.
Results
Exposure to cigarette smoke and LPS resulted in airway inflammation, emphysema and lipid accumulation in wild mice. These mice also exhibited downregulated LXRα and ABCA1 in the lung. Treatment with GW3965 mitigated inflammation, remodeling and lipid deposition, while the deletion of LXRs exacerbated these effects. Furthermore, GW3965 treatment following exposure to cigarette smoke and LPS increased LXRα and ABCA1 expression and attenuated MyD88 expression in wild mice.
Conclusion
LXRs demonstrate the potential to mitigate cigarette smoke and LPS- induced airway inflammation, emphysema and lipid disposition in mice.
Keywords:
1. Introduction
Chronic obstructive pulmonary disease (COPD) stand as the third leading cause of death worldwide.Citation1 Patients grappling with COPD experience persistent respiratory symptoms and a gradual decline in airflow.Citation2 Unquestionably, cigarette smoking is the most prominent risk factor associated with COPD.Citation3 Indeed, statistics reveal that the incidence rate of chronic obstructive pulmonary disease among smokers is as high as 35.5%, significantly surpassing that of nonsmokers (7.8%).Citation4 bacterial and viral infections are also pivotal risk factors exacerbating COPD, often necessitating additional treatment and hospitalization.Citation5 Lipopolysaccharide (LPS) comprises the outer leaflet of the outer membrane of most Gram-negative bacteria and is responsible for the significant bioactivity of bacterial endotoxin. Inhalation of chronic LPS has shown to induce emphysema-like changes in mouse lung.Citation6 It is widely acknowledged that the combination of cigarette smoke (CS) and LPS exposure effectively reproduces the inflammation, airway remodeling, and exacerbation observed in COPD in mouse models.Citation7,Citation8 Current treatment modalities, such as bronchodilators and glucocorticoids, aim to alleviate symptoms and sometimes slow down COPD progression. However, these drugs are typically administered after diagnosis, highlighting the urgent need to comprehend the pathogenesis of COPD and identify novel, portent interventions for early management.
The Liver X receptor (LXR), a nuclear receptor encompassing two subtypes, LXRα (NR1H3) and LXRβ (NR1H2), plays a pivotal role in regulating cholesterol homeostasis. Upon binding to natural ligands like 22 [R] - hydroxycholesterol and 27 hydroxycholesterol,Citation9 or synthetic agonists such as GW3965 and T0901317, activated LXRs initiate the transcriptional expression of genes associated with cholesterol efflux and inflammation, including ATP- binding cassette transporter A1 (ABCA1), ATP-binding cassette transporter G1(ABCG1), among others.Citation10 A growing body of evidence underscores the significant contributions of LXRs in cholesterol metabolism, anti-inflammatory and immune regulation.Citation11–13
In the context of developing airway inflammation and emphysema through cigarette smoke and LPS exposure, the pharmacological properties of LXRs offer promising avenues for COPD treatment. This study aims to assess the impact of LXRs on mice exposed to cigarette smoke and LPS, while also investigating the underlying mechanisms.
2. Materials and methods
2.1. Animals
2. 2.1. Animal preparation
Ten-week-old pathogen-free C57BL/6 wild-type (WT) mice were procured from Nanjing Laboratory Animal Inc. LXR-deficient mice were constructed by Nanjing University-Nanjing Biomedical Research Institute. LXRα−/−/β−/− mice were bred from LXRα−/− mice and LXRβ−/− mice. The mice were raised under controlled conditions, including a temperature range of 21 °C-25 °C, humidity between 40% and 60%, and a strict 12-h light-dark cycle. The mice had ad libitum access to food and water.
2.2. Animal ethics statements
The animal experiment received approved from the Animal Experiment Center of Nanjing First Hospital Affiliated with Nanjing Medical University, following the guidelines of the Animal Care and Use Committee of the institution (license No: SYXK [Su] 2016-0006). Animal experiments were conducted in strict accordance with the Guidance on Feeding and Use of Experimental Animals by the Ministry of Science and Technology of the People’s Republic of China. All procedures were performed under anesthesia, and every effort was made to minimize suffering.
2.3. Experiment protocol
The mice were randomly assigned to four groups:
Control Group: WT mice were exposed to air.
CS + LPS Group: WT mice were exposed to cigarette smoke and received LPS intranasal instillation.
GW3965 Group: WT mice were exposed to cigarette smoke and received LPS intranasal instillation. GW3965 (an LXR agonist, 40mg/kg) was intraperitoneally injected one hour before exposure to cigarette smoke.
LXRα−/−/β−/− Group: LXRα−/−/β−/− mice were exposed to cigarette smoke and received LPS intranasal instillation.
Each group comprised five mice. LPS was intranasally instilled on day 1 and day 21 at a dose of 750 ng/kg dissolved in 50μl saline. Intranasal instillation of LPS has been well-established in prior studies.Citation14–16 Cigarette smoke was generated using 3R4F research cigarettes (Kentucky reference cigarette, University of Kentucky, USA), each containing 11.0 mg of total particulate matter, 9.4 mg of tar, and 0.76 mg of nicotine per cigarette. Except for Day 1 and Day 21, the model groups were exposed to cigarette smoke twice daily for two hours each session, with a 10-minute air exchange in between, once in the morning and once in the afternoon, involving 10 cigarettes per hour, six days a week, for a total duration of 6 weeks. During the exposure period, the oxygen concentration was controlled at (20.5 ± 0.5) %, and the air humidity was controlled at (80 ± 5) %. The control group was placed in the same system under the same conditions for normal air exposure. During the experiment, mice were weighted at intervals every week. No deaths occurred in any group.
2.4. Collection of blood and broncho alveolar lavage fluid (BALF)
For anesthesia, 0.2 ml of 1% Pentobarbital Sodium was intraperitoneally injected into the mice. Blood samples were collected from the orbit and centrifuged at 4000 rpm for 15 min. The supernatant was carefully transferred to a clean 1.5 ml centrifuge tube and stored at − 80° C for subsequent ELISA testing. The mice were euthanized by cervical dislocation, followed by tracheotomy. The bronchoalveolar lavage fluid (BALF) was obtained by injecting cold PBS (0.5 ml) through a tube and repeating this procedure three times (total: 1.5 ml). BALF was then centrifuged at 4 °C and 1100 rpm for 10 min. The cell precipitate was resuspended in 100ul of PBS solution, and 10 μl was used to determine the total cell count. Wright-Giemsa staining (GP1074, Servicebio) was employed to identify different leukocyte cell types. For each mouse, at least 200 cells were counted categorized as macrophages, lymphocytes, or neutrophils based on standard morphology at x400 magnification. The supernatant obtained from BALF was stored at − 80° C for subsequent biochemical analysis.
2.5. Measurement of inflammatory mediators
The levels of IL-6 (KGEMC004-1, KeyGEN BioTECH), IL-8 (KGEMC104-1, KeyGEN BioTECH), and TNFα (KGEMC102a-1, KeyGEN BioTECH) in both BALF and serum were quantified using ELISA, following the manufacturer’s protocols.
2.6. Lung tissue histopathology and masson trichrome staining
Following the collection of BALF samples, the left lung tissue was fixed 4% paraformaldehyde. Subsequently, the tissue was embedded in paraffin, sectioned 5 μ m thick, and stained with hematoxylin and eosin (H&E) solution to assess the inflammatory response. Quantitative analysis was performed as described previously.Citation14,Citation17 The severity of peribronchial inflammation was quantified from 0 to 5 in the lung images stained by HE, where 0 = no cells, 1 = a few cells, 2 = a ring of cells one layer deep, 3 = a ring of cells two layers deep, 4 = a ring of cells three to four layers deep, and 5 = a ring > 4 cell layers deep. The H&E sections were observed under an optical microscope at 200× magnification, with 10 random peripheral visual fields taken from each section. To calculate the average area of per airway, images of the sections were processed in Photoshop. A cross-line was drawn through the center of each positive image, avoiding areas around airways and vessels as much as possible. The number of alveolar spaces (n) intersected by these two lines was counted, and the total length of the cross-line (L) was measured, with Lm calculated as L/N. Additionally, Masson trichrome staining (GP1032, Servicebio) was conducted on the sections to assess collagen deposition. All histological examinations were performed by double-blind assessment at a magnification of 400x. The results were quantified by calculating the average of each airway.
2.7. Western blot
Lung tissues were lysed using RIPA lysis buffer containing protease and phosphatase inhibitor on ice. The resulting tissue homogenate was centrifuged at 12000 g for 15 min, and the supernatant was collected and stored at −80 °C. Total protein content in the lung tissue was determined using BCA protein analysis kit (KGP902, KeyGEN BioTECH). The supernatant was mixed with SDS-PAGE loading buffer at 4:1 ratio and boiled in water for 15 min. Protein separation was achieved through SDS-PAGE electrophoresis, followed by transfer to a PVDF membrane. The membrane, containing the target protein, was blocked with 5% skim milk at room temperature for 2 h. Subsequently, the primary antibodies (LXRα (ab176323, Abcam), ABCG1 (ab52617, Abcam), LXRβ (AP-31123, proteintech), ABCA1 (96292S, CST)), MyD88(CST,4283S) was diluted in TBST buffer and incubated overnight at 4 °C. After washing three times with TBST buffer, the membrane was incubated with the second antibody for 2 h. Proteins visualization was achieved using the ECL Key-GEN system.
2.8. Oil red O assay
Following centrifugation, cells in the BALF were resuspended. A 50 μl cell suspension was seeded on a slide, and an appropriate amount of DMEM was added. The slide was incubated for 4 h to allow macrophages to adhere. After removing other cells by washing with sterile PBS for three times, the cells were fixed with 4% paraformaldehyde for 10 min. Subsequently, the cells were washed three times with PBS and briefly exposed to 60% isopropanol for 15 s. A 200 μl oil red O staining solution was applied to the slide, followed by a 30-minutes incubation. The slide was washed with 60% isopropanol for 10 s (strictly control the time) and immediately washed with PBS for three times. Finally, the slide was sealed with a water-based sealing agent and observed under a microscope. Results were analyzed by calculating the percentage of the stained cells relative to the total number of counted cells counted. The Oil red O staining of frozen sections of lung tissue followed the same protocol.
2.9. Immunostaining for α-SMA
Paraffin slices are dewaxed in water. Subsequently, they were incubated in 3% H2O2 at room temperature for 10 min to neutralize endogenous peroxidase activity. After rinsing with distilled water and soaking in PBS for five minutes twice, the slices were blocked with 5% normal goat serum (diluted with PBS) and incubated at room temperature for 10 min, The serum then poured off without washing. Next, anti-α-SMA (Servicebio GB13044) solutions were added, and the slices were incubated for two hours at 37 °C or overnight at 4 °C. Following incubation, the slices were rinsed with PBS for five minutes, three times. Subsequently, an appropriate HRP-labeled secondary antibody working solution (Servicebio GB23303) was applied, and the slices were incubated at 37 °C for 30 min bdfore being rinsed with PBS for five minutes, three times. An adequate amount of horseradish enzyme-labeled streptomycin working solution was then added, and the slices were incubated at 37 °C for 30 min. The slices were again rinsed with PBS for five minutes, three times. Chromogenic agent (DAB) was applied for eight minutes. Finally, the slices underwent full flushing, re-dyeing, dehydration, transparency, and sealing with tap water. The results were expressed as the average optical density in airway.
2.10. Statistical analysis
Data is presented as mean ± standard error of the mean. Statistical differences between groups were analyzed using one-way ANOVA (with nonparametric or mixed test), followed by multiple comparison tests for all data (GraphPad Prism 8.0). A P-value of <0.05 was considered statistically significant.
3. Result
3.1. Effect of LXRs on inflammatory cells
To assess the impact of LXRs on inflammatory cells in BALF, we performed cytological classification and cell counting. As shown in , compared to the control group, exposure to cigarette smoke and LPS resulted in a significant increase in the number of inflammatory cells in BALF, particularly neutrophils and macrophages (p < 0.05). Treatment with the LXR agonist GW3965 before exposure to cigarette smoke and LPS remarkably reduced the number of neutrophils and macrophages (p < 0.05). In contrast, LXR-deficient mice exhibited a higher count of macrophages, neutrophils, and lymphocytes compared to mice in the cigarette smoke and LPS group (p < 0.05).
Figure 1. Effect of LXRs on inflammatory cells. Total cell counts were determined at 400× magnification using a microscope. Data are presented as means for n = 5 in each group. * p < 0.05 CS + LPS Group vs. Control Group; # p < 0.05 GW3965 Group vs. CS + LPS Group; +p < 0.05 LXRα−/−/β−/− Group vs. CS + LPS Group. Three independent experiments were conducted (5 mice in each group for each experiment). LXR, liver X receptor; BALF, bronchoalveolar lavage fluid.
3.2. Effect of LXRs on inflammatory cytokines
In order to investigate the effect of LXRs on systemic inflammation and local airway inflammation induced by LPS and cigarette smoke in mice, we detected some inflammatory cytokines in the BALF and serum using ELISA method. As depicted in , levels of TNF-α, IL-6, and IL-8 in BALF and serum were elevated in the cigarette smoke and LPS group compared to the control group (p < 0.05). Treatment with the LXR agonist GW3965 led to a significant reduction in the levels of these cytokines (p < 0.05). Conversely, levels of IL-6, TNF-α, and IL-8 were higher in the LXR-deficient group compared to the cigarette smoke and LPS group (p < 0.05).
Figure 2. Effect of LXRs on inflammatory cytokines. Levels of IL-6, IL-8, and TNFα in BALF (a, b and c) and serum (d, e and f) were determined by ELISA. Data are expressed as means for n = 5 in each group. * p < 0.05 CS + LPS Group vs. Control Group; # p < 0.05 GW3965 Group vs. CS + LPS Group; +p < 0.05 LXRα−/−/β−/− Group vs. CS + LPS Group. Three independent experiments were conducted (5 mice in each group for each experiment). LXR, liver X receptor; BALF, bronchoalveolar lavage fluid.
3.3. Effect of LXRs on lung inflammation
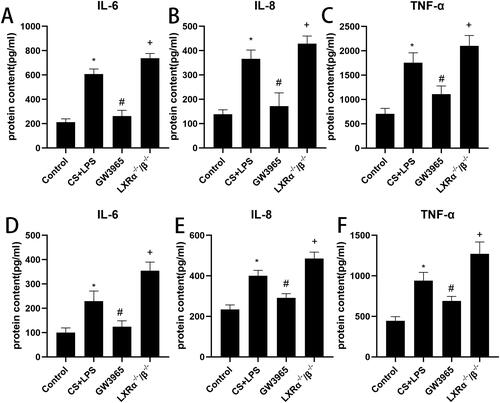
Histological analysis of lung sections () revealed that compared to control group, more inflammatory cells occurred in the airway and interstitial areas in the cigarette smoke and LPS exposure group. GW3965 treatment reduced the presence of inflammatory cells in lung tissue. LXR-deficient mice exhibited more inflammatory cells surrounding small airways and interstitial regions compared to mice in the cigarette smoke and LPS group. illustrates the effect of LXRs on lung inflammation using an inflammation score. Compared to the control group, the smoking group exhibited higher inflammation scores, which were further elevated in the LXRα−/−/β−/− group. However, inflammation scores decreased following GW3965 treatment.
3.4. Effect of LXRs on lung lipid homeostasis
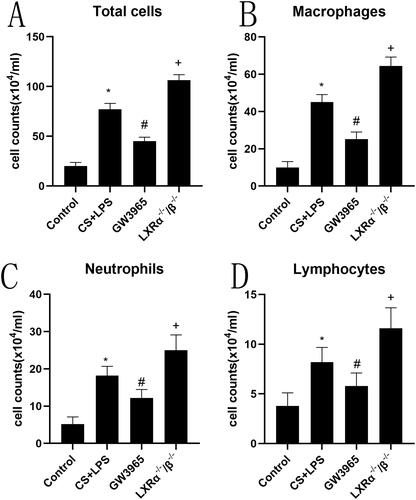
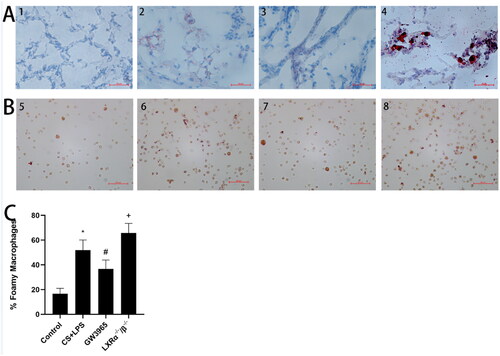
The purpose is to explore the effect of LXRs on lung lipid homeostasis, we employed Oil Red O staining to evaluate lipid deposition in the lung tissues () and the formation of foamy macrophages () in BALF. Significant lipid deposition was observed in lung tissue (small airways and interstitial areas) and an increase in foamy macrophages in both WT mice and LXR-deficient mice after exposure to cigarette smoke and LPS, particularly in the LXR-deficient mice. GW3965 treatment significantly reduced lipid deposition in lung tissue and foamy macrophages in BALF after cigarette smoke and LPS exposure (p < 0.05)().
Figure 3. Effect of LXRs on lung lipid homeostasis. (a) The representative oil red O staining of lung tissues (original magnification, ×200); (b): the representative oil red O staining of macrophages in BALF (original magnification, ×200); (c): Differential BALF macrophage cell count describing regular macrophages and foamy macrophages. 1 and 5:Control Group; 2 and 6: CS + LPS Group;3 and 7:GW3965 Group; 4 and 8:LXRα−/−/β−/− Group. Data are presented as means for n = 5 in each group. * p < 0.05 CS + LPS Group vs. Control Group; # p < 0.05 GW3965 Group vs. CS + LPS Group; +p < 0.05 LXRα−/−/β−/− Group vs. CS + LPS Group. Three independent experiments were conducted (5 mice in each group for each experiment). LXR, liver X receptor; BALF, bronchoalveolar lavage fluid.
3.5. Effect of LXRs on airway remodeling
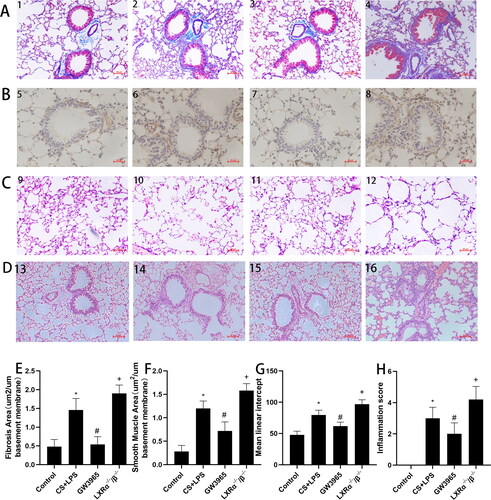
In order to evaluate the role of LXRs in airway remodeling induced by smoking and LPS, we detected related airway remodeling indicators for airway smooth muscle proliferation and collagen extracellular matrix deposition. As shown in , minimal collagen deposition was observed around the airways in the control group. Our results indicate a significant increase in collagen deposition around the airways of mice in the smoking group compared to the control group (p < 0.05). Moreover, when comparing the experimental group, the GW3965 group displayed a reduced collagen deposition area (p < 0.05), while the LXR-deficient group exhibited an increased collagen deposition area (p < 0.05). illustrates that an elevated expression of α-SMA in the peribronchiolar region of cigarette smoke and LPS exposed mice when compared to the control group. Furthermore, the administration of GW3965 notably mitigated the α-SMA-stained smooth muscle layer in contrast to the smoking group. Conversely, in comparison to the smoking group, the LXR-deficient mice exhibited an augmentation in the area of α-SMA stained smooth muscle layer. The extent of lung tissue damage was evaluated by analyzing the mean linear intercept (Lm) of the alveoli through HE staining of lung tissue (). The Lm in the control group was smaller, whereas it exhibited a significant enlargement in the smoking group, which further increased in the LXRα−/−/β−/− group. Furthermore, when compared to the smoking group, the Lm in the GW3965 treatment group showed a significant reduction.
Figure 4. Effect of LXRs on airway remodeling. (a) The representative masson staining of lung tissues (original magnification, ×200). (b) The representative immunohistochemical staining (original magnification, ×200). (c) The representative H&E solution of lung tissues (original magnification, ×200). (d) Representative figures in lung tissues stained with the H&E solution (original magnification, ×200). (e) Image analysis assessing extend of masson’s trichrome staining expressed as means. (f) The area of α-SMA per micrometer length of basement membrane of bronchiole was calculated. (g) Quantitative analysis of the lung destruction, as represented by mean linear intercept. (h) Lung inflammatory scores. Data are expressed as means for n = 5in each group. 1,5,9,13:Control Group; 2,6,10,14: CS + LPS Group; 3,7,11,15:GW3965 Group; 4,8,12,16:LXRα−/−/β−/− Group. Data are presented as means for n = 5 in each group. * p < 0.05 CS + LPS Group vs. Control Group; # p < 0.05 GW3965 Group vs. CS + LPS Group; +p < 0.05 LXRα−/−/β−/− Group vs. CS + LPS Group. Three independent experiments were conducted (5 mice in each group for each experiment). LXR, liver X receptor.
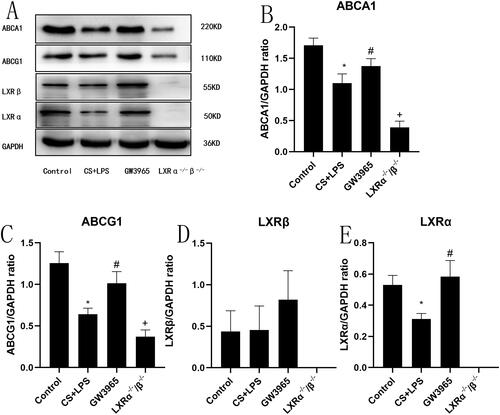
3.6. Effect of LXRs on cholesterol efflux-related protein expression
As described in , mice in the LXR-deficient group exhibited the lowest ABCA1 expression in the lung. In WT mice, cigarette smoke and LPS exposure resulted in a significant reduction in LXRα and ABCA1 in the lung, while GW3965 treatment increased the expression of LXRα and ABCA1(p < 0.05). There were no statistically significant differences in LXRβ between the groups(p > 0.05).
Figure 5. Effect of LXRs on cholesterol efflux-related protein expression. (a) Representative figure of protein expression; (b–e) quantitative analysis of protein expression. Data are presented as means for n = 5 in each group. * p < 0.05 CS + LPS Group vs. Control Group; # p < 0.05 GW3965 Group vs. CS + LPS Group; +p < 0.05 LXRα−/−/β−/− Group vs. CS + LPS Group. Three independent experiments were conducted (5 mice in each group for each experiment). LXR, liver X receptor.
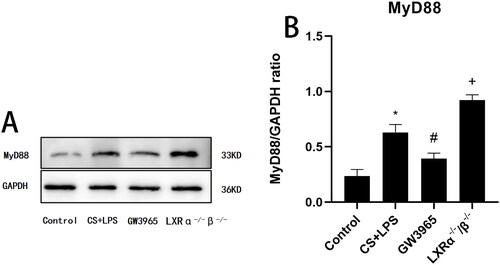
3.7. Effect of LXRs on MyD88 expression
As shown in , after exposure to cigarette smoke and LPS, WT mice exhibited increased expression of MyD88 in the lung compared to the control group. MyD88 expression was significantly suppressed by GW3965 treatment (p < 0.05). Conversely, LXR-deficient mice displayed higher MyD88 expression than WT mice after exposure to cigarette smoke and LPS (p < 0.05).
Figure 6. Effect of LXRs on MyD88 expression. (a) Representative figure of protein expression; (b) quantitative analysis of protein expression. Data are presented as means for n = 5 in each group. * p < 0.05 CS + LPS Group vs. Control Group; # p < 0.05 GW3965 Group vs. CS + LPS Group; +p < 0.05 LXRα−/−/β-/- Group vs. CS + LPS Group. Three independent experiments were conducted (5 mice in each group for each experiment). LXR, liver X receptor.
4. Discussion
In this study, we explored the protective role of LXRs in mitigating lung inflammation and airway remodeling induced by cigarette smoke and LPS in mice. A previous study by Jarrod Sonett reported the inhibitory effect of the LXR agonist T0901317 on cigarette smoke-induced emphysema in mice.Citation23 However, T0901317 lacks high selectivity for LXRs and can activate other nuclear receptors, such as the Farnesoid X receptor (FXR) and Pregnane X receptor (PXR).Citation24,Citation25,Citation26 The highly selective LXR agonist GW3965 can induce LXR transcriptional activity but fails to modulate LXR and PXR.Citation26 Moreover, the use of LXR‐deficient mice to investigate role of LXRs in airway inflammation or emphysema has not been previously reported. In this study, we employed LXR‐deficient mice and GW3965 to provide evidence supporting the protective function of LXRs against cigarette smoke and LPS-induced emphysema.
Alveolar macrophages constitute the predominant cells in the healthy airspaces and serve as the first line defense against infections,Citation27 along with epithelial cells.Citation28 In our emphysema model, we observed lipid deposition in alveolar macrophages and lung tissue. This finding may be correlated with the decreased expression of LXRα and cholesterol efflux transporters. While LXR-ABC transporter signaling plays a pivotal role in regulating inflammation, cholesterol efflux and pulmonary surfactant metabolism,Citation29 its precise role in the alveolar microenvironment during COPD development of remains unclear. Based on existing literature,Citation23 nicotine (a component in cigarette smoke) can reduce the expression of LXRs and ABC transporters in macrophages in vitro which is probably the reason why foamy alveolar macrophages increased in the lungs of smokers and our emphysema model. Fomay macrophages exhibit enhanced inflammatory responses and reduced phagocytic capacity. We hypothesize that GW3965 reduce the lipid burden in macrophages by upregulating the expression of ABCA1 and ABCG1, thereby ameliorating macrophage-mediated inflammation and restore the macrophage phagocytic function, thus reducing the following neutrophilic inflammation in lung.
The MyD88-dependent pathway is utilized by all Toll-like receptors (TLRs) except TLR3, ultimately leading to enhanced expression of proinflammatory cytokine genes. Cigarette smoke active the TLR4-MYD88-NF-κB signaling pathway in vitro,Citation30 which may correlated with the development of COPD. MyD88 also plays an important role in LPS induced lung injury which may be associated with the acute exacerbation of COPD.Citation31,Citation32 We defined in mice that exposure to cigarette smoke and LPS can induce MyD88 protein expression, and this increase was more pronounced in LXR-deficient mice. We propose that that LXR agonists inhibit cigarette smoke and LPS-induced pulmonary inflammation by suppressing MyD88 signaling. But there are some limitations in this study because we didn’t detect the downstream signaling pathway.
Furthermore, we observed that LXRs alleviated peribronchial fibrosis, smooth muscle proliferation, and alveoli enlargement induced by cigarette smoke and LPS exposure. The activation of airway inflammation has been considered a significant contributor to structural damage and airway remodeling caused by smoking.Citation36 Therefore, LXRs may play a role in ameliorating airway remodeling, by restraining airway inflammation. Some studies have shown that LXRs can markedly inhibit the transcription reaction regulated by TGF-β.Citation37,Citation38 Upon binding to its receptor, TGF-β can exacerbate airway remodeling through various mechanisms, such as interfering with normal repair of airway epithelial cells and increasing protease inhibitor expression. Additionally, LXRs can significantly reduce the migration and proliferation of human airway smooth muscle cells(hASM) induced by platelet-derived growth factor-1 (PDGF-1).Citation39 The inhibitory effects of LXRs on TGF-β and hASM proliferation suggest that LXRs may attenuate airway remodeling. But we didn’t test the TGF-β level in BALF and the activation of TGF-β signaling in this study. The mechanism of LXRs affecting airway remodeling requires further study.
It is worth noting that LXRs exhibit different roles in airway inflammation between smoking-induced and antigen-induced allergic airway inflammation. Our previous research has shown that LXRs increased airway inflammation and airway hyperresponsiveness, as well as promote airway remodeling in mice with chronic allergic asthma. it is well known that the STAT signaling pathway is significantly activated in asthma.Citation42,Citation43 LXR has been shown to cooperate with STAT to enhance inflammatory response.Citation44 In addition, nicotine is believed to inhibit the LXR pathway in macrophage.Citation41 We think those are why we have drawn the opposite conclusion in the two animal models.
Although there are many reports that LXR agonists inhibit LPS induced cytokines in macrophages or other cells, there is a published data that GW3965 failed to inhibit LPS induced TNF-α and IL-8 in alveolar macrophages from COPD patients in vitro.Citation45 So we think translation this into human maybe be limited and needs further investigation. In conclusion, our study highlights the protective effects of LXRs against airway inflammation and airway remodeling induced by cigarette smoke and LPS in mice.
Author contributions
Designed this study and revised the article: GW, SY and WBN. Performed the experiment: YFF, ZJR. Analyzed the data: GL, YXB, TY and MZF. Draft this article: YFF.
Disclosure statement
There are no competing financial interests.
Additional information
Funding
References
- Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report: GOLD executive summary. Archivos de Bronconeumologia. 2017;53(3):128–149. doi:10.1016/j.arbr.2017.02.001.
- Pollok J, et al. Psychological therapies for the treatment of depression in chronic obstructive pulmonary disease. 2019.
- Buhl R, Bals R, Baur X, et al. Guideline for the diagnosis and treatment of asthma - guideline of the German Respiratory Society and the German Atemwegsliga in Cooperation with the Paediatric Respiratory Society and the Austrian Society of Pneumology. Pneumologie (Stuttgart, Germany). 2017;71(12):e3.
- Li Y, Li SY, Li JS, et al. A rat model for stable chronic obstructive pulmonary disease induced by cigarette smoke inhalation and repetitive bacterial infection. Biol Pharm Bull. 2012;35(10):1752–1760. doi:10.1248/bpb.b12-00407.
- Sethi S. Bacteria in exacerbations of chronic obstructive pulmonary disease: phenomenon or epiphenomenon? Proc Am Thorac Soc. 2004;1(2):109–114. doi:10.1513/pats.2306029.
- Brass DM, Hollingsworth JW, Cinque M, et al. Chronic LPS inhalation causes emphysema-like changes in mouse lung that are associated with apoptosis. Am J Respir Cell Mol Biol. 2008;39(5):584–590. doi:10.1165/rcmb.2007-0448OC.
- Shin NR, Ko JW, Park SH, et al. Protective effect of HwangRyunHaeDok-Tang water extract against chronic obstructive pulmonary disease induced by cigarette smoke and lipopolysaccharide in a mouse model. J Ethnopharmacol. 2017;200:60–65. doi:10.1016/j.jep.2017.02.027.
- Shin IS, Shin NR, Park JW, et al. Melatonin attenuates neutrophil inflammation and mucus secretion in cigarette smoke-induced chronic obstructive pulmonary diseases via the suppression of Erk-Sp1 signaling. J Pineal Res. 2015;58(1):50–60. doi:10.1111/jpi.12192.
- Janowski BA, Grogan MJ, Jones SA, et al. Structural requirements of ligands for the oxysterol liver X receptors LXRalpha and LXRbeta. Proc Natl Acad Sci U S A. 1999;96(1):266–271. doi:10.1073/pnas.96.1.266.
- Reyes-Quiroz ME, Alba G, Saenz J, et al. Oleic acid modulates mRNA expression of liver X receptor (LXR) and its target genes ABCA1 and SREBP1c in human neutrophils. Eur J Nutr. 2014;53(8):1707–1717. doi:10.1007/s00394-014-0677-0.
- Endo-Umeda K, Makishima M. Liver X receptors regulate cholesterol metabolism and immunity in hepatic nonparenchymal cells. Int J Mol Sci. 2019;20(20):5045. doi:10.3390/ijms20205045.
- Ma Z, Deng C, Hu W, et al. Liver X receptors and their agonists: targeting for cholesterol homeostasis and cardiovascular diseases. Curr Issues Mol Biol. 2017;22:41–64. doi:10.21775/cimb.022.041.
- Hu X, Shen H, Wang Y, et al. Liver X receptor agonist TO901317 attenuates paraquat-induced acute lung injury through inhibition of NF-κB and JNK/p38 MAPK signal pathways. Biomed Res Int. 2017;2017:4652695–4652613. doi:10.1155/2017/4652695.
- Cheng Q, Fang L, Feng D, et al. Memantine ameliorates pulmonary inflammation in a mice model of COPD induced by cigarette smoke combined with LPS. Biomed Pharmacother. 2019;109:2005–2013. doi:10.1016/j.biopha.2018.11.002.
- Liu R, Wang P, Wu C, et al. Therapeutic effects of Hedyotis diffusa Willd in a COPD mouse model challenged with LPS and smoke. Exp Therap Med. 2018;15(4):3385–3391.
- Shin NR, Kim SH, Ko JW, et al. HemoHIM, a herbal preparation, alleviates airway inflammation caused by cigarette smoke and lipopolysaccharide. Lab Anim Res. 2017;33(1):40–47. doi:10.5625/lar.2017.33.1.40.
- Voynow JA, Fischer BM, Malarkey DE, et al. Neutrophil elastase induces mucus cell metaplasia in mouse lung. Am J Physiol Lung Cell Mol Physiol. 2004;287(6):L1293–1302. doi:10.1152/ajplung.00140.2004.
- Jeffery PK. Structural and inflammatory changes in COPD: a comparison with asthma. Thorax. 1998;53(2):129–136. doi:10.1136/thx.53.2.129.
- Fournier M, Lebargy F, Le Roy Ladurie F, et al. Intraepithelial T-lymphocyte subsets in the airways of normal subjects and of patients with chronic bronchitis. Am Rev Respir Dis. 1989;140(3):737–742. doi:10.1164/ajrccm/140.3.737.
- Saetta M, Turato G, Facchini FM, et al. Inflammatory cells in the bronchial glands of smokers with chronic bronchitis. Am J Respir Crit Care Med. 1997;156(5):1633–1639. doi:10.1164/ajrccm.156.5.9701081.
- Wang L, Pelgrim CE, Swart DH, et al. SUL-151 Decreases airway neutrophilia as a prophylactic and therapeutic treatment in mice after cigarette smoke exposure. Int J Mol Sci. 2021;22(9)doi:10.3390/ijms22094991.
- Yu X, Seow HJ, Wang H, et al. Matrine reduces cigarette smoke-induced airway neutrophilic inflammation by enhancing neutrophil apoptosis. Clin Sci (Lond). 2019;133(4):551–564. doi:10.1042/CS20180912.
- Sonett J, Goldklang M, Sklepkiewicz P, et al. A critical role for ABC transporters in persistent lung inflammation in the development of emphysema after smoke exposure. Faseb J. 2018;32(12):fj201701381–6736. doi:10.1096/fj.201701381.
- Zhu C, Di D, Zhang X, et al. TO901317 regulating apolipoprotein M expression mediates via the farnesoid X receptor pathway in Caco-2 cells. Lipids Health Dis. 2011;10(1):199. doi:10.1186/1476-511X-10-199.
- Houck KA, Borchert KM, Hepler CD, et al. T0901317 is a dual LXR/FXR agonist. Mol Genet Metab. 2004;83(1-2):184–187. doi:10.1016/j.ymgme.2004.07.007.
- Mitro N, Vargas L, Romeo R, et al. T0901317 is a potent PXR ligand: implications for the biology ascribed to LXR. FEBS Lett. 2007;581(9):1721–1726. doi:10.1016/j.febslet.2007.03.047.
- Degraaf AJ, Zasłona Z, Bourdonnay E, et al. Prostaglandin E2 reduces Toll-like receptor 4 expression in alveolar macrophages by inhibition of translation. Am J Respir Cell Mol Biol. 2014;51(2):242–250. doi:10.1165/rcmb.2013-0495OC.
- Kirby AC, Raynes JG, Kaye PM. CD11b regulates recruitment of alveolar macrophages but not pulmonary dendritic cells after pneumococcal challenge. J Infect Dis. 2006;193(2):205–213. doi:10.1086/498874.
- Jubinville É, Routhier J, Maranda-Robitaille M, et al. Pharmacological activation of liver X receptor during cigarette smoke exposure adversely affects alveolar macrophages and pulmonary surfactant homeostasis. Am J Physiol Lung Cell Mol Physiol. 2019;316(4):L669–l678. doi:10.1152/ajplung.00482.2018.
- Ou G, Zhu M, Huang Y, et al. HSP60 regulates the cigarette smoke-induced activation of TLR4-NF-κB-MyD88 signalling pathway and NLRP3 inflammasome. Int Immunopharmacol. 2022;103:108445. doi:10.1016/j.intimp.2021.108445.
- Wang Y, Wang Y, Ma J, et al. YuPingFengSan ameliorates LPS-induced acute lung injury and gut barrier dysfunction in mice. J Ethnopharmacol. 2023;312:116452. doi:10.1016/j.jep.2023.116452.
- Ju J, Li Z, Shi Q. Baicalin inhibits inflammation in rats with chronic obstructive pulmonary disease by the TLR2/MYD88/NF-κBp65 signaling pathway. Evid Based Complement Alternat Med. 2022;2022:7273387–7273311. doi:10.1155/2022/7273387.
- Yanagibashi T, Nagai Y, Watanabe Y, et al. Differential requirements of MyD88 and TRIF pathways in TLR4-mediated immune responses in murine B cells. Immunol Lett. 2015;163(1):22–31. doi:10.1016/j.imlet.2014.11.012.
- Putra AB, Nishi K, Shiraishi R, et al. Jellyfish collagen stimulates production of TNF-α and IL-6 by J774.1 cells through activation of NF-κB and JNK via TLR4 signaling pathway. Mol Immunol. 2014;58(1):32–37. doi:10.1016/j.molimm.2013.11.003.
- Sakai J, Cammarota E, Wright JA, et al. Lipopolysaccharide-induced NF-κB nuclear translocation is primarily dependent on MyD88, but TNFα expression requires TRIF and MyD88. Sci Rep. 2017;7(1):1428. doi:10.1038/s41598-017-01600-y.
- Morissette MC, Shen P, Thayaparan D, et al. Disruption of pulmonary lipid homeostasis drives cigarette smoke-induced lung inflammation in mice. Eur Respir J. 2015;46(5):1451–1460. doi:10.1183/09031936.00216914.
- Mo J, Fang SJ, Chen W, et al. Regulation of ALK-1 signaling by the nuclear receptor LXRbeta. J Biol Chem. 2002;277(52):50788–50794. doi:10.1074/jbc.M210376200.
- Cannon MV, Yu H, Candido WM, et al. The liver X receptor agonist AZ876 protects against pathological cardiac hypertrophy and fibrosis without lipogenic side effects. Eur J Heart Fail. 2015;17(3):273–282. doi:10.1002/ejhf.243.
- Delvecchio CJ, Bilan P, Radford K, et al. Liver X receptor stimulates cholesterol efflux and inhibits expression of proinflammatory mediators in human airway smooth muscle cells. Mol Endocrinol. 2007;21(6):1324–1334. doi:10.1210/me.2007-0017.
- Zhang J, Wu Z, Yu F, et al. Role of liver-X-receptors in airway remodeling in mice with chronic allergic asthma. Exp Ther Med. 2021;22(3):920. doi:10.3892/etm.2021.10352.
- Zhang H, Li X, Qian Z. Regulation of macrophage cholesterol efflux and liver X receptor α activation by nicotine. Int J Clin Exp Med. 2015;8(9):16374–16378.
- Vale K. Targeting the JAK-STAT pathway in the treatment of ‘Th2-high’ severe asthma. Future Med Chem. 2016;8(4):405–419. doi:10.4155/fmc.16.4.
- Pernis AB, Rothman PB. JAK-STAT signaling in asthma. J Clin Invest. 2002;109(10):1279–1283. doi:10.1172/JCI0215786.
- Kim SY, Lim EJ, Yoon YS, et al. Liver X receptor and STAT1 cooperate downstream of Gas6/Mer to induce anti-inflammatory arginase 2 expression in macrophages. Sci Rep. 2016;6(1):29673. doi:10.1038/srep29673.
- Higham A, Lea S, Plumb J, et al. The role of the liver X receptor in chronic obstructive pulmonary disease. Respir Res. 2013;14(1):106. doi:10.1186/1465-9921-14-106.
- Shi Y, Xu X, Tan Y, et al. A liver-X-receptor ligand, T0901317, attenuates IgE production and airway remodeling in chronic asthma model of mice. PLoS One. 2014;9(3):e92668. doi:10.1371/journal.pone.0092668.