Abstract
A subset of low-grade fibrosarcomas is composed of CD34-positive spindle cells. These include dermatofibrosarcoma, its morphologic variants, and its associated fibrosarcoma, solitary fibrous tumor, hemangiopericytoma and their malignant counterparts, and some cases of myxoinflammatory fibroblastic sarcoma. Dermatofibrosarcoma and related lesions are characterized by a t(17;22)(q22;q13) rearrangement resulting in fusion of the genes COL1A (17q21-22) and PDGFB1 (22q13). Solitary fibrous tumor displays varying cellularity and fibrosis and a peripheral hemangiopericytomatous pattern; most tumors formerly called hemangiopericytoma are now subsumed into the category of solitary fibrous tumor, although a few strictly defined examples are recognized; however, these are probably not composed of pericytes. Myofibroblastic malignancies are best identified by electron microscopy, with which varying degrees of differentiation, including the presence of fibronexus junctions, can be identified. Low-grade sarcomas showing myofibroblastic differentiation include myofibrosarcomas and inflammatory myofibroblastic tumors. Myofibrosarcomas are spindle cell neoplasms that occur in children or adults in the head and neck, trunk, and extremities as infiltrative neoplasms and that display a fascicular or fasciitis-like pattern with focal nuclear atypia and variable expression of myoid antigens. These sarcomas are prone to recurrence and a small number metastasize. Inflammatory myofibroblastic tumor (synonymous with inflammatory fibrosarcoma) is a neoplasm arising predominantly in childhood, and frequently in intraabdominal locations. It has spindle cells in fascicular, fasciitis-like and sclerosing patterns, with heavy chronic inflammation including abundant plasma cells. Many IMT have clonal chromosomal abnormalities involving 2p22-24, and fusion of the ALK gene with tropomyosin 3 (TPM3-ALK) or tropomyosin 4 (TPM4-ALK) is found in a subset.
LOW-GRADE SARCOMAS COMPOSEDOF CD34-POSITIVE FIBROBLASTS
Most fibroblastic lesions lack specific immunohistochemical markers and express only vimentin, which is of no diagnostic value. A subset of benign and malignant fibroblastic tumors, however, is composed of CD34-positive cells. The human hematopoietic progenitor cell antigen (CD34) is a 110-kDa transmembrane cell surface glycoprotein encoded by a gene in the 1q32 region [Citation[1]]. It reacts with several nonhematopoietic tissues, including vascular endothelium, endoneurial cells, and a subset of dermal dendritic interstitial fibroblastic cells in connective tissue, around blood vessels, nerves, smooth muscle bundles and hair follicles, and their tumors [Citation[1]]. CD34 is also variably expressed in a variety of mesenchymal neoplasms, including some tumors of fibroblasts. These include dermatofibrosarcoma (DFS), fibrosarcoma arising in DFS, solitary fibrous tumor [Citation[2]] and hemangiopericytoma, and some cases of myxofibrosarcoma and myxoinflammatory fibroblastic sarcoma [Citation[3]].
Dermatofibrosarcoma Protuberans (DFS)
DFS is a relatively common lesion, which is more frequent in males, with a peak age incidence of 25–45 years, and which mostly occurs on the trunk and upper limbs. It begins as a dermal plaque or nodule and grows slowly, sometimes becoming multinodular. DFS recurs, especially if incompletely excised; metastasis occurs in fewer than 5% of cases and follows multiple recurrences; exceptionally, there is fibrosarcomatous or MFH-like transformation, which is associated with a more aggressive course.
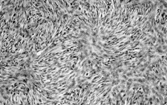
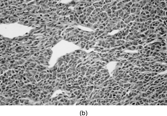
The lesion is composed of uniform, elongated, thin spindle cells with minimal cytoplasm and indistinct margins, in a striking and monotonous “tight” storiform pattern (). The tumor forms a nodule or ill-defined dermal plaque that extends into subcutaneous fat with a characteristic honeycomb pattern including layers of infiltrating tumor parallel to the skin surface. Immunostaining shows strong, diffuse CD34 expression and focal reactivity for smooth muscle actin, but usually no significant staining for S-100 protein. Some cases are CD117 positive, which has led to the suggested use of imatinib (Glivec) in locally advanced DFS or in metastatic disease [Citation[4], Citation[5]].
Fig. 1 Dermatofibrosarcoma with uniform spindle cells with scanty cytoplasm, in characteristic storiform pattern.
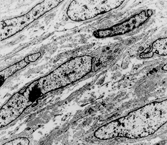
The cell type of DFS is not clearly characterized ultrastructurally: histiocytic, perineural, pericytic, and endothelial origin have all been suggested, but most agree that there are variably developed features of fibroblasts (notably rough endoplasmic reticulum) () [Citation[6]]. The CD34 expression supports the hypothesis that the tumor might be derived from (or differentiating toward) any of the local populations of normal CD34-positive fibroblasts—intradermal, periadnexal, or endoneurial. Occasionally, there are also focal peripheral filament bundles, suggesting myofibroblastic differentiation. In keeping with this, some DFS are actin-positive, and foci of “myoid” differentiation have been described in both DFS and, more frequently, in DFS-FS [Citation[7–11]]. In these, the cells have short, blunt-ended nuclei with eosinophilic cytoplasm that is SMA positive but desmin and CD34 negative, and electron microscopy has shown (mostly) myofibroblastic differentiation. It has been suggested [Citation[12], Citation[13]], however, that this represents a change in the stroma rather than the lesional cells. The feature has no clinical significance. Factor XIIIa (which stains dermal “dendrocytes”) is generally not detectable in the principal lesional cells of DFS [Citation[14], Citation[15]].
Fig. 2 Dermatofibrosarcoma. Electron micrograph showing spindle cells with scanty cytoplasm containing scanty profiles of rough endoplasmic reticulum and no other specific features.
DFS has a reciprocal translocation, t(17;22)(q22;q13) (with a supernumerary ring chromosome) resulting in fusion of the genes COL1A (17q21-22) and PDGFB1 (22q13) [Citation[16–19]]. The same abnormalities have also been shown in fibrosarcoma arising in DFS, in which COL1A1-PDGFB transcripts have been demonstrated [Citation[18], Citation[20]]. Other rearrangements, including t(2;17) and t(9;22), have rarely been found in DFS [Citation[21]]. Gene expression profiling has shown high expression of a group of genes that included PDGFB and osteonectin [Citation[22]].
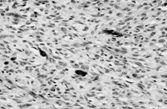
Myxoid and pigmented variants of DFS are described. Rarer atrophic, palisading, and granular cell variants are also recognized but lack clinical significance. Myxoid DFS can be diagnosed by the characteristic cytology as well as the (variable) CD34 positivity (). It must be distinguished from a range of benign myxoid lesions and from myxofibrosarcoma (by absence of nuclear pleomorphism and the different vascular pattern). Pigmented DFS (Bednar tumor) [Citation[23]] has melanin-containing S-100 protein-positive cells singly or in small clusters . This variant is incidentally more common in individuals with pigmented skin. It is cytogenetically identical to regular DFS [Citation[24]], and can also undergo fibrosarcomatous change [Citation[25]].
Fig. 3 Myxoid dermatofibrosarcoma. Other parts of this tumor showed more typical features, with CD34 positivity.
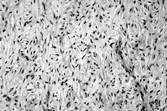
Fig. 4 Bednar tumor (pigmented dermatofibrosarcoma) with scattered melanin-containing cells.
Fibrosarcoma
Fibrosarcoma can arise in DFS, both de novo and in recurrent lesions [Citation[9], Citation[26–31]]. It is characterized by greater cellularity with fascicular architecture and increased mitotic activity (). CD34 can be positive or negative in the fibrosarcomatous area [Citation[32]]. The fibrosarcomatous component behaves relatively aggressively, with local recurrence in over 50% and metastasis in 15% of cases [Citation[9]]. However, adequate local control of DFS with fibrosarcomatous changes, with clear surgical margins, can reduce both local recurrence and the incidence of metastasis [Citation[33]].
Fig. 5 Fibrosarcoma in dermatofibrosarcoma. Fascicles of spindle cells in herringbone pattern. Cells have inconspicuous cytoplasm and increased mitotic index.

Giant Cell Fibroblastoma (GCF)
GCF is a rare childhood (and occasionally adult) lesion of dermis/subcutis, predominantly arising in back of thigh, groin, and chest wall (in a similar distribution to DFS) [Citation[28], Citation[34–41]]. GCF has recurrent potential. It is poorly circumscribed and infiltrative in skin and subcutis, with spindle and bland multinucleate cells in a fibrous and myxoid stroma, and focally forms cystic spaces lined by tumor (not endothelial) cells (). Cellularity varies and both hypercellular and hypocellular (fibrous) areas occur. The multinucleate cells are seen ultrastructurally to have a single markedly indented and convoluted nucleus. The lesional cells are CD34 positive and giant cell fibroblastoma has been recorded as recurring partially or completely as dermatofibrosarcoma protuberans, of either classical or pigmented types. The cytogenetic abnormalities and molecular events in GCF are identical to those in DFS [Citation[42], Citation[43]]. This tumor is therefore currently considered to be a (juvenile) variant of DFS.
Fig. 6 Giant cell fibroblastoma. There are spindle and multinucleate cells in a fibromyxoid stroma. Similar cells line pseudovascular spaces.
Solitary Fibrous Tumor
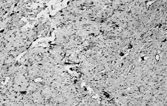
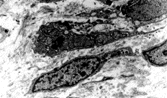
This neoplasm (SFT) was originally described in the pleura and has since been described in almost every site, including peritoneum, pericardium and mediastinum, liver, upper respiratory tract, including nasal cavity, salivary gland, and breast [Citation[44]]. SFT is increasingly recognized in soft tissues [Citation[45–48]]. Typically, the tumor is circumscribed but rarely encapsulated, and is composed of plump or slender spindle or rounded cells in a collagenous background (). The cells are bland and have scanty cytoplasm; multinucleated cells are sometimes present. The lesional cells are arranged in a “patternless” pattern, sometimes with alternating hyper- and hypocellular regions. In the latter, there is variable collagenization, which is focally dense, although stroma is sometimes myxoid. The vessels are focally hemangiopericytomatous, and islands of mature fat can occur (lipomatous hemangiopericytoma [Citation[49–52]]). Mitotic activity varies from 0 to 50 per 10 hpf, and some cases have necrosis or hemorrhage. Immunohistochemically, strong and diffuse CD34 and bcl-2 expression is a feature of this tumor type [Citation[2], Citation[53], Citation[54]]. Indeed, a CD34-negative tumor in an extrapleural location should probably not be diagnosed as solitary fibrous tumor unless other evidence is compelling [Citation[44]]. CD99 is also positive [Citation[55]] and sporadically there is focal positivity for actin, S-100 protein, and (exceptionally) cytokeratin. Electron microscopy shows fibroblastic cells with very occasional myofibroblasts (). In spite of the original reports of pleural tumors, SFT does not show evidence of mesothelial differentiation. A variety of genetic findings has been reported, including loss of 13q, 4q, and 21q; and trisomy 21, and gains at chromosome 8 and at 15q, t(4;15)(q13;q26) [Citation[56]], t(6;17)(p11.2;q23) [Citation[57]], and t(9;22)(q31;p13) [Citation[58]], and also, once again, rearrangements involving 12q13–15 [Citation[57]].
Fig. 7 Solitary fibrous tumor. (a) The tumor has a circumscribed border and a peripheral hemangiopericytic pattern. (b) Fibrous and cellular areas are abruptly juxtaposed. (c) Bland spindle cells are arranged in a patternless pattern in cellular areas. (d) Cords of cells in a collagenous focus.
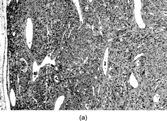
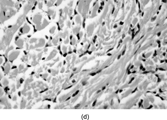


Fig. 8 Solitary fibrous tumor. Cells have ultrastructural features of fibroblasts, with variable amounts of rough endoplasmic reticulum and no external lamina.
Predominantly myxoid SFT (myxoid change exceeding 50% of the tumor) is very uncommon, perhaps representing < 5% of SFT [Citation[59]]. Tumors range between 3 and 14 cm in diameter. Cords of bland spindle cells lie in a vascular myxoid stroma, with focal cellular aggregates, and most also have areas of typical solitary fibrous tumor with variable collagenization and staghorn vessels. Nuclear atypia and large numbers of mitoses are absent. All cases are positive for CD34 and CD99. None of the reported cases experienced recurrence or metastasis.
SFT displays a spectrum of behavior. Recurrence is more likely with larger and histologically aggressive tumors, and atypical and malignant variants in soft tissue have been identified [Citation[47], Citation[60]]. Suggested criteria for malignant SFT [Citation[44]] are (1) presence in an otherwise typical SFT of at least two of the following: high cellularity with nuclear crowding and overlapping, pleomorphism, and mitoses > 4/10 HPF () (and perhaps necrosis); and (2) development of sarcoma in the site of a previous SFT. CD34 expression can be lost in the malignant component.
Fig. 9 Malignant solitary fibrous tumor with cellular crowding and increased numbers of mitoses.
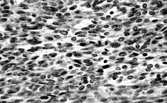
Hemangiopericytoma
Stout and Murray originally described hemangiopericytoma (HPC) in 1942 [Citation[61]] with a further report of 25 cases in 1949 [Citation[62]]. Examination of these series suggests a heterogeneous group of neoplasms, including probable myofibroma. Stout himself stressed that the exact nature of the cells in these tumors had not been proved, and that there was no scientific basis in support of the designation. HPC is, in fact, becoming a diagnosis of exclusion as a wide variety of benign and malignant lesions show at least focally the nonspecific but characteristic branching and staghorn open vascular pattern [Citation[63]]. Many cases that would previously have been termed hemangiopericytoma are now, on the basis of diffuse CD34 expression, diagnosed as solitary fibrous tumors (SFT).
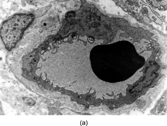
Pericytes are spindle cells surrounding small arterioles, capillaries, and pre- and postcapillary venules, usually in a single complete or incomplete layer. Ultrastructurally, pericytes have overlapping processes and a continuous or interrupted external lamina surrounds the cells and lies between them and the endothelium (). Variable features include micropinocytosis, microfilaments with dense bodies, and junctions. Their immunophenotype varies with location: those around precapillary and postcapillary vessels express smooth muscle actin (SMA), whereas pericytes around capillaries are SMA negative [Citation[64]]. The latter perivascular cell has been considered to represent a persistent primitive mesenchymal cell that can, according to circumstance and specific location, differentiate into (vascular) smooth muscle cells, glomus cells, preadipocytes, and osteoblasts [Citation[65]]. This cell type has also been postulated as the precursor cell of MFH [Citation[66]]. The group of tumors termed PEComas is supposedly derived from a SMA/HMB45-positive perivascular epithelioid cell but the normal counterpart of this cell has not been demonstrated [Citation[67]].
Fig. 10 Pericyte (a) adjacent to blood capillary vessel and (b) enclosed within layers of external lamina, showing pinocytosis, Golgi complex, and abundant cytoplasmic filaments, focally with dense bodies suggesting myoid differentiation.
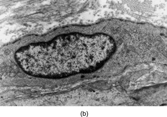
The pericyte lacks specific morphologic features, and it is not clear that hemangiopericytoma, which is usually SMA negative, is composed of pericytes [Citation[68], Citation[69]]. Nevertheless, cases still remain that are diagnosed as HPC after extensive investigation (). A current definition might be a tumor that has (1) spindled, plump, or rounded cells with small to moderate amounts of cytoplasm and indistinct cell margins; (2) a consistent HPC-vascular pattern throughout the entire tumor, with reticulin surrounding individual cells; and (3) the absence of specific differentiation, both morphologically and immunohistochemically. Such tumors usually occur in the deep soft tissue of extremities and retroperitoneum (here associated with hypoglycaemia and hypophosphatemia) but cases are reported in the breast, orbit, nasal cavity, and CNS as well.
Fig. 11 Hemangiopericytoma. (a) Typical appearance, with prominent and widespread typical vascular pattern and uniform plump cells. This tumor is CD34 negative. (b) malignant hemangiopericytoma is a diagnosis of exclusion; many tumors with this appearance prove to be poorly differentiated synovial sarcomas.
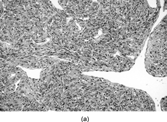
CD34 expression has been reported in apparent HPC, but the majority of such cases formerly called HPC are now regarded as solitary fibrous tumors, especially when the CD34 expression is widespread. However, in SFT the hemangiopericytomatous pattern is usually only focal. There is an occasional report of desmin or smooth muscle actin positivity, and FXIIIa and HLA-DR expression have been reported in a constant subpopulation of HPC cells, as well as a few cells with FVIIIRAg [Citation[70]]. This is not, however, specific, as FXIIIa is present in many spindle cell tumors. Recently, scanty focal CD117 has been described in a small number of HPC [Citation[71]].
Neoplastic cells in HPC are described ultrastructurally as having overlapping processes with poorly developed junctions, focal myoid differentiation, and a fragmented external lamina between tumor cells and between them and the endothelium [Citation[68], Citation[69]]. In SFT, the cells are fibroblast-like without external lamina, with rare myofibroblasts [Citation[46], Citation[60]]. Occasionally myofilaments are seen in pericytes, and recently a group of tumors has been described with perivascular myoid differentiation [Citation[72]]. This group included tumors with histologic features of infantile-type myofibromatosis, tumors with composite features of “hemangiopericytoma” and glomus tumor, and tumors with a distinctive concentric perivascular proliferation of spindle cells. Morphologic overlap among the groups suggests that they are part of a single spectrum, termed perivascular myomas or myopericytomas, and some of these resemble and perhaps overlap with vascular leiomyoma. Strictly speaking, since they show specific differentiation these should be excluded from the HPC category.
Cytogenetic studies can exclude or identify specific tumor types with a hemangiopericytomatous pattern, such as synovial sarcoma with its t(X;18)(p11.2;q11.2) [Citation[73]] and round cell liposarcoma, which predominantly shows t(12;16)(q13;p11). Changes described n HPC have included t(1;3)(q22;q11) and t(7;12)(p22;q13) [Citation[74]]; t(12;19)(q13;q13.3) [Citation[75]] and t(13;22)(q22;q11) [Citation[76]]. Thus, 12q13 is implicated [Citation[77]], but this segment is rearranged in several soft tissue sarcomas and the findings are not specific [Citation[78]].
The behavior of HPC cannot always be predicted from the histology. A benign HPC lacks cellular pleomorphism, necrosis, and hemorrhage and has a low mitotic rate (either < 4/10 hpf [Citation[79]] or < 1/20 hpf [Citation[80]]). With this definition, around 70% of HPC behave in a benign fashion, but some histologically benign cases have metastasized. The existence of a borderline grade (<4 mitoses/10 hpf, <5% necrosis) has been suggested [Citation[81]] but the numbers are too small to assess the validity of this study. Clear-cut malignant cases, with necrosis and mitotic rates higher than above, have 5- and 10-year survival rates of around 40 and 29%, respectively.
In conclusion, most soft tissue tumors with a hemangiopericytomatous pattern can be given a more specific diagnosis. Many are now classified as solitary fibrous tumors, especially if they show varying cellularity and fibrosis and diffuse immunoreactivity for CD34 and bcl2 and often CD99. Some remain that can be termed hemangiopericytoma, although there is no conclusive evidence that the tumor cells are derived from pericytes.
Lipomatous hemangiopericytomas are soft tissue tumors in adults that have been described with an HPC pattern and a component of mature fat [Citation[49–52]], mostly in males, in deep soft tissue, mediastinum, or retroperitoneum. They are circumscribed and display an intricate mix of HPC areas and adipose tissue, with the 2 components occasionally being discrete. Perivascular or stromal hyalinization and focal myxoid change can be seen. Atypia is rare and the reported lesions have been benign. Cases are generally CD99 positive but expression of CD34 and bcl2 has been variable. Folpe et al. found CD34 in 2 of 4 cases [Citation[51]], and in the series of Guillou et al. 7 of 12 were bcl2 positive and 10 of 13 were CD34 positive (one case expressed neither) [Citation[52]], so that at least some of these might represent variants of SFT [Citation[52]]. Sinonasal hemangiopericytoma is a CD34-negative lesion with smooth muscle-like features that might represent differentiation toward a glomus or perivascular myoid cell [Citation[82–84]] and appears to be unrelated to HPC or SFT elsewhere.
LOW-GRADE SARCOMAS OF MYOFIBROBLASTS
Myofibroblasts are modified fibroblasts [Citation[85], Citation[86]] that were first described in granulation tissue [Citation[87]] and have since been identified in normal tissues and as the predominant cell in certain reactive lesions. In granulation tissue, in which they have been most intensively investigated, they are probably derived from local fibroblasts [Citation[87]], in response to mechanical stress, and their functions, appearance, and immunoprofile vary in relation to the phase of activity [Citation[88]]. This is reflected in the variable morphology of reactive lesions, such as nodular fasciitis, and contributes to the range of appearances seen in other pathological conditions. Myofibroblast-like cells are sometimes derived from vascular smooth muscle cells or from pericytes [Citation[86]], or by metaplasia as in spindled carcinomas [Citation[89]], but these phenomena are less well documented. Some fibroblastic lesions have a component of myofibroblasts, but myofibroblasts do not apparently differentiate to smooth muscle cells. In wound healing, as epithelialization is completed, the myofibroblasts are presumed to disappear by apoptosis [Citation[90–92]].
Myofibroblasts are short, bi- or tripolar spindle-shaped or stellate cells, with crenated or ovoid pale-staining nuclei, each of which has a single distinct, punctate nucleolus. There is sparse to moderate amphophilic cytoplasm with indistinct cell margins that can appear to blend with the stroma. In addition to synthesizing collagens and other stromal components, including fibronectin and laminin, myofibroblasts have contractile elements [Citation[88], Citation[91–94]]. By electron microscopy, the cytoplasm has abundant rough endoplasmic reticulum and subplasmalemmal bundles of thin cytoplasmic filaments with dense foci (). This differs from smooth muscle cells in which myofilament bundles with focal dense bodies are distributed throughout the cytoplasm, and which have surface features of pinocytotic vesicles, membrane thickenings, and external lamina. The intracellular microfilament bundles (stress fibers) traverse the cell membrane to join extracellular, fibronectin fibrils as the fibronexus adhesion complex. This is a distinctive and specific cell-to-stromal attachment, not found in smooth muscle cells, which is seen in cells in reactive and other myofibroblastic lesion [Citation[95]]. Other ultrastructural features of myofibroblasts include a prominent Golgi complex and collagen secretion granules in Golgi-derived vesicles.
Fig. 12 (a) Myofibroblast showing fibronexus fibrils in continuity with intracytoplasmic “stress fibers.” (b) Stellate myofibroblast from a case of nodular fasciitis, showing peripheral filament bundles and abundant rough endoplasmic reticulum in each of the arms of the cell.

Fibroblasts have variable amounts of rough endoplasmic reticulum, which can become distended with secretory products, but they lack external lamina, pinocytosis, and organized cytoplasmic filaments. Fibroblasts express only vimentin, whereas myofibroblasts display various combinations of vimentin, actin isoforms (especially α-smooth muscle actin), desmin, and myosin [Citation[96], Citation[97]]. Desmin is found less often in myofibroblastic neoplasms than in smooth muscle tumors [Citation[98]], but both types of neoplasms can have detectable desmin, muscle-specific-actin (MSA), and smooth muscle actin (SMA). Myofibroblasts express calponin but usually lack h-caldesmon, a marker of smooth muscle differentiation [Citation[99]]. Some myofibroblastic lesions can show cytoplasmic (but not usually membranous) immunoreactivity for CD117 (c-kit); this varies with technical factors, including the antibody and whether there is antigen retrieval [Citation[100]]. In intra-abdominal lesions, this can lead to confusion with gastrointestinal stromal tumor [Citation[71]].
Benign neoplasms of myofibroblasts occur in soft tissues, bone, and visceral locations. The term myofibrosarcoma, by analogy with the term fibrosarcoma, indicates a malignant tumor of myofibroblasts [Citation[101]]. Myofibrosarcomas display a range of appearances from fasciitis-like neoplasms to high-grade sarcomas. Low- and intermediate-grade myofibrosarcomas are distinct from pleomorphic myofibrosarcomas, which are malignant fibrous histiocytoma-like tumors. Other low-grade malignant tumors with myofibroblasts include inflammatory myofibroblastic tumor and infantile fibrosarcoma, each of which has characteristic genetic abnormalities.
The myofibrosarcoma concept has been controversial. Many fibroblastic lesions have myofibroblasts, and the proportion of neoplastic myofibroblasts required is not defined. Also, it is difficult to confirm myofibroblastic differentiation without electron microscopy, and there is no universal agreement on the ultrastructural criteria of the neoplastic myofibroblast [Citation[102], Citation[103]]. Some authors require that stress fibers and the fibronexus, a characteristic feature of nonneoplastic myofibroblasts, be present for a diagnosis of myofibrosarcoma [Citation[97]]. Since subcellular features can be incompletely or abnormally developed in malignant cells, the fibronexus has not been an absolute requirement for most investigators. Identification of a cell type depends on several features, and the fibronexus is only one of the features used to define myofibroblastic differentiation. This structure has, however, been reported in several myofibrosarcomas [Citation[104]], and has been illustrated more often in case reports than it has been mentioned by their authors [Citation[98], Citation[105]].
It has also been held that sarcomas supposedly composed of myofibroblasts are actually poorly developed smooth muscle tumors [Citation[106]]. Low-grade leiomyosarcomas, however, usually have well-developed smooth muscle differentiation. Furthermore, although the cytoplasmic filament patterns can be somewhat similar in myofibroblasts and smooth muscle cells, the 2 cell types differ in other respects—unlike myofibroblasts, smooth muscle cells have external lamina and cell membrane plaques as well as pinocytotic vesicles. Leiomyosarcoma and myofibrosarcoma can both display actins and desmin, but the latter is less often seen in myofibroblastic neoplasms. Also, in myofibrosarcomas, the actin staining is often manifested as a peripheral rim rather than throughout the cytoplasm [Citation[107]]. The paucity of h-caldesmon in myofibroblastic lesions additionally supports their separate identity [Citation[99], Citation[108]].
Thus, the ultrastructural features and immunohistochemistry, in conjunction with the morphology, are sufficiently distinctive to identify myofibroblastic differentiation in sarcomas. The 2 principal low-grade lesions in this category are myofibrosarcoma and inflammatory myofibroblastic tumor.
Low-grade Myofibrosarcoma
This is a distinctive neoplasm that occurs in soft tissues and bone as a fasciitis- or fibrosarcoma-like spindle cell sarcoma that infiltrates locally but rarely metastasizes. Myofibroblasts were observed in well-differentiated fibrosarcoma in 1975 [Citation[109]], and the first “sarcoma of myofibroblasts” was reported 3 years later [Citation[110]]. About 50 cases have since appeared in the literature [Citation[104], Citation[111–122]].
Low-grade myofibrosarcomas occur at any age (range 7–85 years, mean 40) with a slight male predominance and tumor size ranging between 1.5 and 17 cm. Tumors can arise in extremities, and trunk including breast, or retroperitoneum, but there is a predilection (up to one-third of cases) for the head and neck region in both soft tissue, including the oral cavity, especially the tongue, face and neck, and bone, notably maxilla and mandible.
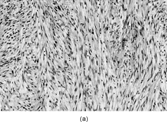
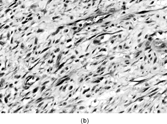
Myofibrosarcomas can be found in subcutis or submucosa, but are mostly deep soft tissue or intraosseous tumors. They can be circumscribed, but mostly infiltrate irregularly along connective tissue septa or into skeletal muscle. The tumors are composed of mostly bland or focally pleomorphic stellate or spindled cells with tapered or ovoid nuclei with small nucleoli, and scanty or moderate amounts of eosinophilic cytoplasm. The patterns include fibrosarcoma-like fascicles, sheets, or storiform whorls, with variable collagenous or myxoid stroma and scanty inflammation (). Nuclear atypia is always present at least focally and usually mildly with scattered enlarged hyperchromatic nuclei, but larger atypical cells are sometimes seen. Mitotic activity is observed but necrosis is unusual. Recurrences tend to be more pleomorphic [Citation[119], Citation[123]], but in one example a breast metastasis from a grade 2 fibrosarcoma-like tumor resembled nodular fasciitis [Citation[119]].
Fig. 13 Low-grade myofibrosarcoma. (a) Moderately cellular tumor with fascicular architecture and scanty lymphocytic infiltrate. (b) Cells are mildly pleomorphic with variable nuclear staining; some have small nucleoli. (c) Myxoid areas resemble nodular fasciitis but differ in displaying nuclear atypia.

Electron microscopy shows myofibroblasts that have variable RER, cytoplasmic filament bundles (), and in some cases minimal external lamina. Fibronexus structures and collagen secretion granules are sometimes found.
Fig. 14 Ultrastructure of low-grade myofibrosarcoma. This neoplastic cell has peripheral filament bundles and plentiful rough endoplasmic reticulum, and stress fibers with fibronexus.
Low-grade myofibrosarcomas express actin (as a peripheral rim, beneath the cell membrane), and about one-half express desmin, usually in fewer cells [Citation[118], Citation[119]]. These antigens can be expressed separately, with either a desmin-positive/SMA-negative or desmin-negative/SMA-positive immunophenotype [Citation[118]]. Calponin is diffusely positive, but h-caldesmon is only focally expressed in an occasional case [Citation[124]]. These findings are similar to those in nodular fasciitis, and differ somewhat from leiomyosarcoma, which usually displays diffuse h-caldesmon as well as calponin. Fibronectin [Citation[125]] has been found in some myofibrosarcomas but not collagen IV or laminin. Rare myofibrosarcomas focally display cytokeratins or CD34 [Citation[118]], and one example had focal S-100 protein positivity [Citation[113]]. ALK-1 expression has not been reported.
Low-grade myofibrosarcomas are indolent, but they can relapse and metastasize even after a long period. In one series, there was local recurrence in 2 of 11 patients and metastasis in one [Citation[118]]. In the series of Montgomery et al. [Citation[119]], 4 of 9 low-grade myofibrosarcomas, and 3 of 4 intermediate-grade tumors recurred (1 twice), and 1 intermediate-grade tumor, which arose in the breast, resulted in pulmonary metastasis after 12 months.
The differential diagnosis of myofibrosarcoma includes benign myofibroblastic lesions, such as nodular fasciitis and fibromatosis, and other sarcomas, notably fibrosarcoma and leiomyosarcoma. Both clinical data and morphology are important for diagnosis, since immunohistochemistry does not discriminate between myofibroblastic lesions and the immunophenotype overlaps with that of smooth muscle ().
Table 1 Some Tumors with Hemangiopericytomatous Pattern
Table 2 Immunohistochemistry of Smooth Muscle and Fibro/Myofibroblastic Tumors
Nodular fasciitis is a mainly subcutaneous lesion that appears suddenly and grows rapidly but does not usually exceed 5 cm in diameter. There are myxoid, cellular, and fibrous areas, often in different parts of the same lesion. Nuclear atypia and necrosis are absent. Myofibrosarcoma is more cellular and uniform than nodular fasciitis, and infiltrates more widely. Fibromatosis has files of slender spindle cells in dense collagen with slit-like blood vessels and mast cells, and lacks nuclear atypia. Fibromatosis also infiltrates skeletal muscle, but muscle fibers show atrophy rather than separation by tumor as in myofibrosarcoma.
Adult-type fibrosarcoma has a more herringbone-like fascicular architecture, and cells with scanty cytoplasm and elongated, tapered nuclei. There is variable intercellular collagen and myoid markers are usually absent. Leiomyosarcoma typically has alternating fascicles of cells that are more parallel-sided with square-ended nuclei scattered paranuclear vacuoles. Caldesmon expression is more widespread (). Other spindle cell sarcomas, such as synovial sarcoma, malignant peripheral nerve sheath tumor, and spindle cell rhabdomyosarcoma, can be recognized by their morphology and by their specific immunophenotype. In head and neck and breast tumors, spindle cell carcinoma has to be excluded by use of multiple cytokeratin antibodies.
Inflammatory Myofibroblastic Tumor
Inflammatory myofibroblastic tumor (IMT) is most commonly seen in the lung in childhood. Extrapulmonary tumors usually arise within the abdomen (retroperitoneum or mesentery) as a solitary or multicentric mass [Citation[126], Citation[127]] with peak incidence in the first or second decades with a slight female predominance. B-type symptoms, anemia and hypergammaglobulinemia, are sometimes present. Similar cases have also been termed inflammatory fibrosarcoma [Citation[128]], but these conditions are very similar and probably represent a single entity with a spectrum of morphology and behavior [Citation[129]], i.e., a low-grade neoplasm of myofibroblasts.
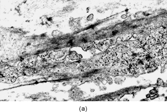
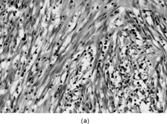
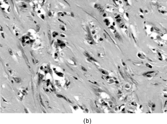
IMT presents with multinodular and sometimes multiple firm, white or yellow tumors up to 10 cm in diameter. Histologically () these are irregularly infiltrative lesions with 3 principal patterns, usually found in combination: (1) fasciitis-like, with bland stellate or short spindle cells in a vascular, myxoid and inflamed stroma, including numerous plasma cells; (2) fascicular and fibrosarcoma- or leiomyosarcoma-like, again with marked inflammation; and (3) hypocellular areas with hyalinization and calcification. The lesional cells are usually uniform, but atypical cells with prominent nucleoli [Citation[128]], and ganglion-like or Reed-Sternberg-like cells can occasionally be seen [Citation[130]].
Fig. 15 Inflammatory myofibroblastic tumor. (a) Long, bland, spindle-shaped cells in fascicular and fasciitis-like patterns with numerous plasma cells. (b) Sclerosing area with scanty lesional cells and persistent plasma cell infiltrate.
Ultrastructurally, there is a mixture of cells with fibroblastic and myofibroblastic features [Citation[95], Citation[127], Citation[128], Citation[131]]. However, there is little documentation of fibronexus structures being identified in inflammatory myofibroblastic tumors. Immunohistochemically, most cases are positive for smooth muscle and muscle-specific actins, and a smaller number for desmin; some cases (especially those in a submesothelial location) express cytokeratins. ALK immunostaining is positive in 36 to 60% [Citation[132–134]] of cases, with a granular pattern in cytoplasm or nucleus, and sometimes cell and nuclear membranous accentuation; the variant patterns possibly relating to different fusion genes. ALK expression, found predominantly in abdominal and pulmonary IMT in childhood, might be associated with an improved outcome.
Genetically, many IMT have clonal chromosomal abnormalities involving 2p22–24, and fusion of the anaplastic lymphoma kinase (ALK) gene, located on 2p23, which encodes a tyrosine kinase receptor, with tropomyosin 3 (TPM3-ALK) or tropomyosin 4 (TPM4-ALK) is found in a subset [Citation[135]]. A transcript involving ALK and CLTC (clathrin heavy chain gene, localized to 17q23) has additionally been reported [Citation[136]].
A third of cases, especially among intra-abdominal tumors, recur at least once. IMT can have increasing atypia with recurrence, and metastasizing sarcomatous change, has been reported [Citation[137]]. However, no factors have been identified that reliably predict behavior in these tumors. It has been suggested that the presence of atypia, ganglion-like cells, p53 expression, and aneuploidy might be useful to identify IMT that might undergo pursue a more aggressive clinical course [Citation[138], Citation[139]]. Surgical resection is the usual treatment for both primary and recurrent tumors.
The differential diagnosis relates to the various histological patterns. Retroperitoneal fibrosis has a distinctive clinical picture, is more inflammatory with mixed cells, and lacks pleomorphism. Fibromatosis has a distinctive architecture (intra-abdominal fibromatosis can have prominent keloidal collagen bundles) and is infiltrated by mast cells rather than plasma cells. Low-grade myxofibrosarcoma has a more monomorphous pattern and lacks the prominent inflammatory component. Inflammatory MFH has atypical xanthomatous cells, while inflammatory leiomyosarcoma is more myoid, although the distinction can sometimes be difficult. Solitary fibrous tumor demonstrates CD34 expression, and sarcomatoid mesothelioma displays epithelial markers. Follicular dendritic cell sarcoma can masquerade as inflammatory pseudotumor in liver or spleen (see above); but is readily identified by expression of CD21, CD23, and CD35. When there is intestinal wall involvement or a prominent fascicular architecture, the possibility of GI stromal tumor may be raised. This has shorter, plumper cells and less inflammation, and immunostaining with CD117 is positive with a membranous accentuation, in a different pattern from the weaker staining seen in myofibroblastic lesions.
Related Research Data
REFERENCES
- van de Rijn M, Rouse RV. CD34: a review. Appl Immunohistochem. 1994; 2: 71–80
- van de Rijn M, Lombard CM, Rouse RV. Expression of CD34 by solitary fibrous tumors of the pleura, mediastinum, and lung. Am J Surg Pathol. 1994; 18: 814–820. [PUBMED], [INFOTRIEVE]
- Meis-Kindblom JM, Kindblom LG.. Acral myxoinflammatory fibroblastic sarcoma: a low-grade tumor of the hands and feet. Am J Surg Pathol. 1998; 22: 911–924. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Rubin BP, Schuetze SM, Eary JF, et al, Molecular targeting of platelet-derived growth factor B by imatinib mesylate in a patient with metastatic dermatofibrosarcoma protuberans. J Clin Oncol. 2002; 20: 3586–3591. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Maki RG, Awan RA, Dixon RH, Jhanwar S, Antonescu CR.. Differential sensitivity to imatinib of 2 patients with metastatic sarcoma arising from dermatofibrosarcoma protuberans. Int J Cancer. 2002; 100: 623–626. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Dominguez-Malagon HR, Ordonez NG, Mackay B.. Dermatofibrosarcoma protuberans: ultrastructural and immunocytochemical observations. Ultrastruct Pathol. 1995; 19: 281–289. [PUBMED], [INFOTRIEVE], [CSA]
- Calonje E, Fletcher CD.. Myoid differentiation in dermatofibrosarcoma protuberans and its fibrosarcomatous variant: clinicopathologic analysis of 5 cases. J Cutan Pathol. 1996; 23: 30–36. [PUBMED], [INFOTRIEVE], [CSA]
- Diaz-Cascajo C.. Myoid differentiation in dermatofibrosarcoma protuberans and its fibrosarcomatous variant. J Cutan Pathol. 1997; 24: 197–198. [PUBMED], [INFOTRIEVE], [CSA]
- Mentzel T, Beham A, Katenkamp D, Dei Tos AP, Fletcher CD.. Fibrosarcomatous (“high-grade”) dermatofibrosarcoma protuberans: clinicopathologic and immunohistochemical study of a series of 41 cases with emphasis on prognostic significance. Am J Surg Pathol. 1998; 22: 576–587. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Morimitsu Y, Hisaoka M, Okamoto S, Hashimoto H, Ushijima M.. Dermatofibrosarcoma protuberans and its fibrosarcomatous variant with areas of myoid differentiation: a report of three cases. Histopathology. 1998; 32: 547–551. [PUBMED], [INFOTRIEVE], [CSA]
- O′Connell JX, Trotter MJ.. Fibrosarcomatous dermatofibrosarcoma protuberans with myofibroblastic differentiation: a histologically distinctive variant [corrected]. Mod Pathol. 1996; 9: 273–278. [CSA]
- Sanz-Trelles A, Ayala-Carbonero A, Rodrigo-Fernandez I, Weil-Lara B.. Leiomyomatous nodules and bundles of vascular origin in the fibrosarcomatous variant of dermatofibrosarcoma protuberans. J Cutan Pathol. 1998; 25: 44–49. [PUBMED], [INFOTRIEVE], [CSA]
- Wang J, Zhu X, Yosuke M, Masanori H, Hiroshi H.. [Myoid/myofibroblastic differentiation in dermatofibrosarcoma protuberans: a clinicopathologic study of six cases]. Zhonghua Bing Li Xue Za Zhi. 2001; 30: 12–15. [PUBMED], [INFOTRIEVE], [CSA]
- Hsi ED, Nickoloff BJ.. Dermatofibroma and dermatofibrosarcoma protuberans: an immunohistochemical study reveals distinctive antigenic profiles. J Dermatol Sci. 1996; 11: 1–9. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Oliveira-Soares R, Viana I, Vale E, Soares-Almeida LM, Picoto A.. Dermatofibrosarcoma protuberans: a clinicopathological study of 20 cases. J Eur Acad Dermatol Venereol. 2002; 16: 441–446. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Pedeutour F, Simon MP, Minoletti F, et al, Translocation, t(17;22)(q22;q13), in dermatofibrosarcoma protuberans: a new tumor-associated chromosome rearrangement. Cytogenet Cell Genet. 1996; 72: 171–174. [PUBMED], [INFOTRIEVE], [CSA]
- Simon MP, Pedeutour F, Sirvent N, et al, Deregulation of the platelet-derived growth factor B-chain gene via fusion with collagen gene COL1A1 in dermatofibrosarcoma protuberans and giant-cell fibroblastoma. Nat Genet. 1997; 15: 95–98. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Wang J, Hisaoka M, Shimajiri S, Morimitsu Y, Hashimoto H.. Detection of COL1A1-PDGFB fusion transcripts in dermatofibrosarcoma protuberans by reverse transcription-polymerase chain reaction using archival formalin-fixed, paraffin-embedded tissues. Diagn Mol Pathol. 1999; 8: 113–119. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Sirvent N, Maire G, Pedeutour F.. Genetics of dermatofibrosarcoma protuberans family of tumors: from ring chromosomes to tyrosine kinase inhibitor treatment. Genes Chromosomes Cancer. 2003; 37: 1–19. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Sheng WQ, Hashimoto H, Okamoto S, et al, Expression of COL1A1-PDGFB fusion transcripts in superficial adult fibrosarcoma suggests a close relationship to dermatofibrosarcoma protuberans. J Pathol. 2001; 194: 88–94. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Sonobe H, Furihata M, Iwata J, et al, Dermatofibrosarcoma protuberans harboring t(9;22)(q32;q12.2). Cancer Genet Cytogenet. 1999; 110: 14–18. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Sandberg AA, Bridge JA.. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: dermatofibrosarcoma protuberans and giant cell fibroblastoma. Cancer Genet Cytogenet. 2003; 140: 1–12. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Dupree WB, Langloss JM, Weiss SW.. Pigmented dermatofibrosarcoma protuberans (Bednar tumor): a pathologic, ultrastructural, and immunohistochemical study. Am J Surg Pathol. 1985; 9: 630–639. [PUBMED], [INFOTRIEVE]
- Nishio J, Iwasaki H, Ishiguro M, et al, Supernumerary ring chromosome in a Bednar tumor (pigmented dermatofibrosarcoma protuberans) is composed of interspersed sequences from chromosomes 17 and 22: a fluorescence in situ hybridization and comparative genomic hybridization analysis. Genes Chromosomes Cancer. 2001; 30: 305–309. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Bisceglia M, Vairo M, Calonje E, Fletcher CD.. [Pigmented fibrosarcomatous dermatofibrosarcoma protuberans (Bednar tumor): 3 case reports, analogy with the “conventional” type and review of the literature]. Pathologica. 1997; 89: 264–273. [PUBMED], [INFOTRIEVE], [CSA]
- Zamecnik M, Michal M.. Myxoid variant of dermatofibrosarcoma protuberans with fibrosarcomatous areas. Zentralbl Pathol. 1993; 139: 373–376. [PUBMED], [INFOTRIEVE]
- Wrotnowski U, Cooper PH, Shmookler BM.. Fibrosarcomatous change in dermatofibrosarcoma protuberans. Am J Surg Pathol. 1988; 12: 287–293. [PUBMED], [INFOTRIEVE]
- Connelly JH, Evans HL.. Dermatofibrosarcoma protuberans: a clinicopathologic review with emphasis on fibrosarcomatous areas. Am J Surg Pathol. 1992; 16: 921–925. [PUBMED], [INFOTRIEVE]
- Diaz-Cascajo C, Weyers W, Borrego L, Inarrea JB, Borghi S.. Dermatofibrosarcoma protuberans with fibrosarcomatous areas: a clinico-pathologic and immunohistochemic study in four cases. Am J Dermatopathol. 1997; 19: 562–567. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Ding J, Hashimoto H, Enjoji M.. Dermatofibrosarcoma protuberans with fibrosarcomatous areas: a clinicopathologic study of nine cases and a comparison with allied tumors. Cancer. 1989; 64: 721–729. [PUBMED], [INFOTRIEVE]
- Lopes JM, Paiva ME.. Dermatofibrosarcoma protuberans: a histological and ultrastructural study of 11 cases with emphasis on the study of recurrences and histogenesis. Pathol Res Pract.. 1991; 187: 806–813. [PUBMED], [INFOTRIEVE]
- Goldblum JR.. CD34 positivity in fibrosarcomas which arise in dermatofibrosarcoma protuberans. Arch Pathol Lab Med. 1995; 119: 238–241. [PUBMED], [INFOTRIEVE], [CSA]
- Goldblum JR, Reith JD, Weiss SW.. Sarcomas arising in dermatofibrosarcoma protuberans: a reappraisal of biologic behavior in eighteen cases treated by wide local excision with extended clinical follow up. Am J Surg Pathol. 2000; 24: 1125–1130. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Terrier-Lacombe MJ, Guillou L, Maire G. Terrier P, et al. Dermatofibrosarcoma protuberans, giant cell fibroblastoma, and hybrid lesions in children: clinicopathologic comparative analysis of 28 cases with molecular data—a study from the French Federation of Cancer Centers Sarcoma Group. Am J Surg Pathol. 2003; 27: 27–39. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Karabela-Bouropoulou V, Liapi-Avgeri G, Mahera H, et al, Giant cell fibroblastoma: an entity or a reactive phenomenon?. Pathol Res Pract.. 1999; 195: 413–419. [PUBMED], [INFOTRIEVE], [CSA]
- Perry DA, Schultz LR, Dehner LP.. Giant cell fibroblastoma with dermatofibrosarcoma protuberans-like transformation. J Cutan Pathol. 1993; 20: 451–454. [PUBMED], [INFOTRIEVE]
- Shmookler BM, Enzinger FM, Weiss SW.. Giant cell fibroblastoma: a juvenile form of dermatofibrosarcoma protuberans. Cancer. 1989; 64: 2154–2161. [PUBMED], [INFOTRIEVE]
- Beham A, Fletcher CD.. Dermatofibrosarcoma protuberans with areas resembling giant cell fibroblastoma: report of two cases. Histopathology. 1990; 17: 165–167. [PUBMED], [INFOTRIEVE]
- Alguacil-Garcia A.. Giant cell fibroblastoma recurring as dermatofibrosarcoma protuberans. Am J Surg Pathol. 1991; 15: 798–801. [PUBMED], [INFOTRIEVE]
- Diaz-Cascajo C, Borrego L, Bastida-Inarrea J, Borghi S.. Giant cell fibroblastoma: new histological observations. Am J Dermatopathol. 1996; 18: 403–408. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Goldblum JR.. Giant cell fibroblastoma: a report of three cases with histologic and immunohistochemical evidence of a relationship to dermatofibrosarcoma protuberans. Arch Pathol Lab Med. 1996; 120: 1052–1055. [PUBMED], [INFOTRIEVE], [CSA]
- Maire G, Martin L, Michalak-Provost S, et al, Fusion of COL1A1 exon 29 with PDGFB exon 2 in a der(22)t(17;22) in a pediatric giant cell fibroblastoma with a pigmented Bednar tumor component: evidence for age-related chromosomal pattern in dermatofibrosarcoma protuberans and related tumors. Cancer Genet Cytogenet. 2002; 134: 156–161. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Vanni R, Faa G, Dettori T, Melis GB, Dumanski JP, O′Brien KP.. A case of dermatofibrosarcoma protuberans of the vulva with a COL1A1/PDGFB fusion identical to a case of giant cell fibroblastoma. Virchows Arch. 2000; 437: 95–100. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Chan JK.. Solitary fibrous tumour—everywhere, and a diagnosis in vogue. Histopathology. 1997; 31: 568–576. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Suster S, Nascimento AG, Miettinen M, Sickel JZ, Moran CA.. Solitary fibrous tumors of soft tissue: a clinicopathologic and immunohistochemical study of 12 cases. Am J Surg Pathol. 1995; 19: 1257–1266. [PUBMED], [INFOTRIEVE], [CSA]
- Hasegawa T, Hirose T, Seki K, Yang P, Sano T.. Solitary fibrous tumor of the soft tissue: an immunohistochemical and ultrastructural study. Am J Clin Pathol. 1996; 106: 325–331. [PUBMED], [INFOTRIEVE], [CSA]
- Vallat-Decouvelaere AV, Dry SM, Fletcher CD.. Atypical and malignant solitary fibrous tumors in extrathoracic locations: evidence of their comparability to intra-thoracic tumors. Am J Surg Pathol. 1998; 22: 1501–1511. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Morimitsu Y, Nakajima M, Hisaoka M, Hashimoto H.. Extrapleural solitary fibrous tumor: clinicopathologic study of 17 cases and molecular analysis of the p53 pathway. Apmis. 2000; 108: 617–625. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Nielsen GP, Dickersin GR, Provenzal JM, Rosenberg AE.. Lipomatous hemangiopericytoma: a histologic, ultrastructural and immunohistochemical study of a unique variant of hemangiopericytoma. Am J Surg Pathol. 1995; 19: 748–756. [PUBMED], [INFOTRIEVE], [CSA]
- Ceballos KM, Munk PL, Masri BA, O′Connell JX.. Lipomatous hemangiopericytoma: a morphologically distinct soft tissue tumor. Arch Pathol Lab Med. 1999; 123: 941–945. [PUBMED], [INFOTRIEVE], [CSA]
- Folpe AL, Devaney K, Weiss SW.. Lipomatous hemangiopericytoma: a rare variant of hemangiopericytoma that may be confused with liposarcoma. Am J Surg Pathol. 1999; 23: 1201–1207. [PUBMED], [INFOTRIEVE], [CSA]
- Guillou L, Gebhard S, Coindre JM.. Lipomatous hemangiopericytoma: a fat-containing variant of solitary fibrous tumor? Clinicopathologic, immunohistochemical, and ultrastructural analysis of a series in favor of a unifying concept. Hum Pathol. 2000; 31: 1108–1115. [PUBMED], [INFOTRIEVE], [CSA]
- Chilosi M, Facchettti F, Dei Tos AP, et al, bcl-2 expression in pleural and extrapleural solitary fibrous tumours. J Pathol. 1997; 181: 362–367. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Suster S, Fisher C, Moran CA.. Expression of bcl-2 oncoprotein in benign and malignant spindle cell tumors of soft tissue, skin, serosal surfaces, and gastrointestinal tract. Am J Surg Pathol. 1998; 22: 863–872. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Renshaw AA.. 013 (CD99) in spindle cell tumors: reactivity with hemangiopericytoma, solitary fibrous tumor, synovial sarcoma and meningioma but rarely with sarcomatoid mesothelioma. Appl Immunohistochem. 1995; 3: 250–256
- Dal Cin P, Pauwels P, Van Den Berghe H.. Solitary fibrous tumour of the pleura with t(4;15)(q13;q26). Histopathology. 1999; 35: 94–95. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Donner LR, Silva MT, Dobin SM.. Solitary fibrous tumor of the pleura: a cytogenetic study. Cancer Genet Cytogenet. 1999; 111: 169–171. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Havlik DM, Farnath DA, Bocklage T.. Solitary fibrous tumor of the orbit with a t(9;22)(q31;p13). Arch Pathol Lab Med. 2000; 124: 756–758. [PUBMED], [INFOTRIEVE], [CSA]
- de Saint Aubain Somerhausen N, Rubin BP, Fletcher CD.. Myxoid solitary fibrous tumor: a study of seven cases with emphasis on differential diagnosis. Mod Pathol. 1999; 12: 463–471. [CSA]
- Nielsen GP, O′Connell JX, Dickersin GR, Rosenberg AE.. Solitary fibrous tumor of soft tissue: a report of 15 cases, including 5 malignant examples with light microscopic, immunohistochemical, and ultrastructural data. Mod Pathol. 1997; 10: 1028–1037. [PUBMED], [INFOTRIEVE], [CSA]
- Stout AP, Murray MR.. Hemangiopericytoma: a vascular tumor featuring Zimmerman's pericytes. Ann Surg. 1942; 116: 26–33
- Stout AP.. Hemangiopericytoma: a study of 25 new cases. Cancer. 1949; 2: 1027–1054. [PUBMED], [INFOTRIEVE]
- Nappi O, Ritter JH, Pettinato G, Wick MR.. Hemangiopericytoma: histopathological pattern or clinicopathologic entity?. Semin Diagn Pathol. 1995; 12: 221–232. [PUBMED], [INFOTRIEVE], [CSA]
- Nehls V, Drenckhahn D.. Heterogeneity of microvascular pericytes for smooth muscle type alpha-actin. J Cell Biol. 1991; 113: 147–154. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Schor AM, Canfield AE, Sutton AB, Arciniegas E, Allen TD.. Pericyte differentiation: clinical. Orthop Related Res. 1995; 81–91
- Iwasaki H, Isayama T, Johzaki H, Kikuchi M.. Malignant fibrous histiocytoma: evidence of perivascular mesenchymal cell origin immunocytochemical studies with monoclonal anti-MFH antibodies. Am J Pathol. 1987; 128: 528–537. [PUBMED], [INFOTRIEVE]
- Bonetti F, Pea M, Martignoni G, et al, The perivascular epithelioid cell and related lesions. Adv Anat Pathol. 1997; 6: 343–358
- Dardick I, Hammar SP, Scheithauer BW.. Ultrastructural spectrum of hemangiopericytoma: a comparative study of fetal, adult and neoplastic pericytes. Ultrastruct Pathol. 1989; 13: 111–154. [PUBMED], [INFOTRIEVE]
- Battifora H.. Hemangiopericytoma: ultrastructural study of five cases. Cancer. 1973; 31: 1418–1432. [PUBMED], [INFOTRIEVE]
- Nemes Z.. Differentiation markers in hemangiopericytoma. Cancer. 1992; 69: 133–140
- Sarlomo-Rikala M, Kovatich AJ, Barusevicius A, Miettinen M.. CD117: a sensitive marker for gastrointestinal stromal tumors that is more specific than CD34. Mod Pathol. 1998; 11: 728–734. [PUBMED], [INFOTRIEVE], [CSA]
- Granter SR, Badizadegan K, Fletcher CD.. Myofibromatosis in adults, glomangiopericytoma, and myopericytoma: a spectrum of tumors showing perivascular myoid differentiation. Am J Surg Pathol. 1998; 22: 513–525. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Fisher C.. Synovial sarcoma. Ann Diagn Pathol. 1998; 2: 401–421. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Perez-Atayde AR, Kozakewich HW, McGill T, Fletcher JA.. Hemangiopericytoma of the tongue in a 12-year-old child: ultrastructural and cytogenetic observations. Hum Pathol. 1994; 25: 425–429. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Henn W, Wullich B, Thonnes M, Steudel WI, Feiden W, Zang KD.. Recurrent t(12;19)(q13;q13.3) in intracranial and extracranial hemangiopericytoma. Cancer Genet Cytogenet. 1993; 71: 151–154. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Limon J, Rao U, Dal Cin P, Gibas Z, Sandberg AA.. Translocation (13;22) in a hemangiopericytoma. Cancer Genet Cytogenet. 1986; 21: 309–318. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Mandahl N, Örndal C, Heim S, et al, Aberrations of chromosome segment 12q13-15 characterize a subgroup of hemangiopericytomas. Cancer. 1993; 71: 3009–3013. [PUBMED], [INFOTRIEVE]
- Jankowski SA, Mitchell DS, Smith SH, Trent JM, Meltzer PS.. SAS, a gene amplified in human sarcomas encodes a new member of the transmembrane 5 superfamily of proteins. Oncogene. 1994; 9: 1205–1211. [PUBMED], [INFOTRIEVE], [CSA]
- Enzinger FM, Smith BH.. Hemangiopericytoma: an analysis of 106 cases. Hum Pathol. 1976; 7: 61–82. [PUBMED], [INFOTRIEVE]
- McMaster MJ, Soule EH, Ivins JC.. Hemangiopericytoma: a clinicopathologic study and long-term followup of 60 patients. Cancer. 1975; 36: 2232–2244. [PUBMED], [INFOTRIEVE]
- Yu CC, Hall PA, Fletcher CD, et al, Haemangiopericytomas: the prognostic value of immunohistochemical staining with a monoclonal antibody to proliferating cell nuclear antigen (PCNA). Histopathology. 1991; 19: 29–33. [PUBMED], [INFOTRIEVE]
- Eichhorn JH, Dickersin GR, Bhan AK, Goodman ML.. Sinonasal hemangiopericytoma: a reassessment with electron microscopy, immunohistochemistry, and long-term follow-up. Am J Surg Pathol. 1990; 14: 856–866. [PUBMED], [INFOTRIEVE]
- Thompson LD, Miettinen M, Wenig BM.. Sinonasal-type hemangiopericytoma: a clinicopathologic and immunophenotypic analysis of 104 cases showing perivascular myoid differentiation. Am J Surg Pathol. 2003; 27: 737–749. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Tse LL, Chan JK.. Sinonasal haemangiopericytoma-like tumour: a sinonasal glomus tumour or a haemangiopericytoma?. Histopathology. 2002; 40: 510–517. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Desmouliere A, Gabbiani G.. The role of the myofibroblast in wound healing and fibrocontractive diseases. Clark RAF. The Molecular and Cellular Biology of Wound Repair, ed 2., 1996; 391–423
- Eyden BP, Ponting J, Davies H, Bartley C, Torgersen E.. Defining the myofibroblast: normal tissues, with special reference to the stromal cells of Wharton's jelly in human umbilical cord. J Submicrosc Cytol PatholNew York, Plenum Press1994; 26: 347–355. [PUBMED], [INFOTRIEVE], [CSA]
- Gabbiani G, Ryan GB, Majno G.. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia. 1971; 27: 549–550. [PUBMED], [INFOTRIEVE]
- Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA.. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002; 3: 349–363. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Balercia G, Bhan AK, Dickersin GR.. Sarcomatoid carcinoma: an ultrastructural study with light microscopic and immunohistochemical correlation of 10 cases from various anatomic sites. Ultrastruct Pathol. 1995; 19: 249–263. [PUBMED], [INFOTRIEVE], [CSA]
- Darby I, Skalli O, Gabbiani G.. α-Smooth muscle actin is transiently expressed by myofibroblasts during experimental wound healing. Lab Invest. 1990; 63: 21–29. [PUBMED], [INFOTRIEVE], [CSA]
- Gabbiani G.. The cellular derivation and the life span of the myofibroblast. Pathol Res Pract. 1996; 192: 708–711. [PUBMED], [INFOTRIEVE], [CSA]
- Gabbiani G.. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol. 2003; 200: 500–503. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G.. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol. 1993; 122: 103–111. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Hinz B, Mastrangelo D, Iselin CE, Chaponnier C, Gabbiani G.. Mechanical tension controls granulation tissue contractile activity and myofibroblast differentiation. Am J Pathol. 2001; 159: 1009–1020. [PUBMED], [INFOTRIEVE]
- Eyden B.. The fibronexus in reactive and tumoral myofibroblasts: further characterisation by electron microscopy. Histol Histopathol. 2001; 16: 57–70. [PUBMED], [INFOTRIEVE], [CSA]
- Skalli O, Gabbiani G, Babaï, Seemayer TA, Pizzolato G, Schürch W.. Intermediate filament proteins and actin isoforms as markers for soft tissue tumor differentiation and origin, II: rhabdomyosarcomas. Am J Pathol. 1988; 130: 515–531. [PUBMED], [INFOTRIEVE]
- Schürch W, Seemayer TA, Gabbiani G.. Myofibroblast. Sternberg SS. Histology for Pathologists, ed 2., New York, Raven Press. 1997; 129–165
- Eyden B.. The myofibroblast: an assessment of controversial issues and a definition useful in diagnosis and research. Ultrastruct Pathol. 2001; 25: 39–50. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Ceballos KM, Nielsen GP, Selig MK, O′Connell JX.. Is anti-h-caldesmon useful for distinguishing smooth muscle and myofibroblastic tumors? An immunohistochemical study. Am J Clin Pathol. 2000; 114: 746–753. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Miettinen M.. Are desmoid tumors kit positive?. Am J Surg Pathol. 2001; 25: 549–550. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Ghadially FN.. Diagnostic Electron Microscopy of Tumours. London, Butterworths. 1980; 136
- Lagace R, Seemayer TA, Gabbiani G, Schurch W.. Myofibroblastic sarcoma. Am J Surg Pathol. 1999; 23: 1432–1435. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Erlandson RA.. Diagnostic Transmission Electron Microscopy of Tumors, with Clinico-pathological, Immunohistochemical and Cytogenetic Correlations. New York, Raven. 1994
- Eyden BP, Christensen L, Tagore V, Harris M.. Myofibrosarcoma of subcutaneous soft tissue of the cheek. J Submicrosc Cytol Pathol. 1992; 24: 307–313. [PUBMED], [INFOTRIEVE]
- Eyden B.. Electron microscopy in the study of myofibroblastic lesions. Semin Diagn Pathol. 2003; 20: 13–24. [PUBMED], [INFOTRIEVE], [CSA]
- Schurch W, Seemayer TA, Gabbiani G.. The myofibroblast: a quarter century after its discovery. Am J Surg Pathol. 1998; 22: 141–147. [PUBMED], [INFOTRIEVE], [CSA]
- Montgomery E, Fisher C.. Myofibroblastic differentiation in malignant fibrous histiocytoma (pleomorphic myofibrosarcoma): a clinicopathological study. Histopathology. 2001; 38: 499–509. [PUBMED], [INFOTRIEVE], [CSA]
- Miettinen MM, Sarlomo-Rikala M, Kovatich AJ, Lasota J.. Calponin and h-caldesmon in soft tissue tumors: consistent h-caldesmon immunoreactivity in gastrointestinal stromal tumors indicates traits of smooth muscle differentiation. Mod Pathol. 1999; 12: 756–762. [PUBMED], [INFOTRIEVE], [CSA]
- Stiller D, Katenkamp D.. Cellular features in desmoid fibromatosis and well-differentiated fibrosarcomas: an electron microscopic study. Virchows Arch A Pathol Anat Histol. 1975; 369: 155–164. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Vasudev KS, Harris M.. A sarcoma of myofibroblasts: an ultrastructural study. Arch Pathol Lab Med. 1978; 102: 185–188. [PUBMED], [INFOTRIEVE]
- Crocker DJ, Murad TM.. Ultrastructure of fibrosarcoma in a male breast. Cancer. 1969; 23: 891–899. [PUBMED], [INFOTRIEVE]
- d'Andiran G, Gabbiani G.. A metastasizing sarcoma of the pleura composed of myofibroblasts. Prog Surg Pathol. 1980; 2: 31–40
- Eyden BP, Banerjee SS, Harris M, Mene A.. A study of spindle cell sarcomas showing myofibroblastic differentiation. Ultrastruct Pathol. 1991; 15: 367–378. [PUBMED], [INFOTRIEVE]
- Smith DM, Mahmoud HH, Jenkins JJ, 3rd, Rao B, Hopkins KP, Parham DM.. Myofibrosarcoma of the head and neck in children. Pediatr Pathol Lab Med. 1995; 15: 403–418. [PUBMED], [INFOTRIEVE], [CSA]
- Taccagni G, Rovere E, Masullo M, Christensen L, Eyden B.. Myofibrosarcoma of the breast: review of the literature on myofibroblastic tumors and criteria for defining myofibroblastic differentiation. Am J Surg Pathol. 1997; 21: 489–496. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Bisceglia M, Magro G.. Low-grade myofibroblastic sarcoma of the salivary gland. Am J Surg Pathol. 1999; 23: 1435–1436. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Gocht A, Bosmuller HC, Bassler R, et al, Breast tumors with myofibroblastic differentiation: clinico-pathological observations in myofibroblastoma and myofibrosarcoma. Pathol Res Pract. 1999; 195: 1–10. [PUBMED], [INFOTRIEVE], [CSA]
- Mentzel T, Dry S, Katenkamp D, Fletcher CD.. Low-grade myofibroblastic sarcoma: analysis of 18 cases in the spectrum of myofibroblastic tumors. Am J Surg Pathol. 1998; 22: 1228–1238. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Montgomery E, Goldblum JR, Fisher C.. Myofibrosarcoma: a clinicopathologic study. Am J Surg Pathol. 2001; 25: 219–228. [PUBMED], [INFOTRIEVE], [CSA]
- Bisceglia M, Tricarico N, Minenna P, Magro G, Pasquinelli G.. Myofibrosarcoma of the upper jawbones: a clinicopathologic and ultrastructural study of two cases. Ultrastruct Pathol. 2001; 25: 385–397. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Watanabe K, Ogura G, Tajino T, Hoshi N, Suzuki T.. Myofibrosarcoma of the bone: a clinicopathologic study. Am J Surg Pathol. 2001; 25: 1501–1507. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Lucin K, Mustac E, Jonjic N.. Breast sarcoma showing myofibroblastic differentiation. Virchows Arch. 2003; 443: 222–224. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Kuhnen C, Homann HH, Mentzel T.. Myofibroblastic sarcoma of the thoracic wall: change in appearance in tumour recurrence. Pathologe. 2003; 24: 128–135. [PUBMED], [INFOTRIEVE], [CSA]
- Fisher C, Goldblum JR, Montgomery E.. Calponin and h-caldesmon in sarcomas of myofibroblasts. Mod Pathol. 2003; 16: 11A
- Eyden BP, Christensen L.. Leiomyosarcoma versus myofibrosarcoma: observations and terminology. Ultrastruct Pathol. 1993; 17: 231–239. [PUBMED], [INFOTRIEVE]
- Coffin CM, Watterson J, Priest JR, Dehner LP.. Extrapulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor): a clinicopathologic and immunohistochemical study of 84 cases. Am J Surg Pathol. 1995; 19: 859–872. [PUBMED], [INFOTRIEVE], [CSA]
- Coffin CM, Humphrey PA, Dehner LP.. Extrapulmonary inflammatory myofibroblastic tumor: a clinical and pathological survey. Semin Diagn Pathol. 1998; 15: 85–101. [PUBMED], [INFOTRIEVE], [CSA]
- Meis JM, Enzinger FM.. Inflammatory fibrosarcoma of the mesentery and retroperitoneum: a tumor closely simulating inflammatory pseudotumor. Am J Surg Pathol. 1991; 15: 1146–1156. [PUBMED], [INFOTRIEVE]
- Coffin CM, Dehner LP, Meis-Kindblom JM.. Inflammatory myofibroblastic tumor, inflammatory fibrosarcoma, and related lesions: an historical review with differential diagnostic considerations. Semin Diagn Pathol. 1998; 15: 102–110. [PUBMED], [INFOTRIEVE], [CSA]
- Mirra M, Falconieri G, Zanconati F, Dibonito L.. Inflammatory fibrosarcoma: another imitator of Hodgkin′s disease?. Pathol Res Pract. 1996; 192: 474–478. [PUBMED], [INFOTRIEVE], [CSA]
- Meis-Kindblom JM, Kjellstrom C, Kindblom LG.. Inflammatory fibrosarcoma: update, reappraisal, and perspective on its place in the spectrum of inflammatory myofibroblastic tumors. Semin Diagn Pathol. 1998; 15: 133–143. [PUBMED], [INFOTRIEVE], [CSA]
- Chan JK, Cheuk W, Shimizu M.. Anaplastic lymphoma kinase expression in inflammatory pseudotumors. Am J Surg Pathol. 2001; 25: 761–768. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Cessna MH, Zhou H, Sanger WG, et al, Expression of ALK1 and p80 in inflammatory myofibroblastic tumor and its mesenchymal mimics: a study of 135 cases. Mod Pathol. 2002; 15: 931–938. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Cook JR, Dehner LP, Collins MH, et al, Anaplastic lymphoma kinase (ALK) expression in the inflammatory myofibroblastic tumor: a comparative immunohistochemical study. Am J Surg Pathol. 2001; 25: 1364–1371. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Lawrence B, Perez-Atayde A, Hibbard MK, et al, TPM3-ALK and TPM4-ALK oncogenes in inflammatory myofibroblastic tumors. Am J Pathol. 2000; 157: 377–384. [PUBMED], [INFOTRIEVE]
- Bridge JA, Kanamori M, Ma Z, Pickering D, Hill DA, Lydiatt W, et al, Fusion of the ALK gene to the clathrin heavy chain gene, CLTC, in inflammatory myofibroblastic tumor. Am J Pathol. 2001; 159: 411–415. [PUBMED], [INFOTRIEVE]
- Donner LR, Trompler RA, White RR.. Progression of inflammatory myofibroblastic tumor (inflammatory pseudotumor) of soft tissue into sarcoma after several recurrences. Hum Pathol. 1996; 27: 1095–1098. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Biselli R, Boldrini R, Ferlini C, Boglino C, Inserra A, Bosman C.. Myofibroblastic tumours: neoplasias with divergent behaviour. Ultrastructural and flow cytometric analysis. Pathol Res Pract. 1999; 195: 619–632. [PUBMED], [INFOTRIEVE], [CSA]
- Hussong JW, Brown M, Perkins SL, Dehner LP, Coffin CM.. Comparison of DNA ploidy, histologic, and immunohistochemical findings with clinical outcome in inflammatory myofibroblastic tumors. Mod Pathol. 1999; 12: 279–286. [PUBMED], [INFOTRIEVE], [CSA]