Abstract
Fibroblastic and myofibroblastic tumors in neonates, infants, and children provide a diagnostic dilemma in surgical pathology due to their relative rarity and similarity in appearances. These tumors may be congenital or occur early during the first years of life or later during the first and second decades of life. The morphologic, immunocytochemical, ultrastructural, cytogenetic, and molecular features of the more “common” pediatric fibroblastic and myofibroblastic tumors are reviewed. In addition, the importance of a multimodal approach to tumor diagnosis is emphasized, with correlation with treatment and outcome differences among these unique fibroblastic and myofibroblastic tumors. The importance of providing an accurate diagnosis with pediatric fibroblastic and myofibroblastic tumors cannot be overstated, because treatment, prognosis, follow-up, and outcome are based on the initial assessment of these fascinating, but oftentimes, perplexing tumors.
Keywords:
When one considers soft tissue tumors in pediatrics, tumors of vascular (29%), neurogenic (15%), and myogenic (striated muscle, 14%) origin occur more often than fibroblastic–myofibroblastic tumors (12%) [Citation[1–8]]. The spectrum of fibroblastic/myofibroblastic tumors is quite divergent from both clinical and histopathologic viewpoints (). These tumors are most commonly benign, but may follow an aggressive course that mimics a malignant process. The only “true” pediatric fibroblastic malignancy is infantile fibrosarcoma, which has a characteristic tumor-defining translocation in most cases. Other fibroblastic/myofibroblastic tumors may be associated with congenital syndromes (inclusion body fibromatosis and fibrous hamartoma of infancy) and a predisposition to adenomatous polyps and colon cancer (Gardner-associated fibromas and desmoids [Citation[7]]). The major problem for the surgical pathologist is the similarity between myofibromas and infantile fibrosarcomas and the vast difference in treatment of myofibromas versus infantile fibrosarcoma. This presentation is limited to infantile fibrosarcoma, myofibroma/myofibromatosis, a newly recognized entity—pericytic tumor with t(7;12), inclusion body fibromatosis, fibrous hamartoma of infancy, juvenile hyaline fibromatosis, and fibrodysplasia ossificans progressiva.
Table 1 Tumors of Fibroblastic and Myofibroblastic Origin in Children and Adolescents
TRIAGING OF FIBROBLASTIC AND MYOFIBROBLASTIC TUMOR TISSUE
A general schema for triaging tissue from fibroblastic/myofibroblastic tumors is presented in [Citation[8]]. Paramount to evaluation of these tumors is providing adequate tissue for intraoperative interpretation and final diagnosis. Perhaps, it should be emphasized that glutaraldehyde-fixed tissue should be set aside for electron microscopy. Often with soft tissue tumors due to nonspecific, aberrant, and spurious immunocytochemical findings, ultrastructural examination will provide the clues necessary to provide a definitive diagnosis. Once it has been determined that adequate tissue has been obtained for diagnosis, residual tissue may be designated for cytogenetics, molecular analysis, and biologic study. In general, submission of fresh tissue for cytogenetics, and retention of the frozen section tissue block at − 70°C for molecular, RT-PCR, biochemical, and microarray gene product analyses will allow for appropriate assessment and further characterization of the tumor. The preparation of cytologic imprints from fresh tissue can be performed prior to submitting the tissue for other protocol studies. Cytologic imprints allow for fluorescent in situ hybridization (FISH) evaluation of mutated genes, tumor-defining translocations, increased oncogene copy number (amplification), and other cytogenetic abnormalities.
Table 2 Triaging of Fibroblastic and Myofibroblastic Tumors in Children and Adolescents
There are several recently initiated protocols by the Children's Oncology Group dealing with infantile fibrosarcoma and various fibroblastic/myofibroblastic tumors. Communication with the local or regional pediatric oncologist and tumor protocol coordinator should ensure submission of the tissue required for protocol enrollment based upon the biopathology of the tumor.
INFANTILE FIBROSARCOMA (CONGENITAL FIBROSARCOMA, CONGENITAL INFANTILE FIBROSARCOMA)
This malignant fibroblastic tumor [Citation[4–6], Citation[9–15]] presents during the first year of life, or even at birth, in the majority of cases and may be detected in utero in some instances. The trunk and extremities are most often involved. Unlike adult fibrosarcoma, this tumor usually involves the distal portions of the extremities. The infant presents with an asymptomatic, rapidly growing, bulky tumor that is located in the deep soft tissues. These tumors tend to be locally invasive and metastasize infrequently. Conservative resection with negative surgical margins is the key to avoiding recurrence. Outcome is dependent on the site of the tumor, with low survival rates for retroperitoneal tumors and high survival rates for extremity tumors. With large nonresectable tumors, chemotherapy based on rhabdomyosarcoma protocols has proven beneficial in reducing tumor size and allowing for adequate resection [Citation[12–15]]. Many pediatric oncologists now consider chemotherapy for infantile fibrosarcoma to reduce tumor volume and limit the need for more radical surgery, especially when amputation may be a concern at initial evaluation.
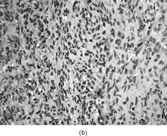
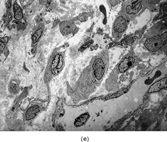
Gross examination reveals an infiltrative fleshy tumor that lacks a well-defined border. The cut surface of the tumor tends to be lobulated with a myxoid to mucinous character with areas of necrosis, cystic degeneration, and hemorrhage. The tumor is a highly cellular neoplasm with a herringbone pattern () [Citation[1], Citation[3–6], Citation[9–11]]. The cells are spindled in outline, but have a high nuclear to cytoplasm ratio. There is considerable nuclear hyperchromasia, tumor necrosis, and frequent mitoses scattered throughout the tumor. The tumor cells are closely packed and overlap one another. Focal hemangiopericytoma-like areas with increased vascularity may be seen. These tumors typically are diffusely positive for vimentin and focally reactive for actin (). Electron microscopy provides evidence for fibroblastic differentiation (, ). The ultrastructural features of lack of to extremely rare extracellular banded collagen, rare to absent basal lamina, branching and markedly dilated rough endoplasmic reticulum, irregular nuclear membranes, and extracellular fibrillogranular material are helpful in separating this malignant fibroblastic tumor from myofibromas. These features are also more characteristic of origin from a primitive mesenchymal spindle cell, representing a precursor cell in the fibroblastic/myofibroblastic cell line. Electron microscopy plays a critical role in distinguishing this malignant tumor from spindle cell rhabdomyosarcoma, infantile rhabdomyosarcoma, undifferentiated sarcoma, and benign fibroblastic/myofibroblastic neoplasms. Ultrastructural examination is particularly important with the approximately 20–30% of infantile fibrosarcomas that immunoreact with desmin and/or muscle specific actin, and may result in confusion with a spindle cell rhabdomyosarcoma [Citation[4],Citation[9],Citation[10]]. An extensive search for striated muscle differentiation (well-formed external lamina, monoparticulate glycogen, myofilaments, and Z-band material) should be undertaken with a spindle cell tumor possessing a predominance of banded collagen and absence of extracellular matrix fibrillogranular material. In addition, the herringbone pattern, nuclear hyperchromasia, necrosis, and cystic degeneration distinguish this tumor from a benign myofibromatous tumor. An unusual childhood tumor that may be confused with infantile fibrosarcoma is the infantile rhabdomyofibrosarcoma () [Citation[4], Citation[9], Citation[10]]. The identification of rhabdomyoblasts by immunocytochemistry, or more likely by electron microscopy, and chromosomal 19 monosomy define the high-grade infantile rhabdomyofibrosarcoma [Citation[4], Citation[9], Citation[10]]. Infantile fibrosarcoma shares a tumor-defining translocation (ETV6-NTRK3) with cellular mesoblastic nephroma () [Citation[4], Citation[6], Citation[15–19]]. In addition, it may have other cytogenetic abnormalities, such as trisomy of certain chromosomes () [Citation[1–6]].
FIG. 1 Infantile fibrosarcoma composed of interlacing fascicles of spindled cells (a, b, original magnification × 100 and 200 × respectively; H & E) with minimal intervening stroma and lack of collagen matrix. The tumor cells have a moderate degree of pleomorphism with frequent mitotic figures (c, original magnification × 400 H & E). Areas within the tumor may show a prominent hemangiopericytoma-like pattern (d, original magnification × 100 H & E). Infantile fibrosarcoma possesses extracellular fibrillogranular material, which is helpful in differentiating this malignant tumor from other fibroblastic/myofibroblastic tumors (e, f, × 3,000 and × 10,000 respectively, transmission electron microscopy). After chemotherapy for a nonresectable infantile fibrosarcoma (g, original magnification × 100 H & E), the spindle cells are conspicuously absent, leaving a fibrous matrix background with a prominent bland vascular pattern.
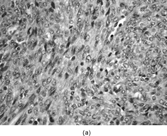
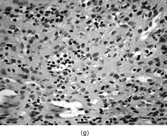
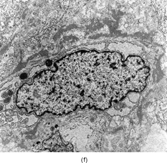
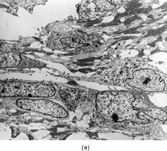
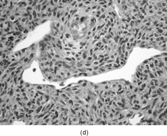


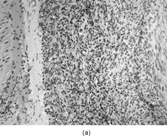
FIG. 2 Infantile rhabdomyofibrosarcoma resembles congenital infantile fibrosarcoma with a highly cellular spindle tumor cell population and frequent mitotic figures (a, b, original magnification × 200 and × 400, respectively). Myogenic immunocytochemical markers may discover rare immunoreactive cells(c, myogenin antibody, original magnification × 400). Ultrastructural examination provides evidence for rhabdomyoblastic differentiation in the spindle cells with detection of thin and thick filaments and z-band material(d, × 4,000, transmission electron microscopy).

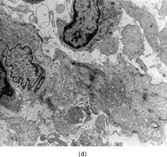
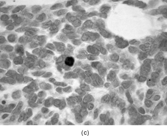

Table 3 Tumors of Fibroblastic and Myofibroblastic Origin in Children and Adolescents: Immunocytochemical, Ultrastructural, and Cytogenetic Features
INFANTILE MYOFIBROMATOSIS (MYOFIBROMATOSIS, MYOFIBROMA)
Although initially described some 5 decades ago as congenital fibrosarcoma, this entity [Citation[1–4], Citation[20–24]] was quickly reclassified as congenital generalized fibromatosis in I954 when it was recognized that this spindle cell tumor lacked malignant potential. Review of a large number of cases resulted in recognition of the myofibroblastic nature of the tumor and the tumor was renamed infantile myofibromatosis [Citation[1–4]]. There are 3 forms: solitary (most common, > 50%), multicentric (less common, 33%), and multicentric with visceral involvement (uncommon, < 15%). The solitary form is usually cutaneous with dermal involvement and extension into the underlying subcutis, muscle, and even bone. Several soft tissue sites may be involved alone (multicentric) or concomitantly with lung, cardiac, gastrointestinal, or even central nervous system involvement (multicentric with visceral involvement). Solitary or multicentric bone lesions may also be present as the sole type of lesions. The prognosis for solitary and multicentric forms is excellent; whereas > 75% of infants with visceral involvement die of disease. Many of the lesions without visceral involvement stabilize and may undergo spontaneous regression. Some of the solitary lesions may become locally aggressive with unremitting gradual destruction of normal tissues. Particularly aggressive tumors in nonresectable sites may require chemotherapy, similar to that for infantile fibrosarcoma to allow for resection or alleviate dysfunction. The head and neck followed by the extremities and trunk are the most common sites of involvement.
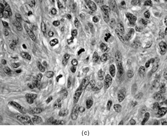
The histopathology of myofibromas [Citation[1–4],Citation[20],Citation[21],Citation[23],Citation[24]] is characterized a nodular or multinodular proliferation with a zoning phenomenon, characterized by peripheral spindle-shaped cells organized into fascicles (). These tend to merge and blend with centrally placed sheets of less differentiated ovoid to polygonal-shaped cells. A prominent pericytoma architecture may be seen throughout the tumor, but more prominently within the center of the tumor. The recognition of this pattern has resulted in inclusion of the tumor previously considered to be infantile hemangiopericytoma into the infantile myofibromatosis category [Citation[4],Citation[23],Citation[24]]. This is appropriate because these tumors lack the cytogenetics features and other features of true adult hemangiopericytoma (solitary fibrous tumor, as reclassified by the WHO) [Citation[4]]. Myofibromas with this pattern are often referred to as myofibromas with a hemangiopericytoma-like pattern [Citation[4],Citation[23],Citation[24]]. Myofibromas may have a relatively high mitotic rate without atypical mitotic figures, areas of necrosis and calcifications, stromal hyalinization, nuclear atypia, and even subendothelial “intravascular” tumor growth. These histopathologic features have no bearing on the clinical outcome of myofibromas. Immunocytochemical staining of the tumor cells reveals vimentin and alpha-smooth muscle actin reactivity, with lack of immunoreactivity with S-100 protein, epithelial membrane antigen, keratin, and desmin (). Ultrastructural examination shows myofibroblastic differentiation (, ) with prominent dilated rough endoplasmic reticulum, longitudinal filaments with dense bodies, and focal basal lamina. Fibronexus structures may be found in a limited number of cases. It should be emphasized that intercellular junctions, pinocytotic junctions, and basal lamina are found in myofibroblastic tumors, but they are found much more commonly in rhabdomyosarcomas. This may give the false sense of a benign fibroblastic lesion when striated muscle differentiation (well-formed external lamina, monoparticulate glycogen, myofilaments, and Z-band material) is expressed ultastructurally in infrequent to rare tumor cells. Immunocytochemical studies should be performed in those cases that do not provide convincing evidence of myofibroblastic differentiation (peripheral cytoplasmic filament arrays).
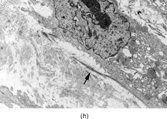
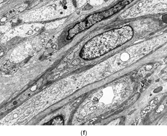
FIG. 3 Myofibromas are often surrounded by a thin fibrous capsule (a, original magnification × 100, H&E) and show increased cellularity at the periphery of the lesion. Toward the central portion of the mass, there is decreased cellularity and increased collagen matrix deposition (b, original magnification × 200, H&E). Mitotic activity is frequently noted with no affect on clinical outcome of the lesion (c, original magnification × 400, H&E). Some myofibromas have a hemangiopericytoma-like pattern (d, original magnification × 100, H&E) with subendothelial proliferation of myofibroblasts. Necrosis with or without dystrophic calcifications is also commonly found (e, original magnification × 200, H&E). Ultrastructural examination reveals spindle cells with myofibroblastic features, including longitudinal filaments (f, original magnification × 4,000), prominent dilated rough endoplasmic reticulum with granular material (g, original magnification × 12,000), and fibronexus structures (arrow, h, original magnification × 10,000).


Cytogenetic analyses of myofibromas have shown nonspecific findings () with chromosome 8 abnormalities [Citation[1–4],[Citation[6],Citation[20],Citation[21],Citation[23],Citation[24]]. A small number of reports of familial myofibromatosis have implicated an autosomal dominant inheritance pattern [Citation[23]]. It must be emphasized that the vast majority of myofibromas are sporadic, isolated occurrences. Most importantly, these tumors lack the tumor-defining translocation (ETV6-NTRK3) found with infantile fibrosarcoma and cellular mesoblastic nephroma [Citation[6],Citation[12–19]]. In particularly troublesome cases, RT-PCR or FISH for the ETV6-NTRK3 translocation should be performed to eliminate infantile fibrosarcoma as a consideration.
PERICYTOMA WITH T(7;12)
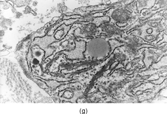
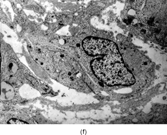
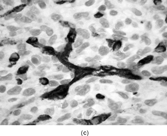
A distinct pericytic tumor [Citation[25]] with a recurrent novel translocation involving the short arm of chromosome 7 and the long arm of chromosome 12 has recently been recognized. This tumor may be mistaken for a myofibroma with a prominent hemangiopericytoma-like pattern or a true adult hemangiopericytoma (solitary fibrous tumor, as reclassified by the WHO). These tumors have involved a spectrum of ages (11–65 years) and sites (tongue, stomach, lower leg). These tumors were characterized by multilobulation and infiltrative growth of spindle-shaped tumor cells arranged around thin-walled small vessels (). The tumor cells were subendothelial. Cytologic atypia and pleomorphism were lacking. Mitoses were infrequent. Immunocytochemistry revealed smooth muscle actin, laminin, and type IV collagen. The tumor cells were not immunoreactive with S-100 protein, keratin, desmin, or CD34. Ultrastructural features supported a pericytic origin with incomplete basal lamina, subplasmalemmal thickenings, and thin filaments with focal dense bodies along the periphery of the cytoplasm (, ). Considering the overall features, this tumor will most likely be classified within the myopericytoma category, according to the WHO soft tissue guidelines.
FIG. 4 Pericytoma with a t(7;12) translocation ischaracterized by plump ovoid to spindled cells in arranged around capillary-sized vessels often containing erythrocytes (a, b, original magnification × 100 and × 400, respectively, H&E). The endothelial cells immunoreact with CD34 antibody (c, CD34 antibody, original magnification × 400); however, the tumor cells are nonreactive. The tumor cells express smooth muscle actin (d, smooth muscle actin antibody, original magnification × 400). Ultrastructural examination reveals plump spindle cells in close proximity to thin-walled vessels lined by bland endothelial cells (e, original magnification × 1,200) with abundant extracellular basal-lamina like material adherent to the cell surfaces (f, original magnification × 8,000).



A novel translocation associated with this tumor () involves the beta-actin gene (ACTB) and GLI oncogene [t(7; 12)(p21–22; q13–15)] [Citation[25]]. This fusion gene may result in overexpression of GLI by the inclusion of a promoter region from ACTB. GLI is essential in the sonic hedgehog signaling pathway, which participates in cell cycle regulation, cell adhesion, apoptosis, signal transduction and cell proliferation.
These pericytic tumors are limited in number [Citation[25]], but 3 of the 5 tumors were resected with negative margins and have not recurred. Two of the tongue tumors required chemotherapy to decrease tumor size and to allow for gross total resection. No recurrences or metastatic disease were reported over a mean follow-up period of 24 months.
INCLUSION BODY FIBROMATOSIS (CONGENITAL INFANTILE DIGITAL FIBROMATOSIS)
Although identified as a distinct entity only in 1965, there are case reports that describe such lesions are early as 1924 [Citation[1–4], Citation[26], Citation[27]]. This lesion occurs almost exclusively on the dorsal aspect of digits in young children ( < 3 years of age) with about one-third of cases being diagnosed shortly after birth. However, cases in adults have also been reported. This typically red to pink lesion is composed of a firm, broad-based, nontender nodule that stretches the overlying skin. The clinical course is a gradual enlargement of the nodule, which may lead to interphalangeal joint deformity. The lesion may erode the underlying bone. Treatment is complete surgical excision. Local recurrence happens in about 50–60% of cases.
The tumor has typical features of a spindle cell proliferation with the neoplastic cells arranged into fascicles and sheets, and embedded in a variable amount of collagen matrix (). The tumor cells infiltrate the adjacent deep dermis and subcutis. There is lack of encapsulation, and the tumor is poorly defined. The spindle cells are fibroblastic/myofibroblastic in origin with eosinophilic inclusions that are negative for PAS, while being positive for trichrome, phosphotungstic acid–hematoxylin, and iron hematoxylin. Immunocytochemistry finds the inclusions to be reactive with smooth muscle actin and vimentin. Ultrastructural examination illustrates the fibroblastic/myofibroblastic nature of the tumor (, ). The spindle cells possess dilated rough endoplasmic reticulum, cytoplasmic thin filaments with dense bodies, and large ovoid aggregates with a granular to filamentous character (inclusions). The cytoplasmic thin filaments extend into the large aggregates.
FIG. 5 Inclusion body fibromatosis (congenitalinfantile digital fibromatosis) is composed of bland fibroblastic cells arranged into sheets and fascicles with abundant collagen matrix (a, original magnification × 200, H&E). Trichrome (b, original magnification × 400) and smooth muscle actin (c, original magnification × 200) staining may reveal oval cytoplasmic bodies with the spindle cells. Upon ultrastructural examination, these cytoplasmic bodies prove to be whorls of actin filaments that are characteristic for this specific fibroblastic tumor (d, original magnification × 5,000).

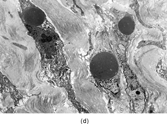

A final consideration with this entity is the recent report of infants with digital fibromas comprising one component of a syndrome [Citation[28], Citation[29]]. Digital fibromas have been associated with facial pigmentary dysplasia, focal dermal hypoplasia, metacarpal and metatarsal disorganization, and limb malformations. The association with these malformations may lead to new insight into the molecular genetics and mechanisms behind infantile digital fibroma formation.
FIBROUS HAMARTOMA OF INFANCY
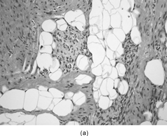
This unique tumor occurs primarily in the first year of life (90%) and there is a male predilection (2.4 M:1.0 F) [Citation[1–4], Citation[30], Citation[31]]. The most common sites for this tumor are axillary regions, upper arms, upper trunk, inguinal regions, and external genitalia. The typical lesion is a rapidly growing, painless nodule that is freely movable, but poorly circumscribed. Most are single lesions. The tumor is composed of 3 components () that form organoid structures: (1) well-defined bundles (trabeculae) of dense, intertwining, fibrous tissue that extends into subcutis adipose tissue; (2) primitive mesenchyme arranged in nests, whorls, and bands in a myxoid matrix; and (3) mature adipose tissue admixed with the other components. The fibrous tissue is composed of myofibroblasts and fibroblasts separated by variable amounts of collagen. The spindle cells react primarily with vimentin and to a lesser extent with smooth muscle actin. Ultrastructural examination reveals an admixture of fibroblastic and myofibroblastic cells (, ). The primitive mesenchymal cells embedded in the myxoid stroma show slender cell processes and a paucity of organelles. Cytogenetic characterization data are lacking. Treatment is surgical excision, with recurrence rates relatively low (12%).
FIG. 6 Fibrous hamartoma of infancy iscomposed of fibrous tissue, primitive mesenchyme within a myxoid matrix and mature adipose tissue (a, original magnification × 100, H&E). There is an abrupt transition from “mature” fibrous tissue to primitive mesenchyme within a myxoid stroma (b, c, original magnification × 200 and × 400, respectively, H&E). The fibroblastic tumor cells with irregular and deeply indented nuclear borders are embedded in a dense haphazardly arranged collagen background (d, original magnification × 3,000, transmission electron microscopy).

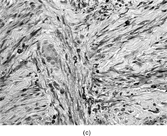
JUVENILE HYALINE FIBROMATOSIS
This hereditary disorder was first described in 1873 as molluscum fibrosum [Citation[1–4], Citation[32], Citation[33]]. In the mid-1960s, it became known as juvenile hyaline fibromatosis. This autosomal recessive disorder is characterized by aberrant collagen synthesis with deposition of hyaline material in the supporting tissues of the skin, gingiva, bone, and joints [Citation[1–4], Citation[32], Citation[33]]. It has been divided into 2 separate categories, a severe form, infantile systemic hyalinosis, and a mild form, juvenile hyaline fibromatosis. The majority of individuals present with soft tissue nodules during infancy or early childhood, but adult onset cases have been reported. Typically, there is a progressive increase in the number and size of subcutaneous and deep nodules, leading to deformity and dysfunction. With infantile and early childhood onset, survival may be into adulthood. The face and neck are frequently involved, as well as the gingiva, skull, long bones, and phalanges. The involvement of periarticular areas results in joint contractures; whereas bony lesions cause osteolysis and osteoporosis. Perianal lesions may resemble genital warts.
The nodules are formed by plump fibroblasts embedded in an abundant nonfibrillary, eosinophilic hyaline matrix (). In active lesions or in younger patients, the nodules may be quite cellular. The fibroblasts have a relatively amphophilic cytoplasm and an indistinct fascicular arrangement. In long-standing lesions, the cellularity is markedly decreased and the fibroblasts appear compressed by the matrix. Some areas of the nodule may have a chondroid character. PAS stain tends to be strong and diastase resistant. Immunocytochemical studies show only vimentin reactivity. Ultrastructural examination reveals the fibroblastic nature of the lesion (, ). The fibroblasts have a unique appearance with frequent markedly dilated membrane-bound vesicles containing granular and filamentous (fibrillogranular) material. The contents of these vesicles are similar in morphology to that for the surrounding ground substance. Occasionally, the vesicles will be in direct continuity with the surrounding ground substance.
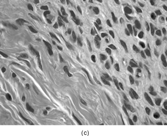
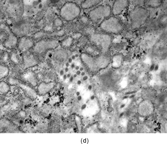
FIG. 7 Juvenile hyaline fibromatosis is composed of bland spindled cells embedded in a dense collagenous matrix lacking a fibrillary character for the most part (a, b, original magnification × 100 and × 200, respectively, H&E). Ultrastructural examination displays the dilated rough endoplasmic reticulum containing granular material (c, original magnification × 3,500) and intracytoplasmic collagen fibers (d, original magnification × 14,000). Often, dilated membrane-bound vesicles with granular material are lined by a thin cell surface membrane and nearly in contact with the extracellular matrix(e, original magnification × 4,500).




Abnormalities on the long arm of chromosome 4 (4q21) in juvenile hyaline fibromatosis have been known for some time () [Citation[34–36]]. Recently, the mutated gene associated with juvenile hyaline fibromatosis has been identified [Citation[35], Citation[36]]. Capillary morphogenesis gene-2 (CMG2) is affected in both the mild and severe forms of juvenile hyaline fibromatosis. This gene encodes a protein that is upregulated in endothelial cells during capillary formation. The resulting protein binds laminin and collagen IV via a von Willebrand factor type A domain. It is believed that a mutation that causes complete interference with binding by the von Willebrand factor A results in the severe form of the disease—infantile systemic hyalinosis. The milder form of the disease—juvenile hyaline fibromatosis—may occur when there is an in-frame mutation that affects a highly conserved cytoplasmic domain with retention of some binding function. Fibroblasts derived from affected patients indicate that CMG2 mutations abrogate normal cell interaction with the extracellular matrix. It is possible that this condition represents a perturbation of the basement membrane matrix assembly apparatus culminating in excessive hyaline deposition. CMG2 also functions as a cellular receptor for anthrax toxin. The role for this anthrax toxin receptor function in juvenile hyaline fibromatosis is unclear.
FIBRODYSPLASIA OSSIFICANS PROGRESSIVA (MYOSITIS OSSIFICANS PROGRESSIVA, STIFF MAN DISEASE)
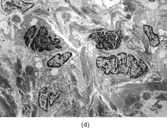
This rare autosomal dominant and sporadic disorder is characterized by congenital malformation of the great toes (hallus valgus, monophalangic great toes) in an otherwise normal-appearing child [Citation[37–41]]. During the first or second decade of life (mean age of onset 5 years), painful fibrous nodules form over the neck, back, and shoulders. These fibroproliferative nodules undergo progressive ossification via an endochondral bone formation mechanism. Restrictive heterotopic ossification is present in about 80% of patients by 7 years of age. By 15 years of age, almost all affected individuals will have severely restricted mobility of their arms. Heterotopic ossification proceeds progressively and unrelentingly from axial to appendicular, from cranial to cauda, and from proximal to distal in direction. There is progressive heterotopic ossification of tendons, ligaments, fasciae, joints, and skeletal muscles. This process may initially mimic aggressive juvenile hyaline fibromatosis. However, the development of mature heterotopic ossification defines the condition rapidly. Death results from the respiratory complications associated with severe chest wall motion restriction. Heterotopic ossification occurs progressively in the absence of trauma; however, minor injury to the soft tissues, including biopsy and surgical procedures, induces and accelerates ossification. In fact, surgical intervention is said to “fuel the fire” of ossification and disease progression.
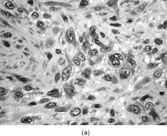
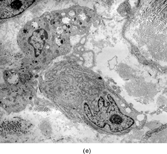
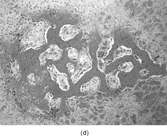
The earliest histopathologic evidence [Citation[37–41]] for this disease process is a perivascular lymphocytic infiltrate that may extend into adjacent skeletal muscle. This explains the initial designation of this disorder as myositis ossificans progressiva. The inflammatory infiltrate is transitory and followed by skeletal myocyte degeneration in concert with a markedly vascularized fibroblastic proliferation that is indistinguishable from aggressive juvenile hyaline fibromatosis and fibromatosis colli. Mast cells are readily recognized within the lesion and are increased by 40- to 150-fold compared with normal fibroconnective tissue and by 10- to 40-fold when compared with inflammatory myopathy [Citation[41]]. This vascular fibroproliferative lesion undergoes endochondral bone formation, which results in mature lamellar bone with bone marrow elements (). The spindle cells in the early lesions resemble active fibroblasts or myofibroblasts with vimentin and smooth muscle actin reactivity [Citation[37–41]]. Of considerable interest is the presence of bone morphogenetic protein 2/4 [Citation[42]]. Ultrastructural features of the fibroproliferative areas reveal active fibroblastic cells with moderately dilated rough endoplasmic reticulum, abundant ground substance, and collagen (, ). Foci of early calcification are not typically seen in the fibroproliferative areas.
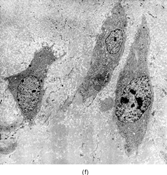
FIG. 8 Fibrodysplasia ossificans progressiva is composed of plump spindled cells in a collagen matrix, which varies from myxoid to chondroid (a, original magnification × 400, H&E). The matrix undergoes hyalinization with “entrapment” of the spindled cells (b, original magnification × 400, H&E). The matrix becomes densely eosinophilic and resembles osteoid with ensuing calcification (c, original magnification × 200, H&E). Areas within the lesional tissue form intersecting trabeculae of calcified bone with adjacent chondroid-like areas (d, original magnification × 100, H&E). Ultrastructural examination of the noncalcified matrix and early lesions reveal plump fibroblastic cells with mildly to moderately dilated rough endoplasmic reticulum and collagen matrix (e, original magnification × 3,000). Elongated spindle cells elaborating collagen matrix and embedded in relatively abundant collagen matrix are also noted (f, original magnification × 2,000). Some of the ground substance is clumped together and has an “amorphous” character similar to that seen in osteogenic tissues.


Cytogenetic and molecular studies have provided several clues to the mechanism behind this rare and unusual disorder [Citation[42–46]]. Disruption in the regulation of the bone morphogenetic protein (BMP) pathway would appear to be involved in the pathogenesis. Genetic linkage of fibrodysplasia ossificans progressiva at 4q27–31 (bone morphogenetic protein signaling pathway gene) and 14q22–23 (BMP-4) have been identified in several patients and pedigrees with fibrodysplasia ossificans progressiva. As noted earlier, BMP-4 expression may be found within the lesions and in particular these are found with the lymphocytic infiltrate and lesional spindle cells [Citation[42]]. BMP-4 is overexpressed in immortalized lymphoblastoid and fibroblastic cells from lesional tissue [Citation[42],Citation[44],Citation[45]]. BMP4 is inactivated by binding to a secreted polypeptide known as noggin. This protein is encoded by the gene NOG (14q22–23) and is expressed in developing bone [Citation[42],Citation[45],Citation[46]]. Murine models in which the NOG gene has been “knocked out” (NOG-/-) expire at birth due to multiple defects, including fusion of the bones of the appendicular skeleton [Citation[45]]. It may not be surprising to realize that mutations and dysregulation of NOG (14q22–23) have been discovered in familial and sporadic cases of fibrodysplasia ossificans progressiva [Citation[46]]. It would appear that dysregulation of bone morphogenetic protein pathways plays a major role in the pathogenesis of fibrodysplasia ossificans progressiva [Citation[42],Citation[44],Citation[45]]. Gene therapy targeted toward regulation of the bone morphogenetic protein pathway would appear to hold promise for individuals affected by fibrodysplasia ossficans progressiva.
SUMMARY
Fibroblastic and myofibroblastic tumors in children span the realm from benign to malignant and from congenital to acquired. Accurate diagnosis is important to determine appropriate therapy, prognosis, long-term follow-up, and genetic counseling. This requires the incorporation of many different tools available to the surgical pathologist. Histochemical, immunocytochemical, ultrastructural, cytogenetic, and diagnostic molecular pathology findings are essential to avoid diagnostic confusion with fibroblastic and myofibroblastic pediatric tumors.
Related Research Data
REFERENCES
- Coffin CM, Dehner LP. Fibroblastic-myofibroblastic tumors in children and adolescents: a clinicopathologic study of 108 examples in 103 patients. Pediatr Pathol. 1991; 11: 569–588. [PUBMED], [INFOTRIEVE]
- Coffin CM, Dehner LP. Soft tissue tumors in first year of life: a report of 190 cases. Pediatr Pathol. 1990; 10: 509–526. [PUBMED], [INFOTRIEVE]
- Coffin CM, Dehner LP. Pathologic evaluation of pediatric soft tissue tumors. Am J Clin Pathol. 1998; 109(4 Suppl 1)S38–S52. [PUBMED], [INFOTRIEVE], [CSA]
- Fletcher CDM, Unni KK, Mertens F. Fibroblastic/myofibroblastic tumors. Fletcher CDM, Unni KK, Mertens F. Tumours of Soft Tissue and Bone. LyonFrance, IARC Press. 2002; 47–108
- Coffin CM, Jaszcz W, O'Shea PA, Dehner LP. So-called congenital-infantile fibrosarcoma: does it exist and what is it?. Pediatr Pathol. 1994; 14: 133–150. [PUBMED], [INFOTRIEVE]
- Sandberg AA, Bridge JA. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumor: congenital (infantile) fibrosarcoma and mesoblastic nephroma. Cancer Genet Cytogenet. 2002; 132: 1–13. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Wherli BM, Weiss SW, Yandow S, Coffin CM. Gardner-associated fibromas (GAF) in young patients: a distinct fibrous lesion that identifies unsuspected Gardner syndrome and risk for fibromatosis. Am J Surg Pathol. 2001; 25: 645–651. [CSA], [CROSSREF]
- Joshi VJ, Balarezo F, Hicks MJ, Mierau GW, Tsongalis GJ. Approach to small round cell tumors of childhood. Pathol Case Rev. 2000; 5: 26–41
- Trobs RB, Meier R, Bennek J, Heinrich S, Willnow U. Fibrosarcoma in infants and children: a retrospective analysis—overdiagnosis in early years. Pediatr Surg Int. 1999; 15: 123–128. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Cecchetto G, Carli M, Alaggio R, et al, Italian Cooperative Group. Fibrosarcoma in pediatric patients: results of the Italian Cooperative Group studies (1975-1995). J Surg Oncol. 2001; 78: 225–231. [PUBMED], [INFOTRIEVE], [CROSSREF]
- O, Malley DP, Mierau GW, Beckwith JB, Weeks DA. Ultrastructure of cellular mesoblastic nephroma. Ultrastruct Pathol. 1996; 20: 417–427. [CSA]
- Loeb DM, Hill DA, Dome JS. Complete response of recurrent cellular congenital mesoblastic nephroma to chemotherapy. J Pediatr Hematol Oncol. 2002; 24: 478–481. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Shetty AK, Yu LC, Gardner RV, Warrier RP. Role of chemotherapy in the treatment of infantile fibrosarcoma. Med Pediatr Oncol. 1999; 33: 425–427. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Loh ML, Ahn P, Perez-Atayde AR, Gebhardt MC, Shamberger RC, Grier HE. Treatment of infantile fibrosarcoma with chemotherapy and surgery: results from the Dana-Farber Cancer Institute and Children's Hospital, Boston. J Pediatr Hematol Oncol. 2002; 24: 722–726. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- McCahon E, Sorensen PHB, Davis JH, Rogers PCJ, Schultz KR. Non-resectable congenital tumors with the ETV6-NTRK3 gene fusion are highly responsive to chemotherapy. Med Pediatr Oncol. 2003; 40: 288–292. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Adem C, Gisselsson D, Cin PD, Nascimento AG. ETV6 rearrangements in patients with infantile fibrosarcomas and congenital mesoblastic nephromas by fluorescence in situ hybridization. Mod Pathol. 2001; 14: 1246–1251. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Argani P, Fritsch M, Kadkol SS, Schuster A, Beckwith JB, Perlman EJ. Detection of ETV6-NTRK3 chimeric RNA of infantile fibrosarcoma/cellular congenital mesoblastic nephroma in paraffin-embedded tissue: application to challenging pediatric renal stromal tumors. Mod Pathol. 1999; 13: 29–36. [CSA], [CROSSREF]
- Bourgeois JM, Knezevich SR, Mathers JA, Sorensen PHB. Molecular detection of the ETV6-NTRK3 gene fusion differentiates congenital fibrosarcoma form other childhood spindle cell tumors. Am J Surg Pathol. 2000; 24: 937–946. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Knezevich SR, McFadden DE, Tao W, Lim JF. Sorensen PH: A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat Genet. 1998; 18: 184–187. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Behar PM, Albritton FD, Muller S, Todd NW. Multicentric infantile myofibromatosis. Int J Pediatr Otorhinolarngol. 1998; 45: 249–254. [CROSSREF]
- Netscher DT, Eladoumikdachi F, Popek EJ. Infantile myofibromatosis; case report of a solitary hand lesion with emphasis on differential diagnosis and management. Ann Plast Surg. 2001; 46: 62–67. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Ikediobi NI, Iyengar V, Hwang L, Collins WE, Metry DW. Infantile myofibromatosis: support for autosomal dominant inheritance. J Am Acad Dermatol. 2003; 49: S148–S150. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Mentzel T, Calonje E, Nascimento AG, Fletcher CDM. Infantile hemangiopericytoma versus infantile myofibromatosis: study of a series suggesting a continuous spectrum of infantile myofibroblastic lesions. Am J Surg Pathol. 1994; 18: 922–930. [PUBMED], [INFOTRIEVE]
- Variend S, Bax NMA, Van Gorp J Are infantile myofibromatosis, congenital fibrosarcoma and congenital haemangiopericytoma histogenetically related?. Histopathology. 1995; 26: 57–62. [PUBMED], [INFOTRIEVE], [CSA]
- Dahlen A, Fletcher CDM, Mertens F, et al, Activation of the GLI oncogene through fusion with the beta-actin (ACTB) gene in a group of pericytic tumor with t(7; 12): pericytoma with (t7;12). Am J Pathol. 2004; 164: 1645–1653
- Falcon NA, Upton J. Infantile digital fibromas. J Hand Surg. 1995; 20A: 1014–1020
- Rimareix F, Bardot J, Andrac L, Vasse D, Galinier P, Magalon G. Infantile digital fibroma: report of eleven cases. Eur J Pediatr Surg. 1997; 7: 345–348. [PUBMED], [INFOTRIEVE], [CSA]
- Horii E, Sugiura Y, Nakamura R. A syndrome of digital fibromas, facial pigmentary dysplasia, and metacarpal and metatarsal disorganization. Am J Med Gen. 1998; 80: 1–5. [CROSSREF]
- Breuning MH, Oranje AP, Langemeijer RA, et al, Recurrent digital fibroma, focal dermal hypoplasia, and limb malformations. Am J Med Gen. 2000; 94: 91–101. [CROSSREF]
- Dickey GE, Sotelo-Avila C. Fibrous hamartoma of infancy: current review. Pediatr Dev Pathol. 1999; 2: 236–243. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Scott DM, Pena JR, Omura EF. Fibrous hamartoma of infancy. J Am Acad Dermatol. 1999; 41(5 Pt2)857–859. [PUBMED], [INFOTRIEVE], [CSA]
- Halem A, Al-Hindi HN, Al-Juboury M, Al-Husseini H, Al-Ajlan A. Juvenile hyaline fibromatosis: morphologic, immunohistochemical, and ultrastructural study of three siblings. Am J Dermatopathol. 2002; 24: 218–224. [CSA], [CROSSREF]
- Breier F, Fang-Kircher S, Wolff K, Jurecka W. Juvenile hyaline fibromatosis: impaired collagen metabolism in human skin fibroblasts. Arch Dis Child. 1997; 77: 436–440. [PUBMED], [INFOTRIEVE]
- Rahman N, Dunstan M, Teare MD, et al, The gene for juvenile hyaline fibromatosis maps to chromosome 4p21. Am J Hum Genet. 2002; 71: 975–980. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Hanks S, Adams S, Douglas J, et al, Mutations in the gene encoding capillary morphogenesis protein 2 cause juvenile hyalinosis fibromatosis and infantile systemic hyalinosis. Am J Hum Genet. 2003; 73: 791–800. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Dowling O, Difeo A, Ramirez MC, et al, Mutations in capillary morphogenesis gene-2 result in the allelic disorders juvenile hyaline fibromatosis and infantile systemic hyalinosis. Am J Hum Genet. 2003; 73: 957–966. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Feldman G, Li, Ming, Martin S, et al, Fibrodysplasia ossficans progressiva, a heritable disorder of severe heterotopic ossification, maps to human chromosome 4p27-31. Am J Hum Genet. 2000; 66: 128–135. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Kaplan FS, Glaser DL, Hebela N, Shore EM. Heterotopic ossification. J Am Acad Orthop Surg. 2004; 12: 116–125. [PUBMED], [INFOTRIEVE], [CSA]
- Blaszczyk M, Majewski S, Brzezinska-Weislo L, Jablonska S. Fibrodysplasia ossificans progressiva. Eur J Dermatol. 2003; 13: 234–237. [PUBMED], [INFOTRIEVE], [CSA]
- Brandao-Paim L, de Lourdes-Liphaus B, da Silva CAA. Fibrodysplasia ossificans progressiva. Indian Pediatr. 2003; 40: 786–788. [CSA]
- Gannon GH, Glaser DL, Caron R, Thompson LD, Shore EM, Kaplan FS. Mast cell involvement in fibrodysplasia ossificans progressiva. Hum Pathol. 2001; 32: 842–848. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Olmsted EA, Kaplan FS, Shore EM. Bone morphogenic protein-4 regulation in fibrodysplasia ossificans progressiva. Clin Orthop. 2003; 408: 331–343. [PUBMED], [INFOTRIEVE]
- Hegyi L, Gannon FH, Glaser DL, Shore EM, Kaplan FS, Shanahan CM. Stromal cells of fibrodysplasia ossificans progressive lesions express smooth muscle lineage markers and the osteogenic transcription factor Runx2/Cbfa-1: clues to a vascular origin of heterotopic ossification?. J Pathol. 2003; 201: 141–148. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Lucotte G, Barthelier C, Mercier G, et al, Localization of the gene for fibrodysplasia ossificans progressive (FOP) to chromosome 17q21-22. Genet Couns. 2000; 11: 329–334. [PUBMED], [INFOTRIEVE], [CSA]
- Glaser DL, Economides AN, Wang L, et al, In vivo somatic cell gene transfer of an engineered Noggin mutein protein prevents BMP4-induced heterotopic ossification. J Bone Joint Surg Am. 2003; 85-A: 2332–2342. [PUBMED], [INFOTRIEVE], [CSA]
- Semonin O, Fontaine K, Daviaud C, Ayuso C, Lucotte G. Identification of three novel mutations of the noggin gene in patients with fibrodysplasia ossificans progressive. Am J Med Genet. 2001; 102: 314–317. [PUBMED], [INFOTRIEVE], [CROSSREF]