
ABSTRACT
The presence of myeloid bodies (MBs) is classically associated with Fabry disease (FD). However, MBs are also identified in patients without clinical evidence of FD. We attempt to further understand the clinicopathologic significance of incidental MBs in those without FD. Among the 4400 renal biopsies accessioned at the University of Rochester Medical Center from 2010 to 2021, we identified 32 cases showing MBs, 6 of which had FD. Medications were compared between a non-FG and a control-group of randomly selected cases without MBs (non-MBs). Both Fabry-group (FG) and non-Fabry-group (non-FG) were predominantly middle-aged (mean 48 years vs 56, respectively). Non-FG had slight female predominance (1:4), while all in FG were female. The majority of both non-FG and non-MBs cohort were on the same medications reported to cause phospholipidosis except sertraline and hydralazine (p = .04), which were more frequent in non-FG. Ultrastructurally, non-FG tended to show focal MBs in predominantly podocytes, while FG showed more extensive MBs in not only podocytes but also parietal, tubular, endothelial, and myocyte cells (p = .03). In addition, half of FG had another superimposed renal disease including kappa-light chain deposition disease, thin-basement membrane nephropathy, and lithium-related changes. MBs are encountered not only in FD but in other settings including CADs, toxins, and other inheritable diseases. Although secondary causes of MBs typically show less extensive involvement compared to FD, these features overlap. Given the challenges in diagnosing female carriers, the finding of MBs, though not specific to FD, may be the only clue that leads to further work-up and timely diagnosis, underscoring the importance of considering FD among other etiologies in differential diagnosis.
Introduction
The presence of myeloid or “zebra” bodies have been classically associated with Fabry disease (FD), a rare X-linked metabolic disorder due to deficient α-galactosidase A activity. In FD, myeloid bodies are concentrically laminated or whorled layers of electron-dense material typically identified within enlarged lysosomes in every cell type in the kidney.
Not uncommonly, myeloid bodies (MB) are seen in biopsies of those without clinical evidence of FD. Similar morphologic changes have been reported in multiple case reports in other settings, notably, secondary to iatrogenic causes of phospholipidosis.Citation1–7 To further understand the significance of MBs in those without FD, we performed the first biopsy-based single-center study of renal biopsies with incidental findings of MBs.
Material and methods
In this study, all native kidney biopsies accessioned at the University of Rochester Medical Center’s (URMC) Pathology Laboratory from 2010 to 2021 were retrospectively reviewed for evidence of MBs identified by electron microscopy. Among those with MBs, we divided cases into two groups: those with FD aka Fabry-group (FG) and those without FD aka non-Fabry-group (non-FG). We also randomly selected 22 cases without both FD and MBs (non-MBs) as a control group to compare medication information with non-FG.
All renal biopsies were processed using standard techniques for light microscopy (LM), immunofluorescence (IF), and electron microscopy (EM), and were interpreted by three renal pathologists. Electronic medical records were used to collect clinical information. Clinical data included laboratory parameters such as serum creatinine, eGFR, proteinuria, urinalysis, abnormal serologies, medications, and whether the patient had a clinical history or diagnosis of FD at presentation or follow-up.
Tissues submitted for electron microscopy were fixed in 2.5% glutaraldehyde in Millonig’s buffer, washed in Millonig’s buffer for fiveCitation5 minutes, and post fixed in 1% osmium tetroxide in Millonig’s buffer for 20–30 minutes. Dehydration of tissue was performed as follows: 25% ethyl alcohol, 5 minutes; 50% ethyl alcohol, 7 minutes, 75% ethyl alcohol, 10 minutes, 95% ethyl alcohol 15 minutes, 100% ethyl alcohol 30 minutes × 3 changes, 100% + propylene oxide, 30 minutes (1:1 ratio), propylene oxide, 20 minutes × 2 changes, and P.O. + Eponate 12- araldite 502, 60 minutes (1:1 ratio – cap off). After the specimen was embedded in epoxy resin, and sectioned using standard techniques, toluidine blue sections were used for selection of appropriate areas for thin section preparations. All renal compartments were carefully examined under a Hitachi HT 7800. Selected photographs were then taken and stored in an electronic folder.
Statistical analysis was performed using SPSS software (version 28). To compare continuous values between the two groups, we performed two-sample tests without assuming equal variances. To compare categorical differences, we created cross tabulations of each variable against the group and used chi-squared tests for equal distribution between the two groups. Statistical significance was assumed at p < .05.
Results
Demographics and clinical data
Between January 2010 and December 2021, 4400 patients underwent native and transplant renal biopsies that were accessioned in the Pathology Laboratory at Rochester University Medical Center. Of those patients, 32 patients had MBs identified by electron microscopy. Six of the 32 patients had FD, while the remaining 26 did not. In the Fabry-group (FG), the mean age was 48 years (range 38–59) and all were female while in the non-Fabry-group (non-FG), the mean age was 56 years (range 11–85) with slight female predominance (F:M ratio: 1.4) (). Of the 26 non-FG patients, 15 had history of hypertension and 8 diabetes mellitus. Proteinuria was the most common biopsy indication in both FG and non-FG (50% & 83%, respectively) followed by acute kidney injury (42% & 50%). The non-FG tended to be present with heavier proteinuria at presentation compared to the FG, but the difference was not statistically significant (mean 3.5 vs 0.2 g/g; p = .26). There were also no statistically significant differences between both group in terms of serum creatinine and eGFR.
Table 1. Renal biopsy findings
Table 2. Clinical and pathologic data of cases with myeloid bodies mean ± SD or N (%)
Eighteen cases in the non-FG had medication information available (), of which 15 were on anti-hypertensive medication. The most common anti-hypertensive medications were amlodipineCitation6 and hydralazine,Citation5 followed by beta blockers,Citation5 and ACE/ARB inhibitors.Citation5 Eight were on anti-cholesterol medication, of which seven were on statins, and one on fenofibrate and ezetimibe. All 4 patients on anti-diabetic medication were taking metformin. Seven cases were on anti-depressants or anti-anxiolytics: 5 sertraline, 1 venlafaxine, and 1 alprazolam. Six patients were on a proton-pump inhibitor, while 3 patients were on allopurinol, cetirizine, and/or hydroxychloroquine. We compared the list of medications in the non-FG with a control group of randomly selected cases without both FD and MBs (non-MBs) () and found no significant differences except for hydralazine and sertraline, which were identified with more frequency in the non-FG (p = .04). Moreover, 83% of patients in non-FG and 86% in non-MBs cohorts were on at least 1 drug reported as a cause of drug-induced phospholipidosis.
Table 3. Medications in non-FG vs non-MBs
Follow-up evaluation
Limited follow-up information was available for 13 patients in non-FG and 2 in FG. The average follow-up for the non-FG was 23 ± 29 months, and for the FG it was 31 ± 36 months.
Among the non-FG patients, three cases had MBs attributed to hydroxychloroquine, while two were suspected to be due to anti-depressants and statins. In one patient in whom no other pathology could explain the heavy proteinuria, sertraline was withheld. Proteinuria subsequently dropped from 18 to 5.5 g/g after 1 year. However, because of the persistent nephrotic range proteinuria, the patient was scheduled for genetic counseling, but unfortunately passed away from complications of infection. Another patient with acute interstitial nephritis due to NSAIDs was given prednisone for 2–3 months with improvement of renal function from serum creatinine of 6.3 to 0.86 mg/dL. The patient was also on rosuvastatin, which was felt to be a potential cause of MBs. The drug was subsequently withheld and replaced with another anti-cholesterol medication. None of the 13 non-FG cases developed clinical signs of Fabry disease. However, only two patients were tested for α-galactosidase A activity, which were normal, and none underwent mutational analysis.
Both FG cases with follow-up data obtained were receiving enzyme replacement therapy and fabrazyme. One patient continued to have stable renal function and persistent but mild proteinuria without other clinical symptoms, while the other developed mild increase in serum creatinine and autonomic insufficiency but resolving proteinuria.
Pathology data
Six patients had Fabry disease confirmed on biopsy, three of which had additional pathology: one had kappa-type light-chain deposition disease (LCCD), one with thin-basement membrane disease (TBMN), and one with lithium-related chronic tubulointerstitial nephropathy (CIN).
The remaining 26 non-FG had heterogeneous biopsy findings (). The most common biopsy diagnosis in non-FG was hypertensive arterionephrosclerosis (53%), followed by acute tubular necrosis (27%), diabetic glomerulosclerosis (19%), ANCA- associated pauci-immune glomerulonephritis (15%), lupus nephritis (12%), and acute interstitial nephritis (8%). In non-FG, MBs were most often seen in podocytes (85%) but were typically either rare or focal. The three non-FG cases had MBs in parietal cells (13%) and/or endothelial cells (11%). None were found in tubular cells or myocytes. In contrast, all the cases in the Fabry group had numerous MBs in podocytes (100%), and the majority involved parietal, tubular, and endothelial cells, and vascular myocytes (67%; p = .03). Foot process effacement (FPE) was less than 50% in all the cases in the FG. Similarly, in most of the non-FG cases, FPE were focal ranging from preserved to up to 50% except in four cases, which had greater than 50% FPE, 1 of which was MCD and 3 SLE nephritis.
Discussion
Fabry disease (FD) (also called Anderson-Fabry disease) was first described in 1898 by two physicians, Johannes Fabry and William Anderson, in patients with a maculopapular rash now recognized as angiokeratoma.Citation8 In 1965, Hashimoto et al. discovered by electron microscopy the presence of MBs in endothelial cells, smooth muscle cells, fibrocytes, and perivascular cells in patients with FD.Citation9 Since then, MBs also called “zebra bodies” or “lamellar bodies,” have been thought to be diagnostic of FD. However, MBs are occasionally encountered in biopsies of patients without clinical evidence of FD. To further understand the clinicopathologic significance of these unique ultrastructural findings, we report the first retrospective study to focus on incidental MBs in biopsies of patients without FD.
As early as 1948, MBs have been reported in animal models and patients treated with cationic amphiphilic drugs (CADs), which are drugs that contain a cationic amphiphilic structure capable of inducing phospholipidosis, such as gentamicin and chloroquine.Citation10–13 Since then, over 50 FDA-approved agents have been associated with the development of lysosomal accumulation of MBs (), also known as drug-induced phospholipidosis (DIP).Citation14 Pathologic features of DIP on renal biopsy are indistinguishable from FD including prominent vacuolization of podocytes by LM and the presence of MBs by EM ().
Table 4. Medications reported to cause DIP.Citation14,Citation19,Citation21,Citation22,Citation30,Citation56
Figure 1. A. Electron microscopy showing myeloid bodies within the podocyte and parietal epithelial cell cytoplasm in a non-FG case (original magnification: 4000×). B. Electron microscopy of the same case shown at higher magnification of myeloid bodies within the podocyte cytoplasm (original magnification: 12000×).
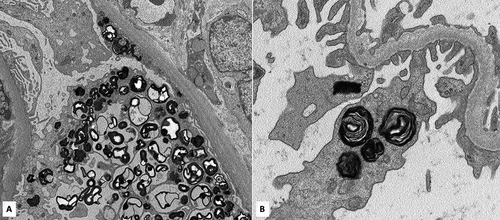
Drugs such as aminoglycosides, hydroxychloroquine, and amiodarone have been well described involving the kidney.Citation1,Citation2,Citation4–7,Citation12,Citation15–17 Sertraline has also been recently reported as a renal allograft adversely affected by DIP.Citation18 Other drugs, including anti-cholesterol medications (e.g. statins and fenofibrate), beta-blockers, anti-depressants (e.g. tricyclic anti-depressants, citalopram, and fluoxetine), macrolides (e.g. azithromycin), and anti-estrogen therapy (e.g. Tamoxifen) have been reported to cause DIP in other organ systems notably the lung and liver, as well as in animal and cell-based models.Citation19–29 Interestingly, the majority in both non-FG and non-MBs cohorts were on the same CADs reported in DIP except for sertraline and hydralazine, both of which were more common in the non-FG cases. Sertraline has been well documented in inducing phospholipidosis but not hydralazine. That said, a recent study demonstrated the ability of hydralazine to inhibit phospholipase resulting in phospholipidosis.Citation30 The same study found that within 163 drugs assayed, fosinopril, an angiotensin-converting enzyme inhibitor also not previously reported to cause DIP, was the most potent inhibitor of phospholipid catabolism.
Several mechanisms as the basis of DIP have been proposed.Citation30 CADs share similar structural properties including a hydrophobic domain consisting of an aromatic or aliphatic ring, and a hydrophilic side chain with a positively charged amine at physiologic pH. Because of their cationic structure, they can easily enter the cell where they are protonated in the lysosome’s acidic environment. These subsequently form complexes with phospholipids that are indigestible for phospholipase resulting in their accumulation in lysosomes. Another theory is that CADs directly bind to and inhibit lysosomal phospholipases through competitive or allosteric mechanisms. Some propose that CADs inhibit phospholipase activity by interfering with the electrostatic charge interaction between the cationic residues found in phospholipases and negatively charged phospholipids.Citation30 Since the activity of phospholipases increases with more negatively charged membrane lipids, CADs can counteract this activity by neutralizing the membrane charge.Citation31 Other study findings also suggest that through this charge interference, CADs cause displacement of phospholipases from the lysosomal membrane leading to their degradation by lysosomal proteases.Citation32 Through these processes, it appears CADs potentially cause cellular toxicity, which has been supported by in-vitro studies and case reports.Citation33–35 Proteinuria and renal dysfunction have been attributed to DIP in a few case reports where no abnormalities were detected in mutational analysis and discontinuation of the drug led to clinical improvement.Citation2,Citation15,Citation18,Citation36,Citation37 Some of these cases even showed decreased α-galactosidase A enzyme activity and corneal deposits similar to those seen in FD, both of which were resolved after drug withdrawal.Citation2,Citation15,Citation38 Remission of MBs has also been reported after cessation of treatment.Citation39 Given that most of the cases in the non-FG cohort had renal disease to explain their clinical presentation, it is unclear if or how much was contributed by DIP. However, in one non-FG patient in whom sertraline was suspected to be a cause of their nephrotic-range proteinuria, drug cessation led to a partial response suggesting a component of DIP-related toxicity.
Noteworthy is that the majority of the non-MBs were on the same CADs as the non-FG patients but did not develop MBs. One possible explanation is that patients in the non-MBs group were on a comparatively lower dosage and for a shorter duration of treatment. This is supported by findings from multiple toxicological studies in which test animals were given higher dosages for longer durations and demonstrated that the development of phospholipidosis was dose-dependent and directly proportional to the accumulation of CAD within the tissue.Citation39–41 Some authors suggest the possibility that individuals who carry polymorphisms resulting in diminished enzyme activity may be susceptible to DIP and that such individuals may potentially benefit from personalized therapy where substitutes for CADs could be considered.Citation34 Although a minority of non-FG cases had no identifiable drug known to cause DIP, this could be explained by a lack of complete or accurate medication list which may not have accounted for over-the-counter medications or other drugs taken previously but recently discontinued.
FD is a diagnosis that is made principally on clinical grounds and mutational analysis. However, there are few pathologic findings that appear to be more characteristic of DIP. The non-FG cohort tended to have focal MBs in mostly podocytes with a minority involving endothelial and parietal cells, while all the FG cases exhibited more extensive involvement by MBs in not only podocytes, but also endothelial cells, parietal cells, myocytes, and tubules. Other reports of iatrogenic phospholipidosis show similar findings and support the observation that patients with MBs attributed to drugs are typically focal and mostly in podocytes compared to those with FD in whom MBs are usually more widespread in the podocytes and present in almost every other cell within the kidney.Citation4,Citation6 A notable exception are aminoglycosides such as gentamicin, which appear to primarily affect tubular cells rather than podocytes.Citation12 Moreover, some cases of DIP exhibit MBs with a similar distribution as those seen in FD, through focal.Citation2,Citation15 Nonetheless, even female heterozygous carriers have also been noted to show limited renal involvement, making exclusion of FD based on these characteristics alone impossible. Another potential differentiating feature of DIP from FD proposed by some authors is the presence of foamy macrophages infiltrating capillaries, but this feature is not always present.Citation2,Citation14,Citation15 In our non-FG cohort, infiltrating foamy macrophages were not appreciated.
The differential diagnosis for MBs has become wider with reports finding additional potential associations including silicone nephropathy and even contrast medium.Citation42,Citation43 Other inheritable diseases such as Niemann-Pick and LMX1B-associated nephropathy (also called Nail-patella-like renal disease) have shown similar osmiophilic lysosomal structures by EM.Citation44–46
Because the pathologic features of FD are not specific, it is almost impossible to make the diagnosis of FD on pathology alone and is rather dependent on clinical history, presentation, and genetics. Renal manifestations in FD most frequently present as subnephrotic-range proteinuria and renal insufficiency is likely related to the accumulation of undigested phospholipids leading to injury to podocytes and tubular epithelium. Patients with FD develop symptoms correlating with damage to the organ system affected by the accumulation of globotriaosylceramide such as those involving nerves (e.g. neuropathies or acroparasthesias), central nervous system (e.g. transient ischemic attacks and strokes), skin (e.g. angiokeratomas, hyperhidrosis), eyes (e.g. ocular opacities), heart, and kidneys. However, an increasing number of reports of atypical FD limited to the heart or kidney have been described Citation47–50 including female carriers who have a heterogeneous presentation due to lionization ranging from asymptomatic or minimal to severe disease.Citation51 To that point, all the biopsies from the FG cohort were middle-aged women and likely reflects the challenges that arise with diagnosing female carriers who typically have an atypical presentation of the disease and are therefore often diagnosed later than males.
Biopsy findings in the non-FG cohort showed a wide array of glomerular pathology, which likely accounted for the heavier proteinuria encountered in this group. Half of the FG patients also demonstrated an eclectic sample of superimposed renal diseases including kappa-LCDD, TBMN, and lithium-related changes. Multiple case reports of patients with FD and other superimposed renal diseases have been reported, including IgA nephropathy, membranous glomerulopathy, and granulomatosis with polyangiitis.Citation52–55. While it is arguable that these disease associations are coincidental, the possibility of a pathogenic link would need further investigation. Regardless, the diverse biopsy findings identified in our cohort underscore the importance of considering FD in cases showing other renal diseases that would explain the clinical presentation and yet demonstrate MBs.
Given these complex considerations, confirmation with genetic testing would be ideal but does not appear to be a realistic option currently. Hopefully with increasing availability and affordability of genetic testing in the future, cases such as these will be less ambiguous.
This study has several limitations due to its retrospective nature including small sample size and lack of clinical and laboratory data at presentation and follow-up. Molecular studies and testing for leukocyte α-galactosidase A enzyme activity was also not available for many of the cases.
In summary, MBs are encountered not only in FD but also in multiple other settings including CADs, toxins, and other inheritable diseases. DIP-related renal injury typically shows less extensive involvement by MBs compared to FD, but there is also overlap between the two entities. Because pathologic features of FD are indistinguishable from other etiologies, diagnosis based on pathology alone is problematic and should be interpreted in the clinical context. Lastly, FD can occur superimposed on various other renal diseases, and therefore should be part of the differential when MBs are identified.
Authors’ contributions
HYC designed the study and wrote the first draft of the manuscript. JJ and BG critically revised the work. All authors read and approved the final manuscript.
Data statement availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Statement of ethics
This research was performed under approval of the University of Rochester’s institutional review board (STUDY#00006760) and all ethical principles and guidelines for the protection of human subjects were followed.
Acknowledgments
The authors are grateful to Karen Vanderbilt, Tracy Fontaine-Matteson, and Michelle L Fuller for their excellent technical support.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Jao W, Manaligod JR, Gerardo LT, Castillo MM. Myeloid bodies in drug-induced acute tubular necrosis. J Pathol. 1983;139(1):33–40. doi:https://doi.org/10.1002/path.1711390104.
- Bracamonte ER, Kowalewska J, Starr J, Gitomer J, Alpers CE. Iatrogenic phospholipidosis mimicking Fabry disease. Am J Kidney Dis. 2006;48(5):844–850. doi:https://doi.org/10.1053/j.ajkd.2006.05.034.
- Mingeot-Leclercq MP, Tulkens PM. Aminoglycosides: nephrotoxicity. Antimicrob Agents Chemother. 1999;43(5):1003–1012. doi:https://doi.org/10.1128/AAC.43.5.1003.
- de Menezes Neves PDM, Machado JR, Custódio FB, et al. Ultrastructural deposits appearing as “zebra bodies” in renal biopsy: Fabry disease?- comparative case reports. BMC Nephrol. 2017;18(1):157. doi:https://doi.org/10.1186/s12882-017-0571-0.
- Cornell LD, Leung N. Myeloid bodies in acute tubular injury. Kidney Int. 2021;99(4):1027. doi:https://doi.org/10.1016/j.kint.2020.08.018.
- Costa RM, Martul EV, Reboredo JM, Cigarrán S. Curvilinear bodies in hydroxychloroquine-induced renal phospholipidosis resembling Fabry disease. Clin Kidney J. 2013;6(5):533–536.doi:https://doi.org/10.1093/ckj/sft089.
- Bojic M, Kozakowski N, Becede M, Kerschbaumer A, Bobacz K. The Case | myeloid bodies in the kidney biopsy of a patient with systemic lupus erythematosus. Kidney Int. 2017;92(1):271–272. doi:https://doi.org/10.1016/j.kint.2016.12.025.
- Mehta A, Beck M, Linhart A, Sunder-Plassmann G, Widmer U. History of lysosomal storage diseases: an overview. Mehta A, Beck M, and Sunder-Plassmann G . eds. Fabry Disease: Perspectives from 5 Years of FOS.Oxford: Oxford PharmaGenesis, Chapter 1. 2006.
- Hashimoto K, Gross BG, Lever WF. Angiokeratoma corporis diffusum (Fabry). Histochemical and electron microscopic studies of the skin. J Invest Dermatol. 1965;44(2):119–128. doi:https://doi.org/10.1038/jid.1965.22.
- Nelson AA, Fitzhugh OG. Chloroquine (SN-7618) pathologic changes observed in rats which for 2 years had been fed various proportions. Arch Pathol (Chic). 1948;45(4):454–462.
- Kosek JC, Mazze RI, Cousins MJ. Nephrotoxicity of gentamicin. Lab Invest. 1974;30:48–57.
- Houghton DC, Campbell-Boswell MV, Bennett WM, Porter GA, Brooks RE. Myeloid bodies in the renal tubules of humans: relationship to gentamicin therapy. Clin Nephrol. 1978;10:140–145.
- Gray JE, Purmalis A, Purmalis B, Mathews J. Ultrastructural studies of the hepatic changes brought about by Clindamycin and Erythromycin in animals. Toxicol Appl Pharmacol. 1971;19(2):217–233. doi:https://doi.org/10.1016/0041-008X(71)90108-6.
- Reasor MJ, Hastings KL, Ulrich RG. Drug-induced phospholipidosis: issues and future directions. Expert Opin Drug Saf. 2006;5(4):567–583. doi:https://doi.org/10.1517/14740338.5.4.567.
- Pintavorn P, Cook WJ. Progressive renal insufficiency associated with amiodarone-induced phospholipidosis. Kidney Int. 2008;74(10):1354–1357. doi:https://doi.org/10.1038/ki.2008.229.
- De Broe ME, Paulus GJ, Verpooten GA, et al. Early effects of gentamicin, tobramycin, and amikacin on the human kidney. Kidney Int. 1984;25(4):643–652. doi:https://doi.org/10.1038/ki.1984.69.
- Spaet RH, Sullivan DJ, Diener RM. Occurrence of myeloid bodies in rats following two-year administration of imipramine hydrochloride. Toxicol Pathol. 1983;11(1):3–11. doi:https://doi.org/10.1177/019262338301100102.
- Naseer MS, Chand R, Coppola S, Abreo A, Sharma M, Singh N. Post-transplant de-novo renal phospholipidosis in a kidney transplant recipient: Fabry disease or something else? Clin Nephrol Case Stud. 2020;8(1):46–48. doi:https://doi.org/10.5414/CNCS110131.
- Hanumegowda UM, Wenke G, Regueiro-Ren A, Yordanova R, Corradi JP, Adams SP. Phospholipidosis as a function of basicity, lipophilicity, and volume of distribution of compounds. Chem Res Toxicol. 2010;23(4):749–755. doi:https://doi.org/10.1021/tx9003825.
- Lantuejoul S, Brambilla E, Brambilla C, Devouassoux G. Statin-induced fibrotic nonspecific interstitial pneumonia. Eur Respir J. 2002;19(3):577–580. doi:https://doi.org/10.1183/09031936.02.00258802.
- Fischer H, Atzpodien EA, Csato M, et al. In silico assay for assessing phospholipidosis potential of small drug like molecules: training, validation, and refinement using several data sets. J Med Chem. 2012;55(1):126–139. doi:https://doi.org/10.1021/jm201082a.
- Sawada H, Takami K, Asahi S. A toxicogenomic approach to drug-induced phospholipidosis: analysis of its induction mechanism and establishment of a novel in vitro screening system. Toxicol Sci. 2005;83(2):282–292. doi:https://doi.org/10.1093/toxsci/kfh264.
- Huang LK, Tsai MJ, Tsai HC, Chao HS, Lin FC, Chang SC. Statin-induced lung injury: diagnostic clue and outcome. Postgrad Med J. 2013;89(1047):14–19. doi:https://doi.org/10.1136/postgradmedj-2011-130209.
- Rainey MM, Korostyshevsky D, Lee S, Perlstein EO. The antidepressant sertraline targets intracellular vesiculogenic membranes in yeast. Genetics. 2010;185(4):1221–1233. doi:https://doi.org/10.1534/genetics.110.117846.
- Shahane SA, Huang R, Gerhold D, Baxa U, Austin CP, Xia M. Detection of phospholipidosis induction: a cell-based assay in high-throughput and high-content format. J Biomol Screen. 2014;19(1):66–76.
- Ferslew BC, Brouwer KL. Identification of hepatic phospholipidosis inducers in sandwich-cultured rat hepatocytes, a physiologically relevant model, reveals altered basolateral uptake and biliary excretion of anionic probe substrates. Toxicol Sci. 2014;139(1):99–107. doi:https://doi.org/10.1093/toxsci/kfu033.
- Vitovic P, Alakoskela JM, Kinnunen PK. Assessment of drug-lipid complex formation by a high-throughput Langmuir-balance and correlation to phospholipidosis. J Med Chem. 2008;51(6):1842–1848. doi:https://doi.org/10.1021/jm7013953.
- Joshi UM, Rao P, Kodavanti S, Lockard VG, Mehendale HM. Fluorescence studies on binding of amphiphilic drugs to isolated lamellar bodies: relevance to phospholipidosis. Biochim Biophys Acta. 1989;1004(3):309–320. doi:https://doi.org/10.1016/0005-2760(89)90078-7.
- Van Bambeke F, Montenez JP, Piret J, Tulkens PM, Courtoy PJ, Mingeot-Leclercq MP. Interaction of the macrolide azithromycin with phospholipids. I. Inhibition of lysosomal phospholipase A1 activity. Eur J Pharmacol. 1996;314(1–2):203–214. doi:https://doi.org/10.1016/S0014-2999(96)00552-3.
- Hinkovska-Galcheva V, Treadwell T, Shillingford JM, et al. Inhibition of lysosomal phospholipase A2 predicts drug-induced phospholipidosis. J Lipid Res. 2021;62:100089. doi:https://doi.org/10.1016/j.jlr.2021.100089.
- Mingeot-Leclercq MP, Brasseur R, Schanck A. Molecular parameters involved in aminoglycoside nephrotoxicity. J Toxicol Environ Health. 1995;44(3):263–300. doi:https://doi.org/10.1080/15287399509531960.
- Hurwitz R, Ferlinz K, Sandhoff K. The tricyclic antidepressant desipramine causes proteolytic degradation of lysosomal sphingomyelinase in human fibroblasts. Biol Chem Hoppe Seyler. 1994;375(7):447–450.doi:https://doi.org/10.1515/bchm3.1994.375.7.447.
- Nord JE, Shah PK, Rinaldi RZ, Weisman MH. Hydroxychloroquine cardiotoxicity in systemic lupus erythematosus: a report of 2 cases and review of the literature. Semin Arthritis Rheum. 2004;33(5):336–351. doi:https://doi.org/10.1016/j.semarthrit.2003.09.012.
- Shayman JA, Abe A. Drug induced phospholipidosis: an acquired lysosomal storage disorder. Biochim Biophys Acta. 2013;1831(3):602–611. doi:https://doi.org/10.1016/j.bbalip.2012.08.013.
- Liu FL, Cohen RD, Downar E, Butany JW, Edelson JD, Rebuck AS. Amiodarone pulmonary toxicity: functional and ultrastructural evaluation. Thorax. 1986;41(2):100–105. doi:https://doi.org/10.1136/thx.41.2.100.
- Muller-Hocker J, Schmid H, Weiss M, Dendorfer U, Braun GS. Chloroquine-induced phospholipidosis of the kidney mimicking Fabry’s disease: case report and review of the literature. Hum Pathol. 2003;34(3):285–289. doi:https://doi.org/10.1053/hupa.2003.36.
- Albay D, Adler SG, Philipose J, Calescibetta CC, Romansky SG, Cohen AH. Chloroquine-induced lipidosis mimicking Fabry disease. Mod Pathol. 2005;18(5):733–738. doi:https://doi.org/10.1038/modpathol.3800344.
- Chew E, Ghosh M, McCulloch C. Amiodarone-induced cornea verticillata. Can J Ophthalmol. 1982;17:96–99.
- Reasor MJ, Kacew S. Drug-induced phospholipidosis: are there functional consequences? Exp Biol Med (Maywood). 2001;226(9):825–830.doi:https://doi.org/10.1177/153537020122600903.
- Reasor MJ, Ogle CL, Kacew S. Amiodarone-induced pulmonary toxicity in rats: biochemical and pharmacological characteristics. Toxicol Appl Pharmacol. 1989;97(1):124–133. doi:https://doi.org/10.1016/0041-008X(89)90061-6.
- Wilson BD, Clarkson CE, Lippmann ML. Amiodarone-induced pulmonary inflammation. Correlation with drug dose and lung levels of drug, metabolite, and phospholipid. Am Rev Respir Dis. 1991;143(5 Pt 1):1110–1114. doi:https://doi.org/10.1164/ajrccm/143.5_Pt_1.1110.
- Su H, Ye C, Wen Q, Zhu HY, Yi LX, Zhang C. Case report: lipid inclusion in glomerular endothelial and mesangial cells in a patient after contrast medium injection. BMC Nephrol. 2018;19(1):53. doi:https://doi.org/10.1186/s12882-018-0844-2.
- Banks DE, Milutinovic J, Desnick RJ, Grabowski GA, Lapp NL, Boehlecke BA. Silicon nephropathy mimicking Fabry’s disease. Am J Nephrol. 1983;3(5):279–284. doi:https://doi.org/10.1159/000166730.
- Lei L, Oh G, Sutherland S, et al. Myelin bodies in LMX1B-associated nephropathy: potential for misdiagnosis. Pediatr Nephrol. 2020;35(9):1647–1657. doi:https://doi.org/10.1007/s00467-020-04564-w.
- Pinto EVF, Pichurin PN, Fervenza FC, et al. Nail-patella-like renal disease masquerading as Fabry disease on kidney biopsy: a case report. BMC Nephrol. 2020;21(1):341. doi:https://doi.org/10.1186/s12882-020-02012-3.
- Grafft CA, Fervenza FC, Semret MH, Orloff S, Sethi S. Renal involvement in Niemann-Pick Disease. NDT Plus. 2009;2(6):448–451.doi:https://doi.org/10.1093/ndtplus/sfp101.
- Meehan SM, Junsanto T, Rydel JJ, Desnick RJ. Fabry disease: renal involvement limited to podocyte pathology and proteinuria in a septuagenarian cardiac variant. Pathologic and therapeutic implications. Am J Kidney Dis. 2004;43(1):164–171. doi:https://doi.org/10.1053/j.ajkd.2003.09.022.
- Von Scheidt W, Eng CM, Fitzmaurice TF, et al. An atypical variant of Fabry’s disease with manifestations confined to the myocardium. N Engl J Med. 1991;324(6):395–399. doi:https://doi.org/10.1056/NEJM199102073240607.
- Sakuraba H, Oshima A, Fukuhara Y, et al. Identification of point mutations in the alpha-galactosidase A gene in classical and atypical hemizygotes with Fabry disease. Am J Hum Genet. 1990;47(5):784–789.
- Nakao S, Kodama C, Takenaka T, et al. Fabry disease: detection of undiagnosed hemodialysis patients and identification of a “renal variant” phenotype. Kidney Int. 2003;64(3):801–807. doi:https://doi.org/10.1046/j.1523-1755.2003.00160.x.
- Mehta A, Ricci R, Widmer U, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest. 2004;34(3):236–242. doi:https://doi.org/10.1111/j.1365-2362.2004.01309.x.
- Hanaoka H, Hashiguchi A, Konishi K, Ishii T, Kuwana M. A rare association between Fabry’s disease and granulomatosis with polyangiitis: a potential pathogenic link. BMC Nephrol. 2014;15(1):157. doi:https://doi.org/10.1186/1471-2369-15-157.
- Ren H, Li L, Yu J, et al. Fabry disease and immunoglobulin A nephropathy presenting with Alport syndrome-like findings: a case report. Medicine (Baltimore). 2019;98(28):e16256. doi:https://doi.org/10.1097/MD.0000000000016256.
- Chao CT, Lin WC, Kao TW. Fabry disease and immunoglobulin A nephropathy. Nephrology (Carlton). 2012;17(8):782–783. doi:https://doi.org/10.1111/j.1440-1797.2012.01594.x.
- Liu Y, Xie H, Lin HL, et al. Coexistence of Fabry disease and membranous nephropathy. Iran J Kidney Dis. 2016;10(1):48–50.
- Breiden B, Sandhoff K. Emerging mechanisms of drug-induced phospholipidosis. Biol Chem. 2019;401(1):31–46. doi:https://doi.org/10.1515/hsz-2019-0270.