Abstract
Rhabdoid tumors of kidney and extrarenal rhabdoid tumors are identified by a round-epithelioid cell morphology and a bland immunophenotype, but a distinctive ultrastructure dominated by paranuclear whorls of intermediate filaments, most usually of vimentin. These tumors are also known to be highly aggressive malignancies, which, typically, bear a poor prognosis, frequently measured in months following initial presentation. The authors record the case a soft-tissue rhabdoid tumor in a 12-year-old boy with a unique long-term survival in excess of 16 years. The features of this case are documented, with a brief summary of histological, immunohistochemical, ultrastructural, and genetic characteristics of this entity.
Rhabdoid tumor was first categorized by Beckwith and Palmer in 1978 [Citation[1]], although the term itself was coined by Haas et al. in 1981 [Citation[2]]. Early on, this renal tumor was designated in the National Wilms' Tumor Study I as a rhabdomyosarcomatous variant of Wilms tumor. However, within a few years, renal rhabdoid tumor was recognized as a clinicopathological entity distinct from Wilms tumor [Citation[3]]. Although uncommon, both renal rhabdoid tumor and extrarenal variants are characterized well enough to indicate that these are highly aggressive malignancies with survival times after initial presentation being typically in the range of months rather than years. Longer survivals have been reported, however, of between 72 and 156 months [Citation[4–8]]. We describe a unique 16-year survival in a boy with a gluteal soft-tissue rhabdoid tumor diagnosed at the age of 12.
CASE REPORT
A 12-year-old boy from India presented with a persistent right-sided limp. Clinical examination revealed swelling and fullness in the right iliac region, anterior to the iliac crest. Review of the initial X-ray confirmed a large, soft-tissue swelling with scalloping of the iliac blade. A biopsy of this mass revealed a necrotic tumor with hemorrhagic areas. The iliac apophysis was split to confirm nonosseous involvement. The tumor was excised from the gluteus medius muscle, and the postoperative course was uneventful. The child was treated with 16 weeks of adjuvant chemotherapy over a 2-year period, consisting of iv cyclophosphamide (300 mg, 3 times weekly), vincristine (1.5 mg, twice weekly), and actinomycin D (2.3 mg once weekly). Radiotherapy was not given. The child is alive and well with no evidence of disease recurrence 16 years post- operatively.
RESULTS
Gross Findings
The surgical specimen consisted of irregular brown- and gray-colored soft friable tissue measuring 15 × 10 cm. The specimen was fixed in histological formalin for histology and immunohistochemistry, and tissue from formalin was taken for electron microscopy.
Histology
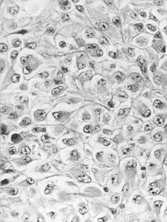
The tumor consisted of a lobulated proliferation of fairly uniform, mostly rounded cells with occasional spindled forms in sheets or clusters. Tumor cells were of medium size with eccentric vesicular nuclei and prominent nucleoli (). Mitoses were 8 in 10 high-power fields. The cytoplasm was abundant, eosinophilic and contained diastase-resistant PAS- positive inclusions.
Fig. 1 Sheets of typical rhabdoid cells with eccentric nuclei, prominent nucleolus and abundant eosinophilic cytoplasm. H&E, × 250.
Immunohistochemistry
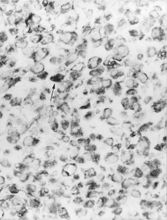
Sections of paraffin-embedded tissue at 4–5 µm were immunostained according to a standard 3-layered streptavidin–biotin peroxidase procedure. Tumor cells expressed vimentin [Dako 1:100] (). The following markers were negative: cytokeratin (Dako, dilution 1:50), EMA (Novocastra, 1:50), S-100 (Novocastra, 1:500), myoglobin (Dako, 1:600), smooth-muscle actin (Novocastra, 1:50), desmin (Novocastra, 1:100), CD99 (Dako, 1:60), and HMB45 (ID Labs, 1:10).
Fig. 2 Positive cytoplasmic immunostaining for vimentin. The poor tissue preservation probably accounts for the rather small number of positively staining vimentin-filament aggregates (arrow), × 250.
Electron Microscopy
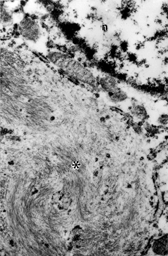
Tissue was embedded in epoxy resin following conventional processing procedures. Tumor cells contained prominent paranuclear spheroidal aggregates of intermediate filaments in a whorled pattern, largely excluding membranous organelles (). Otherwise, the cytoplasm contained little other than a few cisternae of rough endoplasmic reticulum and mitochondria.
Fig. 3 Electron micrograph of tumor cell showing prominent paranuclear aggregate of intermediate filaments (*). n, nucleus, × 12,800.
DISCUSSION
Rhabdoid tumor was originally described as a distinctive, highly malignant neoplasm of the kidney [Citation[1]]. Extrarenal rhabdoid tumors have now been reported in many organs, including the brain [Citation[9–11]], eye [Citation[12]], heart [Citation[13]], liver [Citation[14]], colon [Citation[15]], tongue [Citation[16]], bladder [Citation[17]], prostate [Citation[18]], vulva [Citation[19]], uterus [Citation[20]], skin [Citation[21]], and soft-tissue from many sites such as the extremities [Citation[22]], neck, mediastinum, retroperitoneum, pelvis, and paraspinal regions [Citation[23]]. Some reported cases of extrarenal rhabdoid tumor have the same clinical pattern as their renal counterparts, including young age, early dissemination, and lethal outcome [Citation[5]]. A few have been associated with a second brain primary tumor [Citation[24]].
The diagnosis of rhabdoid tumor is based on the histological recognition of large epithelioid “rhabdomyoblast-like” cells with large nuclei and abundant eosinophilic cytoplasmic inclusions, which, ultrastructurally, consist almost exclusively of large numbers of intermediate filaments [Citation[1]]. However, while characteristic, the latter feature is nonspecific and has been described in other neoplastic entities [Citation[24–27]]. It is therefore important to make the distinction between extrarenal rhabdoid tumor and extrarenal tumor with rhabdoid-cell phenotype having diverse types of differentiation. Among these so-called pseudo-rhabdoid tumors are neuroendocrine tumors, carcinoma, malignant fibrous histiocytoma, epithelioid sarcoma and rhabdomyosarcoma [Citation[27]], myoepithelial tumor [Citation[26]], and peripheral primitive neuroectodermal tumor (PNET) [Citation[24]]. While these, like true rhabdoid tumors, have paranuclear whorls of intermediate filaments, they also betray evidence of other lines of differentiation through immunostaining and/or ultrastructure. The case presented here showed no immunohistochemical or ultrastructural evidence of a specific line of differentiation, and was therefore regarded as a true extrarenal rhabdoid tumor.
While their immunophenotype is rather bland and their ultrastructure shows a limited number of nonetheless distinctive features, there is a growing body of cytogenetic information to characterize these tumors. Cytogenetic studies have shown evidence of a common genetic basis for CNS, renal, and extrarenal rhabdoid tumors [Citation[28]]. Chromosomal abnormalities of rhabdoid tumor involve chromosomal region 22q11 and 11p15.5 [Citation[29–32]]. Deletions in the chromosome 22p11 region suggest the loss or inactivation of the hSNF5/INI1 tumor-suppressor gene that is responsible for the progression of rhabdoid tumor in CNS and other sites [Citation[33], Citation[34]]. However, hSNF5/INI1 mutations have also been detected in PNET, choroid plexus carcinomas, and medulloblastomas [Citation[35]]. A recent study of a congenital extrarenal rhabdoid tumor in a premature newborn baby [Citation[36]] suggests a pluripotent cell with a possible myogenic differentiation, as Inv (11)(p13p15), PAX3 expression (also seen in early skeletal muscle progenitor cells and in the developing nervous system) and Myf3 expression (myogenic determination factor regulating muscle differentiation) were observed.
One of the most important features of rhabdoid tumor, however, is the clinical one, that they are known to be highly aggressive with a lethal outcome in most cases. The longest survival reported to date is 156 months [Citation[7]]. Overall, less than 50% of cases have survived without tumor recurrences, regardless of the therapy employed (Table). Our case is unique in that the patient is alive and well with no evidence of disease recurrence 16 years after the initial diagnosis. At present, the mechanism of long-term survival is not known, but understanding this is likely to come with detailed cytogenetic and genetic investigations, provided they are implemented in the context of a diagnosis based on classical morphology combined with ancillary techniques of immunohistochemistry and electron microscopy to avoid confusion with pseudo-rhabdoid tumors.
TABLE Survival of Extrarenal Rhabdoid Tumors
REFERENCES
- Beckwith JB, Palmer NF.. Histopathology and prognosis of Wilms' tumors: results from the First National Wilms' Tumor Study. Cancer. 1978; 41: 1937–1948. [PUBMED], [INFOTRIEVE]
- Haas JE, Palmer NF, Weinberg AG, Beckwith JB.. Ultrastructure of malignant rhabdoid tumor of the kidney: a distinctive renal tumor of children. Hum Pathol. 1981; 12: 646–657. [PUBMED], [INFOTRIEVE]
- Weeks DA, Beckwith JB, Mierau GW, Luckey DW.. Rhabdoid tumor of kidney: a report of 111 cases from the National Wilms' Tumor Study Pathology Center. Am J Surg Pathol. 1989; 13: 439–458. [PUBMED], [INFOTRIEVE]
- Fanburg-Smith JC, Hengge M, Hengge UR, Smith JS, Jr, Miettinen M.. Extrarenal rhabdoid tumors of soft tissue: a clinicopathologic and immunohistochemical study of 18 cases. Ann Diagn Pathol. 1998; 2: 351–362. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Parham DM, Weeks DA, Beckwith JB.. The clinicopathologic spectrum of putative extrarenal rhabdoid tumors: an analysis of 42 cases studied with immunohistochemistry or electron microscopy. Am J Surg Pathol. 1994; 18: 1010–1029. [PUBMED], [INFOTRIEVE], [CSA]
- Gururangan S, Bowman LC, Parham DM, et al, Primary extracranial rhabdoid tumors: clinicopathologic features and response to ifosfamide. Cancer. 1993; 71: 2653–2659. [PUBMED], [INFOTRIEVE]
- Kodet R, Newton WA, Jr, Sachs N, et al, Rhabdoid tumors of soft tissues: a clinicopathologic study of 26 cases enrolled on the Intergroup Rhabdomyosarcoma Study. Hum Pathol. 1991; 22: 674–684. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Sotelo-Avila C, Gonzalez-Crussi F, deMello D, et al, Renal and extrarenal rhabdoid tumors in children: a clinicopathologic study of 14 patients. Semin Diagn Pathol. 1986; 3: 151–163. [PUBMED], [INFOTRIEVE]
- Bonnin JM, Rubinstein LJ, Palmer NF, Beckwith JB.. The association of embryonal tumors originating in the kidney and in the brain: a report of seven cases. Cancer. 1984; 54: 2137–2146. [PUBMED], [INFOTRIEVE]
- Biggs PJ, Garen PD, Powers JM, Garvin AJ.. Malignant rhabdoid tumor of the central nervous system. Hum Pathol. 1987; 18: 332–337. [PUBMED], [INFOTRIEVE]
- Cooley L, Vogel H.. Primary malignant rhabdoid tumor of the central nervous system. Ultrastruct Pathol. 1997; 21: 361–368. [PUBMED], [INFOTRIEVE], [CSA]
- Gunduz K, Shields JA, Eagle RC, Jr, Shields CL, De Potter P, Klombers L.. Malignant rhabdoid tumor of the orbit. Arch Ophthalmol. 1998; 116: 243–246. [PUBMED], [INFOTRIEVE], [CSA]
- Small EJ, Gordon GJ, Dahms BB.. Malignant rhabdoid tumor of the heart in an infant. Cancer. 1985; 55: 2850–2853. [PUBMED], [INFOTRIEVE]
- Di Cori S, Oudjhane K, Neilson K.. Primary malignant rhabdoid tumour of the liver. Can Assoc Radiol J. 1993; 44: 52–54. [PUBMED], [INFOTRIEVE]
- Nakamura I, Nakano K, Nakayama K, et al, Malignant rhabdoid tumor of the colon: report of a case. Surg Today. 1999; 29: 1083–1087. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Patron M, Palacios J, Rodriguez-Peralto JL, Burgos E, Contreras F.. Malignant rhabdoid tumor of the tongue: a case report with immunohistochemical and ultrastructural findings. Oral Surg Oral Med Oral Pathol. 1988; 65: 67–70. [PUBMED], [INFOTRIEVE]
- Carter RL, McCarthy KP, al-Sam SZ, Monaghan P, Agrawal M, McElwain TJ.. Malignant rhabdoid tumour of the bladder with immunohistochemical and ultrastructural evidence suggesting histiocytic origin. Histopathology. 1989; 14: 179–190. [PUBMED], [INFOTRIEVE]
- Ekfors TO, Aho HJ, Kekomaki M.. Malignant rhabdoid tumor of the prostatic region: immunohistological and ultrastructural evidence for epithelial origin. Virchows Arch A Pathol Anat Histopathol. 1985; 406: 381–388. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Brand A, Covert A.. Malignant rhabdoid tumor of the vulva: case report and review of the literature with emphasis on clinical management and outcome. Gynecol Oncol. 2001; 80: 99–103. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Hsueh C.. Malignant rhabdoid tumor of the uterine corpus. Gynecol Oncol. 1996; 61: 142–146. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Hsueh C, Kuo TT.. Congenital malignant rhabdoid tumor presenting as a cutaneous nodule: report of 2 cases with review of the literature. Arch Pathol Lab Med. 1998; 122: 1099–1102. [PUBMED], [INFOTRIEVE], [CSA]
- Kent AL, Mahoney DH, Jr, Gresik MV, Steuber CP, Fernbach DJ.. Malignant rhabdoid tumor of the extremity. Cancer. 1987; 60: 1056–1059. [PUBMED], [INFOTRIEVE]
- Tsuneyoshi M, Daimaru Y, Hashimoto H, Enjoji M.. The existence of rhabdoid cells in specified soft tissue sarcomas: histopathological, ultrastructural and immunohistochemical evidence. Virchows Arch A Pathol Anat Histopathol. 1987; 411: 509–514. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Chang CH, Ramirez N, Sakr WA.. Primitive neuroectodermal tumor of the brain associated with malignant rhabdoid tumor of the liver: a histologic, immunohistochemical, and electron microscopic study. Pediatr Pathol. 1989; 9: 307–319. [PUBMED], [INFOTRIEVE]
- McNutt MA, Bolen JW, Gown AM, Hammar SP, Vogel AM.. Coexpression of intermediate filaments in human epithelial neoplasms. Ultrastruct Pathol. 1985; 9: 31–43. [PUBMED], [INFOTRIEVE]
- Eyden B, Dardick I, Bishop P.. Filamentous inclusions of unusual composition and architecture in a metastatic tumor showing myoepithelial differentiation. Ultrastruct Pathol. 1991; 15: 663–670. [PUBMED], [INFOTRIEVE]
- Erlandson RA.. Diagnostic Transmission Electron Microscopy of Tumors. New York, Raven Press. 1996
- Ho DM, Hsu CY, Wong TT, Ting LT, Chiang H.. Atypical teratoid/rhabdoid tumor of the central nervous system: a comparative study with primitive neuroectodermal tumor/medulloblastoma. Acta Neuropathol. 2000; 99: 482–488
- Sait SN, Nowak NJ, Singh-Kahlon P, et al, Localization of Beckwith-Wiedemann and rhabdoid tumor chromosome rearrangements to a defined interval in chromosome band 11p15.5. Genes Chromosomes Cancer. 1994; 11: 97–105. [PUBMED], [INFOTRIEVE], [CSA]
- Shashi V, Lovell MA, von Kap-herr C, Waldron P, Golden WL.. Malignant rhabdoid tumor of the kidney: involvement of chromosome 22. Genes Chromosomes Cancer. 1994; 10: 49–54. [PUBMED], [INFOTRIEVE], [CSA]
- White FV, Dehner LP, Belchis DA, et al, Congenital disseminated malignant rhabdoid tumor: a distinct clinicopathologic entity demonstrating abnormalities of chromosome 22q11. Am J Surg Pathol. 1999; 23: 249–256. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Zhou J, Fogelgren B, Wang Z, Roe BA, Biegel JA.. Isolation of genes from the rhabdoid tumor deletion region in chromosome band 22q11.2. Gene. 2000; 241: 133–141. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Biegel JA, Allen CS, Kawasaki K, Shimizu N, Budarf ML, Bell CJ.. Narrowing the critical region for a rhabdoid tumor locus in 22q11. Genes Chromosomes Cancer. 1996; 16: 94–105. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Bhattacharjee M, Hicks J, Dauser R, et al, Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. 1999; 59: 74–79
- Sevenet N, Sheridan E, Amram D, Schneider P, Handgretinger R, Delattre O.. Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am J Hum Genet. 1999; 65: 1342–1348. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Staehelin F, Bissig H, Hosli I, et al, Inv(11)(p13p15) and myf-3(MyoD1) in a malignant extrarenal rhabdoid tumor of a premature newborn. Pediatr Res. 2000; 48: 463–467. [PUBMED], [INFOTRIEVE], [CSA]