
ABSTRACT
Huntington´s disease (HD) is a progressive neurodegenerative disease with onset in adulthood that leads to a complete disability and death in approximately 20 years after onset of symptoms. HD is caused by an expansion of a CAG triplet in the gene for huntingtin. Although the disease causes most damage to striatal neurons, other parts of the nervous system and many peripheral tissues are also markedly affected. Besides huntingtin malfunction, mitochondrial impairment has been previously described as an important player in HD. This study focuses on mitochondrial structure and function in cultivated skin fibroblasts from 10 HD patients to demonstrate mitochondrial impairment in extra-neuronal tissue. Mitochondrial structure, mitochondrial fission, and cristae organization were significantly disrupted and signs of elevated apoptosis were found. In accordance with structural changes, we also found indicators of functional alteration of mitochondria. Mitochondrial disturbances presented in fibroblasts from HD patients confirm that the energy metabolism damage in HD is not localized only to the central nervous system, but also may play role in the pathogenesis of HD in peripheral tissues. Skin fibroblasts can thus serve as a suitable cellular model to make insight into HD pathobiochemical processes and for the identification of possible targets for new therapies.
Introduction
Mitochondria are essential eukaryotic organelles composed of an outer membrane, inner membrane arranged into cristae, and an internal matrix. More than 90% of cellular ATP is synthesizedCitation1 in the oxidative phosphorylation system (OXPHOS) localized in the inner mitochondrial membrane.Citation2 ATP synthesis along with many other functions like purine synthesis, amino acid metabolism, fatty acid oxidation, or urea cycle running in mitochondria make them central metabolic organelles in eukaryotic cells.
Mitochondria create dynamic networks and their arrangement reflects the metabolic state of the cell and any disturbances can indicate pathological changes in mitochondrial functions.Citation3 Similarly, ultrastructural changes (low cristae number, mitochondrial swelling) also indicate mitochondrial disruption.Citation4 Besides primary mitochondrial disorders, caused by mutations in mitochondrial genes coded by mitochondrial DNA (mtDNA)Citation5 or nuclear DNA (nDNA),Citation6 there are disorders caused by mutations in non-mitochondrial genes, secondarily affecting mitochondrial metabolism. Such mitochondrial dysfunctions may act as important factors in the development of a disease pathologyCitation7 like Huntington’s chorea or Alzheimer’s disease.Citation8
The autosomal dominant Huntington’s disease (HD) is caused by an expansion of a CAG triplet (>36 repeats) in the first exon of the gene for the huntingtin protein (Htt).Citation9 The basic pathology mechanism is the misfolding of Htt and a consequent loss of its function caused by an extended N-terminal polyglutamine sequence.Citation10
Htt is expressed in all tissues; nevertheless, the highest levels were found in the brain and testes.Citation11 Although the function of Htt is still not fully understood, it has been shown to participate in synaptic transduction and neuronal stability, vesicular and organelle trafficking,Citation12 regulation of gene expression,Citation13 mitochondrial calcium handlingCitation14 or anti-apoptotic defense.Citation15 Moreover, in the mouse model, a confirmed Htt knockout leads to early embryonic lethality.Citation16
Symptoms of HD include progressive motor dysfunctions (chorea, dyskinesia, and dystonia) and psychiatric disturbances (cognitive decline, changes in personality, mood, and aggression).Citation17
Despite the intensive research of HD, there is not yet a known cure. Recently, the new gene therapy for the disease is tested in experimental animalsCitation18 and at present under investigation in humans.Citation19 Several independent clinical trials for HD gene therapy are ongoing in Phase III.Citation20 Unfortunately, some of them had to be stopped due to the negative impact of the normal htt protein.Citation21 All therapeutic approaches will require time-consuming testing, where all possible tools will be needed, such as human cultured cells of various origins, including skin fibroblasts.
Htt malfunction affects a broad spectrum of cellular pathways with the most pathologic impact on the central nervous system, especially in the striatum.Citation22 HD pathology is detectable also in peripheral tissues, for example, in the testes, blood, skeletal muscle, or fibroblasts.Citation23,Citation24
The connection between HD and mitochondrial impairment was demonstrated on a decrease of mitochondrial energy metabolism,Citation25,Citation26 higher level of reactive oxygen species,Citation27 calcium imbalance,Citation28 and a malfunction of the pyruvate dehydrogenase complex (PDHc).Citation29 Moreover, these mitochondrial changes in HD are detectable not only in the brain, but also in many other non-neural tissues like muscle,Citation30 heart,Citation31 testesCitation32 in TgHD models, or in muscle,Citation33 heart,Citation34 and peripheral bloodCitation35 in HD patients, respectively.
Publications focusing on the mitochondrial status in HD human peripheral tissue, such as muscle,Citation36 blood (lymphocytes),Citation35 or fibroblastsCitation37,Citation38 are scarce since these tissues from HD patients are not readily available. We studied mitochondrial impairment in cultivated skin fibroblasts from a group of 10 symptomatic heterozygous HD patients with the classical form of the disease, comparing them to healthy age-matched controls.
We observed decreased cristae number, lower mitochondrial density and a decreased amount of selected mitochondrial proteins (Optic atrophy protein 1 (Opa1), PDH 1-α subunit and Complex I NDUFA9 subunit) in patients’ fibroblasts. In concordance with the ultrastructural changes, we also detected functional impairment of the mitochondria.
Material and methods
Patient group
Ten lines of cultivated skin fibroblasts from symptomatic patients (P1 – P10) with confirmed HD were included in the experimental group (8 men, 2 women – P4, P6). All patients were heterozygotes with the classical form of the disease. CAG repeat number at the mutated allele varied between 42 and 62. Clinical symptoms, age of onset and sex are summarized in .
Table 1. Main characteristics of HD patients analyzed in experimental group.
The control group consists of 15 fibroblast lines obtained from healthy volunteers (9 men, 6 women aged between 21 and 50 years).
All work involving human samples was carried out in accordance with the Declaration of Helsinki of 1975 and was approved by the Ethical Committee of the General University Hospital in Prague, approval No. 115/15 Grant COST MSMT-COST 1. LF UK 29.9.2015 and No. 14/19 Grant AZV VES 2020 VFN 23.5.2019. The written informed consent was obtained from patients as well as control subjects prior to inclusion in the study.
Cultured fibroblasts
Cultivated skin fibroblasts were established from skin biopsy. Fibroblasts of all patients and controls were cultivated in Dulbecco’s modified Eagle’s medium (DMEM, Sigma Aldrich – Merck, Darmstadt, Germany) with 10% fetal bovine serum (FBS, Thermo Scientific, Waltham, MA) at 37°C under 5% CO2 atmosphere. Fibroblasts for enzymatic measurements were grown to approximately 80% confluence, harvested by incubation (10 min, 37°C) with TE (Trypsin, 0.05% (w/V); EDTA (0.02%, w/V)), washed and suspended in PBS (1.5 mM KH2PO4; 8 mM Na2HPO4 · 12H2O; 5.4 mM KCl; pH 7,2) and stored at −80°C until use.
Structural analyses
Transmission electron microscopy (TEM)
Fibroblasts were cultivated as mentioned previously to 90% confluence and fixed in phosphate-buffered saline (PBS) containing 2% (w/V) potassium permanganate for 15 min at room temperature (RT). After fixation, cells were dehydrated in an ethanol series (for 10 min in 50%, 70%, and 90% ethanol, for 30 min in 100% ethanol) at RT. Dehydrated cells were incubated in propylenoxide (2x15 min) and embedded in Durcupan Epon (Electron Microscopy Sciences, Hatfield, PA) (Durcupan Epon:Propyleonxide 1:1 for 2 hours, Durcupan Epon:Propylenoxide 1:3 overnight at 60°C). Polymerized blocks were sectioned by Ultracut III ultramicrotome (Reichert, Depew, NY) and 600–900 Å thick slices were stained with lead citrate and uranyl acetate.Citation4 Pictures were taken on a transmission electron microscope JEOL JEM-1200 EX.
The electron microscopic images were analyzed semi-quantitatively and results were organized into five different categories of pathologies observed. The data for analysis were collected from approximately 100 cells on one grid for each sample (the cell represents a compact unit with a nucleus). Studied categories were: Cristae disruption,Citation39 Low cristae number,Citation40 Mitochondrial swelling,Citation41 Degradation bodies,Citation23 Golgi complex structural abnormalities.Citation42
Fluorescence microscopy
Mitochondrial network staining: Fibroblasts from seven patients (P1-P7) and four control lines were cultivated on cover glasses in the DMEM medium and 10% FBS as described above. Mitochondria were stained using vital probe 10 nM MitoTracker Red CMX Ros (Molecular Probes, Eugene, OR) for 15 min, at 37°C. Cells were washed with PBS, mitochondrial network was detected by Nikon Diaphot 200 inverted microscope (Nikon, Tokyo, Japan) and pictures were taken with an Olympus DP50 CCD camera and Viewfinder Lite 1.0 software (Pixera, Santa Clara, CA). Mathematical analysis was carried out in the Fiji program (http://fiji.sc/Fiji). Two factors – mitochondrial density (MD) and the branching of mitochondria (branching factor, BF) were tested. MD characterizes the density of mitochondria per cells, BF compares the level of branching between mitochondrial networks in patients’ and control cells. The analysis was performed as published previously.Citation43 Obtained data were statistically assessed using a T-test.
Protein analysis (SDS-PAGE, Western blot, immunodetection)
Tricine SDS-PAGE was carried out under standard conditions with 12% polyacrylamide, 0.1% (w/v) SDS gels. Details are described in supplementary materials.
Functional analyses
Measurement of oxygen consumption in fibroblasts
The measurement was performed following a modified protocol of Kuznetsov et al.,Citation44 for details, see supplementary material. Results were normalized on protein concentration measured by the Lowry protocol.Citation45
Measurement of mitochondrial energy generating system capacity (MEGS)
The analysis was performed according to the Janssen et al. protocolCitation46 with [1−14C]pyruvate, [U-14C]malate and [1−14C]succinate as substrates. The composition of individual incubations is summarized in Table S1 (Supplementary material). Freshly harvested cells (cca 50 mg of wet pellet) were resuspended in 200 ul PBS and permeabilized by digitonin (0.3 µg/1 mg wet pellet, 20 min, 4°C). For one MEGS analysis, 30–50 ug of permeabilized cell suspension was used. The produced 14CO2 was collected on a filter paper soaked with NaOH and measured in a liquid scintillation counter.
Measurement of enzymatic activity of selected mitochondrial enzymes
Basal and activated enzymatic activity of the PDHc with 5 mM DCA was assayed as the release of 14CO2 from [1−14C]pyruvate.Citation47 The activities of NADH:coenzyme Q10 reductase (NQR, complex I), succinate:coenzyme Q10 reductase (SQR, complex II) and cytochrome c oxidase (COX, complex IV) and citrate synthase (CS) were measured spectrophotometrically according to Rustin et al.Citation48 About 80–100 µg of protein was used per one enzymatic assay. Total protein amount was determined by the method of LowryCitation45.
Statistics
MEGS and enzyme activity data were statistically analyzed by a linear model (Software R, version 3.2.3, R Core Team (2015). Three parameters were analyzed: HD, gender and passage of fibroblasts cultivation. For the quantification of mitochondrial network parameters student T-test was used.
Results
Structural analyses
Mitochondrial ultrastructure
Samples from all 10 patients and 5 control lines were analyzed by TEM to study the ultrastructure of cells. There were five cellular and/or mitochondrial abnormalities evaluated in the patient's cell lines compared to the controls (arranged in the representative ), controls in ). The three main pathological phenotypes across all patient's samples were: Cristae alteration (10/10), Low cristae number (10/10), Mitochondrial swelling (10/10) (). Furthermore, a higher number of degradation bodies (autophagic vacuoles) and lysosomes with accumulated material were detected in P2, P3, P4, and P7 cell lines, and abnormalities of Golgi complex architecture were observed in P2, P3, P5, P7, and P8. In the latter samples, the typical Golgi stacks of cisternae were disorganized into a higher number of vesicles (Golgi bodies) (). This semi-quantitative analysis was performed by comparing approximately 100 cells from each sample (1 grid); the presence and frequency of pathological markers in patient's fibroblasts are summarized in .
Figure 1. Ultrastructural alterations in fibroblasts of HD patients in comparison to controls. Pathological changes in ultrastructure of mitochondria and Golgi complex in fibroblasts of patients with HD visualized by transmission electron microscopy (TEM). Pictures were taken on a transmission electron microscope JEOL JEM-1200 EX. a) Normal mitochondria in adult control fibroblasts, b) Cristae disruption (P7), c) Low cristae number, marked by # (P3), d) Mitochondrial swelling, marked by * (P8), e) Degradation bodies, marked by < (P2), f) Structural abnormalities of Golgi complex (P2). Original magnification: 15 000 x (A-E), 25 000 x (f).
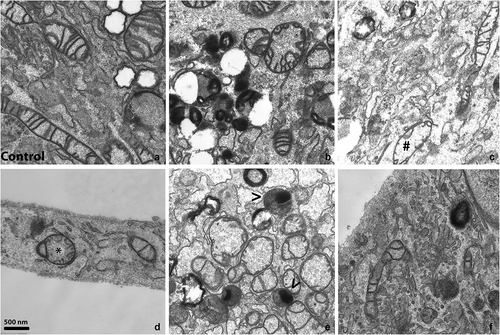
Table 2. Semi-quantitative analysis of ultrastructural alterations in fibroblasts of 10 HD patients. Columns present most common disturbances detected in patient’s fibroblasts. The number of crosses (+) defines the level of impairment in category, (-) the phenotype was not detected in the sample. Data were obtained in approximately 100 cells on one grid, where a compact unit with nucleus was considered as a cell.
Mitochondrial network
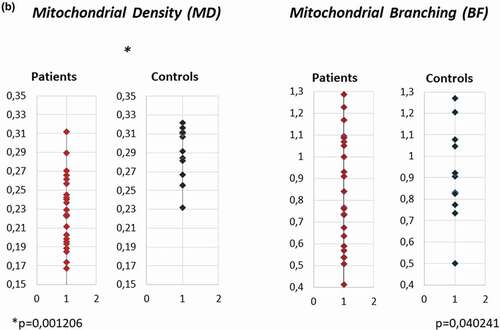
Native dying of the mitochondrial network in fibroblasts from P1-P7 and four controls showed unequally distributed mitochondria in patient's samples. A mild network dissipation with isolated mitochondrial spots (in , P2, P6, P7) was present. Mitochondrial density per cell (MD) analyzed in the Fiji program in patient's and control samples showed a significant decrease of MD in patient's cells (p = 0.0012). The branching factor (BF) was mildly decreased in patient's samples (). a)
Figure 2. Mitochondrial network visualization and quantitative analysis of mitochondrial density (MD) and branching factor (BF) in fibroblasts of 7 HD patients (P1 – P7) and 4 controls. a) Native dying of mitochondrial network in fibroblasts of HD patients P1-P7 and 4 controls: Cultivated skin fibroblasts were visualized in 10 nM MTR (MitoTracker RED, Molecular probes, Eugene, OR) 15 min at 37°C. Unequal distribution of mitochondrial network and mild fragmentation were detected in all 7 patients in comparison to controls, although the level of disruption was different in different lines. Original magnification: 600x. b) Mitochondrial density (MD) and Branching factor (BF) analysis in mitochondrial network in fibroblasts of 7 HD patients (P1 – P7) and 4 controls. quantitative analysis was provided in Fiji program. T-test was used for statistical analysis. MD and BF were significantly decreased in patient´s cells compared to controls (p = 0.0051) (p = 0.014),respectively.b)
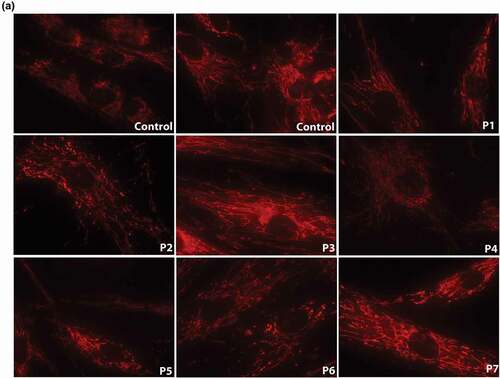
Figure 2. (Continued).
Protein analysis of selected mitochondrial proteins
A spectrum of selected subunits from OXPHOS complexes (Complex I: NDUFA9, NDUFB6, Complex II: SDH70, SDH30, Complex III: Core2, Complex IV: Cox1, Cox2, CV: F1- α), PDH complex (E2, 1-α) were immunodetected in whole-cell lysates after SDS PAGE/WB (supplementary Figure S1). Evaluation of the results is summarized in . Patient's samples showed remarkably decreased levels of the NDUFA9, PDH 1- α subunit and the Opa 1 protein in comparison to controls. The complex I subunit NDUFA9 is reduced in P2-P8 and P10 (26–78% of control values). P1 and P9 showed normal levels of NDUFA9 subunit. The PDH complex protein analysis showed the decrease of PDH E1-α subunit in P1-P8 fibroblasts (34–72% of control values). P9 level was normal and P10 was not analyzed for this protein.
Table 3. Quantification of selected mitochondrial proteins expression in fibroblasts of HD patients and controls. SDS PAGE/WB signal (see Supplementary figure 1) in whole-cell lysates was quantified in G:Box (SynGene imaging system, SynGene UK). Values are in percentage, the average of signal of two adult controls is expressed as 100% of control value. Patient’s values ≤ 80% of controls are red, 120% ≥ in green. Results were normalized to total protein loading and α-tubulin protein. nd – not analyzed.
Three proteins important for the modulation of mitochondrial structure and the fusion/fission process were also analyzed: mitofilin, opa1 and drp1. Mitofilin showed normal levels except for P2, P9, and P10, where the levels decreased to as low as to 23.5% of control values (). Opa1 levels varied between 66.5% and 79.5% of the control value in 8 cases (P1-P3, P5-P7, P9, and P10). Drp1 protein levels were increased in P3, P4, P5, P7, and P8. Although the expression of Htt in fibroblasts was demonstrated earlier,Citation49 we verified this using an anti-Htt antibody (Mab-2166, Milipore), confirming the presence of the Htt protein in two of our control lines (data not shown).
Functional analyses
Measurement of oxygen consumption
Mitochondrial respiration of digitonin-permeabilized fibroblasts was measured in six patients (P1, P3, P4, P5, P6, and P10) and three adult controls. Results were normalized to protein concentration. A mild decrease of respiration after the addition of substrates for complex I (glutamate + malate) and complex II (succinate) and complex IV were detected in fibroblasts from P3, P4, and P5 compared to controls. The oxygen consumption parameters in various respiratory conditions are summarized in .
Figure 3. Alteration of mitochondrial functional parameters in fibroblasts from HD patients. a) Measurement of oxygen consumption. The ratios of average values of oxygen consumption in fibroblasts showed lower values in most of tested patient´s lines in comparison to controls Mitochondrial energy generating system capacity, oxidation of substrate in reaction containing [U-14C]malate+acetylcarnitine+malonate+ADP (b) and [1–414C]succinate+acetylcarnitine+ADP (c) were decreased in HD cells; d) The activity of succinate – coenzyme Q reductase (SQR, complex II of respiratory chain) measured by spectrophotometry, showed a significant interaction of sex, HD and age (p = 0.0078), comparison related to HD and sex are shown. F- female, M- male. Ee) The activity of succinate – coenzyme Q reductase (SQR, complex II of respiratory chain) normalized to activity of citrate synthase (SQR/CS) was associated with sex (p = 0.0213) having a significant interaction of sex, HD and age (p = 0.0188). Comparison related to HD and sex are shown. Details of the effect of age are given in supplement, see Figure S4 and S5.
![Figure 3. Alteration of mitochondrial functional parameters in fibroblasts from HD patients. a) Measurement of oxygen consumption. The ratios of average values of oxygen consumption in fibroblasts showed lower values in most of tested patient´s lines in comparison to controls Mitochondrial energy generating system capacity, oxidation of substrate in reaction containing [U-14C]malate+acetylcarnitine+malonate+ADP (b) and [1–414C]succinate+acetylcarnitine+ADP (c) were decreased in HD cells; d) The activity of succinate – coenzyme Q reductase (SQR, complex II of respiratory chain) measured by spectrophotometry, showed a significant interaction of sex, HD and age (p = 0.0078), comparison related to HD and sex are shown. F- female, M- male. Ee) The activity of succinate – coenzyme Q reductase (SQR, complex II of respiratory chain) normalized to activity of citrate synthase (SQR/CS) was associated with sex (p = 0.0213) having a significant interaction of sex, HD and age (p = 0.0188). Comparison related to HD and sex are shown. Details of the effect of age are given in supplement, see Figure S4 and S5.](/cms/asset/828af83e-66e1-4792-a176-9aad3bf5cf99/iusp_a_2100951_f0003_b.gif)
Mitochondrial energy generating system capacity (MEGS)
MEGS analysis in cultivated skin fibroblasts from 7 HD patients (P1, P2, P4, P6, P7, P9, and P10) revealed a significantly decreased oxidation rate in an incubation containing [U-14C] malate + acetylcarnitine + malonate + ADP (). Moreover, we have verified that this parameter in the controls and patient lines does not differ between different passages (Supplementary Figure S2).
Measurement of OXPHOS and PDH complexes activity
Values of SQR (Complex II) activity and its normalized activity expressed as the ratio of SQR/CS measurement were lower in HD fibroblast samples in comparison to controls. Moreover, SQR showed a significant interaction of sex, HD, and age (p = 0.0078), (). Normalized activity SQR/CS was associated with sex (p = 0.0213), and had a significant interaction of sex, HD, and age (p =0.0188 ()). No correlation between enzyme activity and HD for complex NQR, COX and CS, and ratios COX/CS and NQR/CS was found. Correlation with age and sex was found for COX (supplementary Figure S3).
In contrast to lowered PDH E1-α subunit, measurement of basal and DCA activated PDHc activity did not show significant differences in patients compared to controls (data not shown).
Discussion
Our study presents a comprehensive analysis of cultured skin fibroblasts from 10 HD patients and 15 healthy controls to map how a mutated Htt influences mitochondrial structure and function in a non-neural tissue with a low energy demand.
Skin biopsy of our HD patients was done at different times after disease onset, between 1 year (P8) and 14 years (P5) after the first recorded symptom. Specific mitochondrial dysfunctions or structural changes were well detectable in all patient lines.
Changes in mitochondrial ultrastructure or network organization reflecting functional disturbances of these organelles are usually observed in primary mitochondrial diseases.Citation3 Dissipation of mitochondrial network in neurons was already described in HD,Citation50 Alzheimer’s diseaseCitation51 and mentioned in other neurodegenerative diseases, like Charcot-Marie-Tooth type 2A,Citation52 a peripheral neuropathy or dominant optic atrophy.Citation53 Squitieri’s work on mitochondrial ultrastructural changes in myoblasts and fibroblasts from 4 HD patients (2 homozygous, 2 heterozygous) described similar results and concluded that structural abnormalities are dose-dependent, because changes in the homozygous samples were more profound than in the heterozygous ones.Citation23
Consistent with these facts, we detected structural discrepancies, like mitochondrial swelling, decreased cristae number or cristae alteration by TEM in all HD patients’ fibroblasts but not in control lines. Patients’ mitochondria were distributed unequally through cells and their density (the area of mitochondria per cell) was lower, which was in accordance with previously published data from striatal cells from HD knock-in mice HdhQ111.Citation54 Some patients (P2, P5 in ) showed a slightly dissipated mitochondrial network, usually associated with pro-apoptotic signals in cells.Citation7 Additionally, TEM analysis showed that lines from P2-4 and P7 contained an increased level of degradation bodies, where cellular waste material is stored before its degradation. This fact, together with a mildly elevated drp1 (dynamin related protein 1; ), responsible for mitochondrial fission, and a decreased level of opa1 (), responsible for mitochondrial fusion, suggests that HD patients’ mitochondria in fibroblasts increase pro-apoptotic stimuli. Both drp1 and opa1 are proteins maintaining the integrity of the mitochondrial membrane.Citation55 Increased drp1 causes mitochondrial fragmentation, which produces ultrastructural changes in mitochondrial cristae promoting, cytochrome c release, and increasing sensitivity to apoptosis.Citation39,Citation56 Mitochondrial cristae are dynamic bioenergetic compartments whose shape determines the assembly and stability of respiratory chain complexes and therefore mitochondrial respiratory efficiency.Citation57 Thus, perturbations of the cristae integrity as a consequence of an imbalanced drp1, found in our cohort, could be responsible for the observed alterations in oxidative phosphorylation function. A similar situation was already described in TgHD minipig muscle, where the drp1 protein increased with development of the disease with parallely decreased respiratory chain complexes II and IV and a decreased oxidation rate of different substrates.Citation30 Aside from the pro-apoptotic signs, TEM revealed mitochondrial swelling and a decreased number of mitochondrial cristae. Our findings are in accordance with a previous report of mitochondrial swelling and cristae disruptions in primary striatal YAC128 mouse neurons and human HD neuronal cells.Citation39
Clearly, mitochondria do not function in isolation, but interact with other cell compartments, such as Golgi or endoplasmic reticulum through specialized membrane domains, signaling molecules (Ca2+) or proteins (drp1).Citation58 Mitochondrial disruption may thus affect other cellular compartments and the Golgi complex is no exception. A recent study, for example, described a reduced ATP production (induced by 2-deoxy-D-glucose treating) leading to reversible remodeling of Golgi architecture from compact cisternae to vesicular Golgi bodies in HepG2 cells.Citation42
Similarly to the mitochondrial network, structural changes of the Golgi apparatus during physiological/pathological processes like cell cycle,Citation59 neurodegenerationCitation60 or cellular ATP productionCitation42 were described. Research in the last decade has shown that the Golgi complex is responsible not only for the protein secretory pathway, but is also a highly dynamic organelle influenced by various cellular stimuli and also playing a role in HD pathogenesis. Dysfunctional mHtt disrupts vesicular transport from Golgi along microtubules and prevents the fusion of vesicles with the plasma membrane.Citation61 Lower ATP production or increased cellular stress lead to modulation of steroidogenesis or membrane lipid homeostasis via the Golgi protein ACBD3.Citation62 In HD, increased levels of the ACBD3 protein raise the levels of the ACBD3/mHtt/Rhes complex (Rhes, Ras homolog enriched in striatum, is a small GTPase), causing a higher cytotoxicity in HD via Rhes-induced sumoylation of mHtt.Citation63 We suggest that the observed changes in Golgi architecture in fibroblasts complement the ultrastructural and functional alteration of mitochondria caused by HD pathogenesis.
We analyzed a broad spectrum of mitochondrial proteins in fibroblasts by SDS PAGE/WB. The complex I subunit NDUFA9 (8/10) and the PDH complex subunit PDH 1-α (8/10) were decreased in most patients. Decreased NDUFA9, a subunit of complex I, together with decreased oxidation rate in the reaction containing malate as substrate () would suggest a possible impairment in the first step of the oxidative phosphorylation system in HD fibroblasts.
Similarly, a deficiency of complex I was described in the brain of 3-nitropropionic acid-induced rat model of Huntington’s disease,Citation64 HD patients,Citation65 as well as in the TgHD minipig and the skeletal muscle of R6/2 mice.Citation30,Citation66
Surprisingly, the decreased PDH 1-α subunit levels were not associated with a reduction in enzymatic activity of the PDH complex (Data not shown). We can thus assume that the threshold for PDH protein expression has not been exceeded and that PDH in the fibroblasts has a sufficient reserve in enzymatic capacity, able to cover the cell’s demands even with a reduced content of some subunits.
Decreased expression of the SDH30 complex II subunit was observed in 5/10 HD patients, together with a decreased oxidation rate in the incubation containing succinate () compared to controls. Complex II of the respiratory chain has been shown affected several times in various tissues of HD patients – in the striatum,Citation67–69 peripheral lymphocytes,Citation35 and skeletal muscle,Citation33 as well as in HD models – in the mouse brainCitation70,Citation71 and skeletal muscle.Citation30 However, our study has shown that the enzymatic activity of Complex II in fibroblasts significantly depends on sex and age (), so similar studies would need to be performed on a larger group of patients to support the significance of our findings. Functional insufficiency of mitochondria also causes lower respiration after the addition of substrates for complexes I, II, and IV of OXPHOS in most patients compared to control lines ().
The number of patients and some technique used represent the limitations of the study. 3D images from confocal microscopy could provide considerably more precise information about the mitochondrial network than only 2D section analysis. The classical techniques we used could perhaps benefit from a contribution from other multi-omics data or additional clinical patient data. A model study in human fibroblasts has been already used in patients with Parkinson’s disease to identify complex disease signatures.Citation72 The diverse intensity of mitochondrial ultrastructural and functional defects in HD fibroblasts analyzed in this work can also be partially attributed to different genetic backgrounds of individual lines. Using a larger set of lines could reveal individual variation suggesting the presence of morphological features inherent to specific individuals. Such insights may uncover individual or patient-specific cellular phenotypes, with valuable implications for personalized medicine.
Conclusion
In conclusion, we have shown ultrastructural impairment and functional insufficiency of mitochondria in human cultured HD fibroblasts. Results of our study are valuable for the investigation of the particular pathobiochemical processes in Huntington’s disease in which mitochondria play a significant role, such as affected autophagy/mitophagy.Citation73,Citation74 Our results may also serve as the pilot for an evaluation of possible biomarkers that may either be helpful to monitor the course of the disease or serve as a target for the new therapies.
Authors contributions
Acquisition, analysis, and interpretation of data MV, HS, MK, JK, TD; drafting the manuscript MV, HH; drafting figures MV, HS, others – recruitment of patients, collection of biological material JK, IR, JR; revising the manuscript JZ, HH
Suppl_Figure_S5i.tif
Download TIFF Image (1.1 MB)Suppl_Figure_S4i.tif
Download TIFF Image (1.2 MB)Suppl_Figure_S3ii.tif
Download TIFF Image (939.8 KB)Suppl_Figure_S2i.tif
Download TIFF Image (1.1 MB)Suppl_Figure_S1i.tif
Download TIFF Image (3.9 MB)Suppl_Table_S1i.tif
Download TIFF Image (1.6 MB)Supplementary_material_methods.docx
Download MS Word (18.7 KB)Acknowledgments
The authors are grateful to Dr. Vaclav Capek for the statistical analysis of the data.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary Material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/01913123.2022.2100951
Additional information
Funding
References
- Scheffler IE. Structure and Morphology. Integration into the Cell. Mitochondria: John Wiley & Sons, Inc; 2007:18–59.
- Zalman LS, Nikaido H, Kagawa Y. Mitochondrial outer membrane contains a protein producing nonspecific diffusion channels. J Biol Chem. 1980;255(5):1771–1774. doi:10.1016/S0021-9258(19)85942-2.
- Koopman WJ, Visch HJ, Verkaart S, van den Heuvel LW, Smeitink JA, Willems PH. Mitochondrial network complexity and pathological decrease in complex I activity are tightly correlated in isolated human complex I deficiency. Am J Physiol Cell Physiol. 2005;289(4):C881–90. doi:10.1152/ajpcell.00104.2005.
- Brantova O, Tesarova M, Hansikova H, Elleder M, Zeman J, Sladkova J. Ultrastructural changes of mitochondria in the cultivated skin fibroblasts of patients with point mutations in mitochondrial DNA. Ultrastruct Pathol. 2006;30(4):239–245. doi:10.1080/01913120600820112.
- Zhu DP, Economou EP, Antonarakis SE, Maumenee IH. Mitochondrial DNA mutation and heteroplasmy in type I Leber hereditary optic neuropathy. Am J Med Genet. 1992;42(2):173–179. doi:10.1002/ajmg.1320420208.
- DiMauro S, Bonilla E, Zeviani M, Nakagawa M, DeVivo DC. Mitochondrial myopathies. Ann Neurol. 1985;17(6):521–538. doi:10.1002/ana.410170602.
- Lionaki E, Markaki M, Palikaras K, Tavernarakis N. Mitochondria, autophagy and age-associated neurodegenerative diseases: new insights into a complex interplay. Biochim Biophys Acta. 2015;1847(11):1412–1423. doi:10.1016/j.bbabio.2015.04.010.
- Hedskog L, Zhang S, Ankarcrona M. Strategic role for mitochondria in Alzheimer’s disease and cancer. Antioxid Redox Signal. 2012;16(12):1476–1491. doi:10.1089/ars.2011.4259.
- Trottier Y, Lutz Y, Stevanin G, et al. Polyglutamine expansion as a pathological epitope in Huntington’s disease and four dominant cerebellar ataxias. Nature. 1995;378(6555):403–406. doi:10.1038/378403a0.
- MacDonald ME, Ambrose CM, Duyao MP, et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72(6):971–983. doi:10.1016/0092-8674(93)90585-E.
- Sharp AH, Loev SJ, Schilling G, et al. Widespread expression of Huntington’s disease gene (IT15) protein product. Neuron. 1995;14(5):1065–1074. doi:10.1016/0896-6273(95)90345-3.
- Trushina E, Dyer RB, Badger JD 2nd, et al. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol Cell Biol. 2004;24(18):8195–8209. doi:10.1128/MCB.24.18.8195-8209.2004.
- Huang B, Wei W, Wang G, et al. Mutant huntingtin downregulates myelin regulatory factor-mediated myelin gene expression and affects mature oligodendrocytes. Neuron. 2015;85(6):1212–1226. doi:10.1016/j.neuron.2015.02.026.
- Giacomello M, Hudec R, Lopreiato R. Huntington’s disease, calcium, and mitochondria. Biofactors. 2011;37(3):206–218. doi:10.1002/biof.162.
- Hansson O, Castilho RF, Korhonen L, Lindholm D, Bates GP, Brundin P. Partial resistance to malonate-induced striatal cell death in transgenic mouse models of Huntington’s disease is dependent on age and CAG repeat length. J Neurochem. 2001;78(4):694–703. doi:10.1046/j.1471-4159.2001.00482.x.
- Nasir J, Floresco SB, O’Kusky JR, et al. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81(5):811–823. doi:10.1016/0092-8674(95)90542-1.
- Roos RAC. Huntington’s disease: a clinical review. Orphanet J Rare Dis. 2010;5(1):40. doi:10.1186/1750-1172-5-40.
- Valles A, Evers MM, Stam A, et al. Widespread and sustained target engagement in Huntington’s disease minipigs upon intrastriatal microRNA-based gene therapy. Sci Transl Med. 2021;13(588):588. doi:10.1126/scitranslmed.abb8920.
- Shapiro L. First 2 groups enrolled in phase 1/2 trial of gene therapy AMT-130.March 22 2022. Available from: https://huntingtonsdiseasenews.com/2022/03/22amt-130-trial-huntingtons-gene-therapy-fully-enrolled-first-2-groups/.
- Byun S, Lee M, Kim M. Gene therapy for Huntington’s disease: the final strategy for a cure? J Mov Disord. 2022;15(1):15–20. doi:10.14802/jmd.21006.
- Gardner J. Roche stops giving Huntington’s disease drug in closely watched study.[cited 2021 Mar 23] Available from: www.biopharmadive.com/news/rochestop-dosing-huntington-tominersen-ionis/597171.
- Vonsattel JPG, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57(5):369–384. doi:10.1097/00005072-199805000-00001.
- Squitieri F, Falleni A, Cannella M, et al. Abnormal morphology of peripheral cell tissues from patients with Huntington disease. J Neural Transm. 2010;117(1):77–83. doi:10.1007/s00702-009-0328-4.
- Ciammola A, Sassone J, Sciacco M, et al. Low anaerobic threshold and increased skeletal muscle lactate production in subjects with Huntington’s disease. Mov Disord. 2011;26(1):130–137. doi:10.1002/mds.23258.
- Milakovic T, Johnson GV. Mitochondrial respiration and ATP production are significantly impaired in striatal cells expressing mutant huntingtin. J Biol Chem. 2005;280(35):30773–30782. doi:10.1074/jbc.M504749200.
- Guidetti P, Charles V, Chen EY, et al. Early degenerative changes in transgenic mice expressing mutant huntingtin involve dendritic abnormalities but no impairment of mitochondrial energy production. Exp Neurol. 2001;169(2):340–350. doi:10.1006/exnr.2000.7626.
- Browne SE, Ferrante RJ, Beal MF. Oxidative stress in Huntington’s disease. Brain Pathol. 1999;9(1):147–163. doi:10.1111/j.1750-3639.1999.tb00216.x.
- Jiang R, Diaz-Castro B, Looger LL, Khakh BS. Dysfunctional calcium and glutamate signaling in striatal astrocytes from huntington’s disease model mice. J Neurosci. 2016;36(12):3453–3470. doi:10.1523/JNEUROSCI.3693-15.2016.
- Sorbi S, Bird ED, Blass JP. Decreased pyruvate dehydrogenase complex activity in Huntington and Alzheimer brain. Ann Neurol. 1983;13(1):72–78. doi:10.1002/ana.410130116.
- Rodinova M, Krizova J, Stufkova H, et al. Deterioration of mitochondrial bioenergetics and ultrastructure impairment in skeletal muscle of a transgenic minipig model in the early stages of Huntington’s disease. Dis Model Mech. 2019;12:7.
- Mielcarek M. Huntington’s disease is a multi-system disorder. Rare Diseases. 2015;3(1):e1058464.doi:10.1080/21675511.2015.1058464.
- Macakova M, Bohuslavova B, Vochozkova P, et al. Mutated huntingtin causes testicular pathology in transgenic minipig boars. Neurodegener Dis. 2016;16(3–4):245–259. doi:10.1159/000443665.
- Gehrig SM, Petersen JA, Frese S, et al. Skeletal muscle characteristics and mitochondrial function in Huntington’s disease patients. Mov Disord. 2017;32(8):1258–1259. doi:10.1002/mds.27031.
- Cankar K, Melik Z, Kobal J, Starc V. Evidence of cardiac electrical remodeling in patients with Huntington disease. Brain Behav. 2018;8(8):e01077–e. doi:10.1002/brb3.1077.
- Askeland G, Dosoudilova Z, Rodinova M, et al. Increased nuclear DNA damage precedes mitochondrial dysfunction in peripheral blood mononuclear cells from Huntington’s disease patients. Sci Rep. 2018;8(1):9817. doi:10.1038/s41598-018-27985-y.
- Kosinski CM, Schlangen C, Gellerich FN, et al. Myopathy as a first symptom of Huntington’s disease in a Marathon runner. Movement Disorders. 2007;22(11):1637–1640. doi:10.1002/mds.21550.
- Gardiner SL, Milanese C, Boogaard MW, et al. Bioenergetics in fibroblasts of patients with Huntington disease are associated with age at onset. Neurol Genet. 2018;4(5):e275–e. doi:10.1212/NXG.0000000000000275.
- Jędrak P, Mozolewski P, Węgrzyn G, Więckowski MR. Mitochondrial alterations accompanied by oxidative stress conditions in skin fibroblasts of Huntington’s disease patients. Metab Brain Dis. 2018;33(6):2005–2017. doi:10.1007/s11011-018-0308-1.
- Costa V, Giacomello M, Hudec R, et al. Mitochondrial fission and cristae disruption increase the response of cell models of Huntington’s disease to apoptotic stimuli. EMBO Mol Med. 2010;2(12):490–503. doi:10.1002/emmm.201000102.
- Olichon A, Baricault L, Gas N, et al. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem. 2003;278(10):7743–7746. doi:10.1074/jbc.C200677200.
- Park JY, Jang SY, Shin YK, et al. Mitochondrial swelling and microtubule depolymerization are associated with energy depletion in axon degeneration. Neuroscience. 2013;238:258–269. doi:10.1016/j.neuroscience.2013.02.033.
- Ranftler C, Meisslitzer-Ruppitsch C, Neumüller J, Ellinger A, Pavelka M. Golgi apparatus dis- and reorganizations studied with the aid of 2-deoxy-D-glucose and visualized by 3D-electron tomography. Histochem Cell Biol. 2017;147(4):415–438. doi:10.1007/s00418-016-1515-7.
- Sladkova J, Spacilova J, Capek M, et al. Analysis of mitochondrial network morphology in cultured myoblasts from patients with mitochondrial disorders. Ultrastruct Pathol. 2015;39(5):340–350. doi:10.3109/01913123.2015.1054013.
- Kuznetsov AV, Veksler V, Gellerich FN, Saks V, Margreiter R, Kunz WS. Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat Protoc. 2008;3(6):965–976. doi:10.1038/nprot.2008.61.
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem. 1951;193(1):265–275. doi:10.1016/S0021-9258(19)52451-6.
- Janssen AJ, Trijbels FJ, Sengers RC, et al. Measurement of the energy-generating capacity of human muscle mitochondria: diagnostic procedure and application to human pathology. Clin Chem. 2006;52(5):860–871. doi:10.1373/clinchem.2005.062414.
- Magner M, Vinsova K, Tesarova M, et al. Two patients with clinically distinct manifestation of pyruvate dehydrogenase deficiency due to mutations in PDHA1 gene. Prague Medical Report. 2011;112(1):18–28.
- Rustin P, Chretien D, Bourgeron T, et al. Biochemical and molecular investigations in respiratory chain deficiencies. Clin Chim Acta. 1994;228(1):35–51. doi:10.1016/0009-8981(94)90055-8.
- Marques Sousa C, Humbert S. Huntingtin: here, there, everywhere! Journal of Huntington’s Disease. 2013;2(4):395–403. doi:10.3233/JHD-130082.
- Shirendeb U, Reddy AP, Manczak M, et al. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: implications for selective neuronal damage. Hum Mol Genet. 2011;20(7):1438–1455. doi:10.1093/hmg/ddr024.
- Sheng B, Wang X, Su B, et al. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J Neurochem. 2012;120(3):419–429. doi:10.1111/j.1471-4159.2011.07581.x.
- Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130(3):548–562. doi:10.1016/j.cell.2007.06.026.
- Chen H, Chan DC. Mitochondrial dynamics-fusion, fission, movement, and mitophagy-in neurodegenerative diseases. Hum Mol Genet. 2009;18(R2):R169–76. doi:10.1093/hmg/ddp326.
- Jin YN, Yu YV, Gundemir S, et al. Impaired mitochondrial dynamics and Nrf2 signaling contribute to compromised responses to oxidative stress in striatal cells expressing full-length mutant huntingtin. PLoS One. 2013;8(3):1.
- Hering T, Kojer K, Birth N, Hallitsch J, Taanman J-W, Orth M. Mitochondrial cristae remodelling is associated with disrupted OPA1 oligomerisation in the Huntington’s disease R6/2 fragment model. Exp Neurol. 2017;288:167–175. doi:10.1016/j.expneurol.2016.10.017.
- Jodeiri Farshbaf M, Ghaedi GK. Huntington’s disease and mitochondria. Neurotox Res. 2017;32(3):518–529. doi:10.1007/s12640-017-9766-1.
- Cogliati S, Enriquez JA, Scorrano SL. Mitochondrial cristae: where beauty meets functionality. Trends Biochem Sci. 2016;41(3):261–273. doi:10.1016/j.tibs.2016.01.001.
- Bravo-Sagua R, Torrealba N, Paredes F, et al. Organelle communication: signaling crossroads between homeostasis and disease. Int J Biochem Cell Biol. 2014;50:55–59. doi:10.1016/j.biocel.2014.01.019.
- Villeneuve J, Scarpa M, Ortega-Bellido M, Malhotra V. MEK1 inactivates Myt1 to regulate golgi membrane fragmentation and mitotic entry in mammalian cells. EMBO J. 2012;32(1):72–85. doi:10.1038/emboj.2012.329.
- Fan J, Hu Z, Zeng L, et al. Golgi apparatus and neurodegenerative diseases. International Journal of Developmental Neuroscience. 2008;26(6):523–534. doi:10.1016/j.ijdevneu.2008.05.006.
- Brandstaetter H, Kruppa AJ, Buss F. Huntingtin is required for ER-to-Golgi transport and for secretory vesicle fusion at the plasma membrane. Dis Model Mech. 2014;7(12):1335–1340. doi:10.1242/dmm.017368.
- Yue Y, Qian Q, Gim G, Acyl-CoA-Binding Domain-Containing LI. 1School of Life Science and Technology, ShanghaiTech University, Pudong, Shanghai 201210, China; [email protected] (X.Y.); [email protected] (Y.Q.). Int J Mol Sci. 2019;20(8):2028. doi:10.3390/ijms20082028.
- Subramaniam S, Sixt KM, Barrow R, Snyder SH. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science (New York, NY). 2009;324(5932):1327–1330. doi:10.1126/science.1172871.
- Pandey M, Varghese M, Sindhu KM, et al. Mitochondrial NAD+-linked State 3 respiration and complex-I activity are compromised in the cerebral cortex of 3-nitropropionic acid-induced rat model of Huntington’s disease. J Neurochem. 2008;104(2):420–434.
- Arenas J, Campos Y, Ribacoba R, et al. Complex I Defect in muscle from patients with Huntington’s disease. Ann Neurol. 1998;43(3):397–400. doi:10.1002/ana.410430321.
- Kojer K, Hering T, Bazenet C, et al. Huntingtin aggregates and mitochondrial pathology in skeletal muscle but not heart of late-stage R6/2 mice. Journal of Huntington’s Disease. 2019;8(2):145–159. doi:10.3233/JHD-180324.
- Benchoua A, Trioulier Y, Zala D, et al. Involvement of mitochondrial complex II defects in neuronal death produced by N-terminus fragment of mutated huntingtin. Mol Biol Cell. 2006;17(4):1652–1663. doi:10.1091/mbc.e05-07-0607.
- Browne SE, Bowling AC, Macgarvey U, Browne SE, Bowling AC, MacGarvey U, Baik MJ, Berger SC, Muqit MM, et al. Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann Neurol. 1997;41(5):646–653. doi:10.1002/ana.410410514.
- Gu M, Gash MT, Mann VM, Javoy-Agid F, Cooper JM, Schapira AH. Mitochondrial defect in Huntington’s disease caudate nucleus. Ann Neurol. 1996;39(3):385–389. doi:10.1002/ana.410390317.
- Damiano M, Diguet E, Malgorn C, et al. A role of mitochondrial complex II defects in genetic models of Huntington’s disease expressing N-terminal fragments of mutant huntingtin. Hum Mol Genet. 2013;22(19):3869–3882. doi:10.1093/hmg/ddt242.
- Naseri NN, Xu H, Bonica J, et al. Abnormalities in the tricarboxylic acid cycle in Huntington disease and in a Huntington disease mouse model. J Neuropathol Exp Neurol. 2015;74(6):527–537. doi:10.1097/NEN.0000000000000197.
- Schiff L, Migliori B, Chen Y, et al. Integrating deep learning and unbiased automated high-content screening to identify complex disease signatures in human fibroblasts. Nat Commun. 2022;13(1):1590. doi:10.1038/s41467-022-28423-4.
- Sonsky I, Vodicka P, Vodickova Kepkova K, Hansikova H. Mitophagy in Huntington’s disease. Neurochem Int. 2021;149:105147. doi:10.1016/j.neuint.2021.105147.
- Traa A, Machiela E, Rudich PD, Soo SK, Senchuk MM, Van Raamsdonk JM. Identification of novel therapeutic targets for polyglutamine diseases that target mitochondrial fragmentation. Int J Mol Sci. 2021;22(24):24. doi:10.3390/ijms222413447.