Abstract
Low-grade fibrosarcomas have recently gained increasing attention in the literature, especially with the fall in popularity polls of the ubiquitous malignant fibrous histiocytoma (MFH). Firstly, most tumors previously known as myxoid MFH are labeled presently as myxofibrosarcomas. Secondly, the recognition and better understanding of a family of fibrosing-type fibrosarcoma, encompassing 3 members: fibromyxoid sarcoma (FMS), hyalinizing spindle cell tumor with giant rosettes (HSTGR), and sclerosing epithelioid fibrosarcoma (SEF). To expand further their understanding of the overlapping and distinct features of members included in the spectrum of low-grade fibrosarcoma, the authors carried out a comparative ultrastructural study among 15 low-grade myxofibrosarcomas (MFS) and 12 fibromyxoid sarcomas (FMS), after review of pathology and confirmation of diagnosis. The ultrastructural findings of the LG MFS identified spindle to plump cells, with abundant cytoplasm, rich in well-developed RER cisternae, often distended and sometimes cystically dilated, containing an electronlucent granular material. These results were in keeping with a well-differentiated fibroblastic-type cell phenotype. In addition, a less prominent cellular component included cells with RER, well-developed Golgi apparatus, lysosomes, and filopodia. These latter features define a fibroblastic variant with histiocytic-like properties, also known as histiofibroblasts. Myofibroblastic differentiation was quite limited and mostly absent in most of the cases. In summary, these findings recapitulate a similar spectrum with the cell constituents of so-called MFH. In contrast, the fine microscopic findings of the 12 FMS cases showed an inactive or more primitive form of fibroblastic type cells. The RER cisternae were generally underdeveloped, as expected for a generic fibroblastic-type proliferation. The cytoplasm was scant and showed a paucity of organelles, with the exception of abundant arrays of vimentin-type intermediate filaments. The very long, thin cell processes, sometimes associated with pinocytotic vesicles, were reminiscent of perineurioma ultrastructure.
Pathologists have come to appreciate that tumors composed of malignant fibroblastic cells and their variants, once thought to be rare, represent a large group of soft tissue sarcomas. Factors contributing to this conclusion include the following: ultrastructural demonstration of neoplastic fibroblastic cells in so-called malignant fibrous histiocytoma (MFH), recognition that a significant number of myxoid soft tissue sarcomas of the extremities and trunk are fibrous tumors (e.g. myxofibrosarcomas) [Citation[1], Citation[2]], and the recent identification of some fibrosarcomas that can be classified as low-grade fibrosing subset of fibrosarcomas [Citation[3–9]].
Myxofibrosarcoma (MFS) is the most common sarcoma affecting limbs of old patients [Citation[1], Citation[2]], and it high-grade end of the spectrum is considered as a myxoid variant of malignant fibrous histiocytomas (MFH) by some authors [Citation[10]]. Owing to its ultrastructural features closely resembling ordinary fibroblasts, other investigators favored the nomenclature MFS instead of myxoid MFH and regarded it as a distinct fibroblastic neoplasm characterized by the myxoid nodular appearance and curvilinear vasculatures with a considerably broad spectrum of nuclear pleomorphism, cellularity, and mitoses [Citation[11]].
Fibrosing fibrosarcomas are a group of predominantly fibrosed or sclerosed tumors primarily affecting the extremities of adults of either sex. Recognition of this group started with the description by Evans of low-grade fibromyxoid sarcoma [Citation[3], Citation[4]], a deceptively bland fibrous-tumor-simulting desmoid tumor, but which had focal myxoid changes and surprisingly a definite metastatic potential. Identification of this tumor has been also hampered by its histologic resemblance to myxofibrosarcoma [Citation[2]]. More recently, Folpe et al. [Citation[5]], in a large study encompassing 73 cases, showed that up to 30% of low-grade fibromyxoid sarcomas show overlapping histologic features with hyalinizing fibrosarcoma with giant rosettes [Citation[6], Citation[7]]. Furthermore, a very recent cytogenetic report demonstrated an identical recurrent t(7;16)(q34;p11) translocation in both of these subtypes of fibrosarcoma, thereby providing genetic proof that these 2 tumors are morphologic variants of the same entity [Citation[12]]. The third member of this group is sclerosing epithelioid fibrosarcoma (SEF) [Citation[8], Citation[9]], which is characterized by a diffuse, dene sclerosis, separating closely packed strands and nests of uniform hyperchromatic epithelioid cells.
In this study we performed a comparative ultrastructural analysis of the 2 most common members of low-grade fibrosarcoma, myxofibrosarcoma and fibromyxoid sarcoma, which share morphologic features at microscopic level relatively frequent. A better knowledge of the fine details of these 2 entities might provide us insight on the degree of fibroblastic modulations within the spectrum of low-grade fibrosarcoma.
MATERIALS AND METHODS
The Electron Microscopy Laboratory files of the Department of Pathology of MSKCC were searched for the diagnosis of low-grade fibrosarcoma, myxoid MFH, myxofibrosarcoma, and fibromyxoid arcoma. In all cases microscopic slides were reviewed and only the cases that fit into either low-grade myxofibrosarcoma or fibromyxoid sarcoma were selected for the study.
For ultrastructural analysis, fresh tumor tissue was immersed in a 3% formaldehyde–3% glutaraldehyde fixative, postfixed in 1% osmium tetroxide, dehydrated in graded ethanol, embedded in epoxy resin, and stained with uranyl acetate–lead citrate by using standard protocol. In each case, thick sections were cut and stained with toluidine blue to select suitable areas of ultrastructural analysis under Philips-410 transmission electron microscope.
The ultrastructural features examined included the amount and degree of development of the cytoplasm, RER, intermediate filaments, Golgi apparati, lysosomes, pinocytotic vesicles, etc. We also searched for features to suggest myofibroblastic differentiation, such as ectoplasmic densities of actin microfilaments. The extracellular matrix was examined for the presence of amorphous deposits and also for the presence of a well-defined external lamina.
RESULTS
Upon histologic review, we have identified 15 cases of low-grade myxofibrosarcoma (MFS) and 12 cases of low-grade fibromyxoid sarcoma (FMS) with available material for electron microscopy.
Low-grade Myxofibrosarcoma (MFS)
The clinical and pathologic findings of MFS are listed in . There were 9 females and 6 males, ranging from 27 to 81 years (mean 61.6). The anatomic distribution was predominantly in the soft tissues of the limbs (12/15; 80%), with 7 cases arising in the upper limb (shoulder 1, arm 3, forearm 1, hand 2) and 5 cases in the lower limb (groin 1, buttock 1, thigh 2, ankle 1). Two cases presented in the neck and one case in the trunk (chest wall). The clinical behavior was characterized in the majority of cases by local recurrences (12/15; 80%). In 2 patients there were as many as 5 episodes of local recurrences. Two patients also developed distant recurrences to the lung. At last follow-up only 1 patient was dead of disease, after multiple local and distant metastases. Thirteen patients were alive, with no evidence of disease.
Table 1 Clinicopathologic and Ultrastructural Findings in 15 Low-grade Myxofibrosarcomas
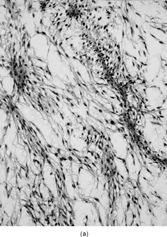
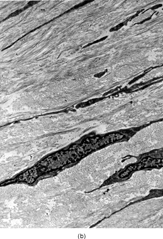
Microscopically, the review of the histologic slides showed tumors with a multinodular growth pattern, with a prominent myxoid stroma. The tumor cells were spindled, with hyperchromatic, irregular nuclei, with variable pleomorphism, and with scattered multinucleated or giant tumor cells. A consistent finding was the presence of a rich vasculature network, with abundant curvilinear-type vessels and a perivascular condensation of the tumor cells ().
FIG. 1 Microscopic appearance of low-grade myxofibrosarcoma: (A) low power with spindle cells separated by abundant myxoid stroma and showing condensation around curvilinear vessels (×200;MFS6); (B) detail of a pseudo-lipoblast showing multivacuolated cytoplasm, with similar appearance as the extracellular matrix (×400;MFS15).
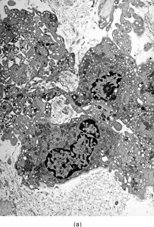
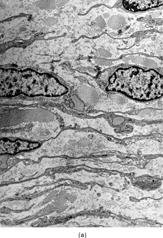
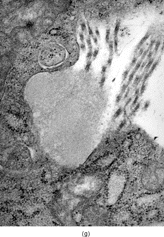
Ultrastructurally, the hallmark of low-grade MFS was spindle cells with irregular nuclei and a well developed RER network, with branching or distended cisternae filled with a proteinaceous material of medium low density (see ) (). These features were in keeping with a well-differentiated fibroblastic cell phenotype. In two cases marked distension and cystic dilatation of the endoplasmic reticulum cisternae were observed. These cells were plumper and showed identifiable filopodia (). Some of these distended RER were filled with a granular material, similar to that seen in the extracellular matrix (). The H&E photos taken from the same case match the vacuolated cytoplasm of what we typically designate as pseudo-lipoblasts ().
FIG. 2 Ultrastructure of myxofibrosarcoma: (A) low-power showing fusiform cells separated by myxoid stroma; abundant cytoplasm is filled with numerous dilated RER cisternae (×4, 320;MFS13);(B) 2 “pseudo-lipoblasts”-type cells showing marked distension and cystic dilatation of RER cisternae (×5, 400;MFS7);(C, D) detail of the distended cisternae, which appear either empty or filled with a granular electronuclent material (×7, 929and ×18,720; MFS 15);(E) cytoplasmic detail with abundant RER, prominent Golgi, and lysosomes (×12, 780;MFS1);(F) plump spindle cells with abundant cytoplasm rich in vimentin-type intermediate filaments, RER, and dense mitochondriae. In addition focal subectoplasmic actin condensation and short filopodia-type cell processes are also evident (×7, 920;MFS9).
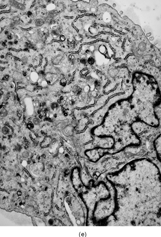

In the plumper cells, with more abundant cytoplasm, the RER complexes were sometimes pushed to the periphery of the cell, while a well-developed Golgi apparatus associated with variable number of lysosomes and vesicles occupied the central portion of the cell (). These cells, with prominent Golgi and abundant secondary lysosomes, were diagnostic for a fibroblastic variant with histiocytic-like features, previously defined as histiofibroblastic cells.
As indicated in the Materials and Methods section, a careful search was undertaken for the presence of myofibroblastic features in this neoplasm. Only in 1 case focal ectoplasmic condensations of actin microfilaments were noted (). In 2 additional cases ill-defined and small ectoplasmic thickenings were detected, possibly representing actin. But the majority of cases and cells examined lacked features suggestive of a myofibroblastic component.
Low-grade Fibromyxoid Sarcoma (FMS)
The clinical and pathologic findings of FMS are listed in . There were 7 males and 5 females, ranging from 11 to 53 years (mean 28.4). The anatomic distribution was predominantly in the deep soft tissues of the limbs (8/12; 67%), 6 cases arising in the lower limb (foot 2, knee 1, thigh 2, groin 1) and 2 cases in the upper limb (forearm and hand). The remaining 4 cases were equally distributed among the neck (2 cases) and trunk (axilla 1, flank 1). Long clinical follow-up was obtained in all cases, except for the 2 recently diagnosed tumors, with follow-up information of less than 6 months. There were 5 local recurrences (42%) and 3 distant metastases (25%). At last follow-up, ranging from 20 to 252 months (mean of 97 months), no patient was dead of disease, 10 were alive no evidence of disease (NED), and 2 were alive with disease (AWD).
Table 2 Clinicopathologic and Ultrastructural Findings in 12 cases of Low-grade Fibromyxoid Sarcoma
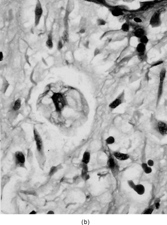
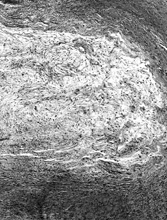
Microscopically, all tumors had a classic appearance of bland spindle cell proliferation embedded in alternating fibrosing and slightly myxoid stroma (). The cells showed dense hyperchromatic nuclei with ill-defined cell borders, and surrounded by a wavy collagen stroma, reminiscent of the shredded carrot pattern of neurofibroma. S-100 protein staining can be very helpful, since it was completely negative in these cases. The nuclear pleomorphism was minimal and only rare cells with slightly larger nuclei were seen. No areas of necrosis were noted. The mitotic activity was low to null. Focal areas of more increased cellularity was noted. Rare and somewhat ill-defined hyalinized rosette-like structures were noted in 2 cases, one in the primary and the other only in the lung metastasis. Interestingly, in 5 cases a different diagnosis was rendered for the primary tumor, either in the biopsy, resection, or both. A correct diagnosis was established either upon resection or after developing subsequent recurrences. The original rendered diagnosis included mainly benign conditions, such as desmoid tumor in 2 cases, neurofibroma in 2 cases, and fibroma of tendon sheath in 1 case. The first lung metastatic event in one of the cases, which was initially thought to represent a neurofibroma, was also misdiagnosed as a solitary fibrous tumor. The majority of these misdiagnoses were encountered in older cases, rendered before or in close proximity to the original report by Evans, when this entity was not well recognized by practicing pathologists (3).
FIG. 3 Microscopic appearance of low-grade fibromyxoid sarcoma: showing bland uniform spindle cells arranged in a swirling pattern and embedded in an alternating myxoid and fibrosing stroma (×40;FMS9).
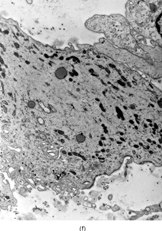
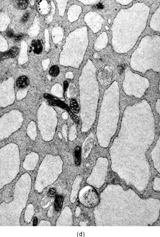
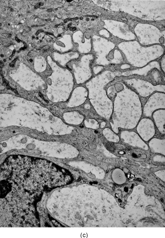
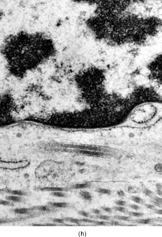
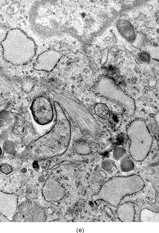
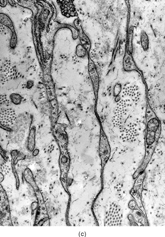
Ultrastructurally, the detailed findings for each FMS are listed in . In summary, the electron microscopic hallmark of this tumor included slender and delicate spindle cells, with limited amount of cytoplasm and long or very long, mostly nonbranching delicate cell processes, embedded in a variable amount of collagenous stroma (). Most cells had a modest number of cytoplasmic organelles, including RER cisternae, which were mostly ill developed or only mildly well developed. Only in 2 cases was the RER network moderately well developed, but lacking the typical branching or dilated cisternae seen in other types of fibroblastic tumors (). Interestingly, 3 cases showed a large number of ovoid cytoplasmic structures with a fibrillary content, consistent with angulate lysosomes (). A consistent finding was the presence of abundant arrays of vimentin intermediate filaments. The striking and distinct ultrastructural feature was the presence of narrow, very long cell processes, which in some cases showed the presence of rare or abundant pinocytotic vesicles (). The combination of these 2 findings was reminiscent of perineurioma. The only distinguishing feature between these 2 lesions was the absence of a well-developed external lamina in FMS, which is typically seen in perineumona. In addition, 9 cases showed variable amount of flocculent, granular extracellular matrix, seen either in proximity of the cell membrane or away, admixed with the collagen stroma (). In one case intracisternal collagen was noted ().
FIG. 4 Ultrastructure of fibromyxoid sarcoma: (A, B) low power showing delicate spindle cells with long, narrow and nonbranching cell processes arranged in a loose (×5, 400;FMS4) or collagenized stroma (×5, 400;FMS8);(C) detail of cell processes showing sparse organelles and small pinocytotic vesicles (×18, 720; FMS 4);(D) linear arrangement of numerous pinocytotic vesicles (×30, 420; FMS 9);(E) cytoplasmic detail with moderately developed RER as well as an ovoid fibrillary structure consistent with angulate lysosomes (×25, 200;FMS10);(F) detail of an angulate lysosomes associated with scattered dense deposits (×17, 000; FMS 12);(G) extracellular deposit of amorphous flocculent substance (×37, 800; FMS 9);(H) intracisternal collagen (×30, 000;FMS3).


DISCUSSION
The term “myxofibrosarcoma” was coined back in 1977 by Angervall, Kindblom, and Merck [Citation[1]]. Their study, including 30 cases of myxofibrosarcoma, showed a predominance of these tumors in the subcutis of extremities of elderly patients. In the same year, 1977, Weiss and Enzinger described the myxoid variant of MFH, defined as having ≥ 50% myxoid areas, to distinguish it from more solid and pleomorphic-type MFH [Citation[10]]. The distinction from the MFH, NOS type, was also based on the observation that the amount of myxoid stroma is inversely related to the metastatic potential of the tumor, which remains quite valid today. The tumor, the way both Angerval et al. [Citation[1]] and Weiss and Enzinger [Citation[10]] described it with 2 different names, is just one: a myxoid lesion, that is typically superficially located and can look very bland and hypocellular microscopically. The very low-grade forms can be misdiagnosed with myxoma, a lesion less commonly located in the subcutis and less vascular. One of the characteristic features of this tumor is the presence of the curvilinear vessels, with perivascular condensation of the tumor cells. But the hallmark of this tumor is the cytology of these cells, which are spindled-shaped, with hyperchromatic and variably pleomorphic nuclei.
For almost 2 decades, the term myxoid-type MFH prevailed over myxofibrosarcoma, both in clinical practice as well as in the published literature. One of the reasons for the wide acceptance of this term was that a significant number of MFS have solid areas microscopically, indistinguishable from a storiform/pleomorphic MFH. A continuity between low-grade and high-grade areas of MFS was indicated by the presence of solid high-grade components within the low-grade tumors as well as the progression of a subset of low-grade MFS into high-grade tumors in local recurrences [Citation[13], Citation[14]]. Presently, the myxoid-type MFH terminology faces tremendous loss in popularity, especially after the reappraisal for the term “myxofibrosarcoma” by Mentzel et al. [Citation[2]].
In 1979, the same 3 Swedish pathologists, Kindblom, Angerval, and Merck [Citation[11]], described the ultrastructure signature of MFS, indicating that the lower-grade tumors were composed mainly of fibroblastic and myofibroblastic-like cells, while the higher-grade tumors had more of the histiocyte-like cells. The findings of the present study are in agreement with their original observations, identifying predominantly fibroblastic type cells, that seem to have a very well-developed RER cisternae, quite often distended and less frequently cystically dilated and filled with an electronlucent granular material. The latter findings correlate with the presence of multivacuolated, so-called “pseudo-lipoblasts,” on light microscopic examination. A less prominent cellular component in these low-grade tumors were cells, which, in addition to a well-developed RER, had an expanded Golgi apparati and numerous lysosomes and filopodia-type cell processes. These findings define a fibroblastic variant mimicking true histiocytes, so-called histiofibroblasts. The third cellular component that is commonly seen in the conventional MFH, the myofibroblast-type cells, is very infrequently noted in this subset.
In contrast to MFS, FMS is a relatively recently described tumor, which due to its deceptively bland appearance was confused with other benign spindle cell lesions. Recognition of this tumor started with the description by Harry Evans in 1987 of 2 metastasizing neoplasms having a misleading benign appearance [Citation[3]]. Both tumors occurred in women patients in their late twenties and were located in the shoulder/trunk area. Lung metastases were present in both cases; one patient died of disease 94 months after presentation and the other was alive with disease 82 month later. Six years later, in 1993, Evans expanded his original series with 10 more cases, also noting that dedifferentiation occurred in one of the cases [Citation[4]].
As indicated from the challenging H&E diagnosis, the morphologic appearance is deceivingly bland and composed of a proliferation of uniform delicate spindle cells arranged in a swirling pattern in a fibromyxoid stroma. In a number of cases the myxoid component can be more prominent, forming myxoid nodules, which can suggest a myxofibrosarcoma diagnosis. The main distinction between the 2 is the paucity of blood vessels in this tumor, as well as lack of the bizarre and multinucleated giant cells of myxofibrosarcoma. More fibrotic examples of FMS can be misinterpreted as fibromatosis, but they lack the straight elongated vessels or the open chromatin nuclei with small nucleoli typically seen in fibromatosis.
In 2000, Folpe et al. suggested a closer link between FMS and another less-common member of this family, hyalinizing spindle cell tumor with giant rosettes (HSTGR)[Citation[5]]. They postulated that based on overlapping morphologic features these represent 2 ends of a common spectrum. Overlapping histologic features of sclerosing epithelioid fibrosarcoma (SEF) with the other 2 members have been only marginally suggested in different reports [Citation[4], Citation[9]], but further genetic analysis is needed to unequivocally confirm this hypothesis. The earliest observation of increased cellularity and anaplastic round cell change in low-grade fibromyxoid sarcoma, as seen routinely in SEF, was provided by Evans in 1993 [Citation[4]], which was interpreted as evidence of “dedifferentiation.” Furthermore, Folpe et al. [Citation[5]] described epithelioid cell foci in up to 45% of their tumors having low-grade fibromyxoid sarcoma/hyalinizing fibrosarcoma with giant rosettes features.
Ultrastructurally, the unifying concept of fibrosing fibrosarcomas is based on well-developed fibroblastic features of the neoplastic cells embedded in a dense collagenous extracellular stroma and admixed with a peculiar amorphous granular material. Nielsen et al. [Citation[15]] reported the ultrastructural findings of 3 cases of hyalinizing spindle cell tumor with giant rosettes. The neoplastic cells showed fibroblastic features with long branching RER complexes. In all tumors in addition to abundant extracellular collagen fibers, aggregates of granular amorphous material resembling “basement membrane-like” substance were noted admixed with the collagen fibers, but also seen within the dilated cisternae. More recently, Franchi et al. [Citation[17]] carried out an elegant comparative electron microscopic analysis on FMS and HSTGR, demonstrating overlapping features at the fine microscopic level as well. Both tumor types were composed of cells with fibroblastic features, embedded in a collagenous stroma with irregular deposits of amorphous basal lamina-like substance.
In the present study, the ultrastructural analysis of 12 FMS, provided further insight into its cell constituents, relative to other types of fibrosarcomas. The electron microscopic features of FMS appeared quite distinct from MFS, suggesting a more inactive or more primitive form of fibroblastic cells. The very scant cytoplasm with a paucity of cell organelles, including a poorly developed RER cisternae, was a common finding in FMS. In addition, the long, thin cell processes, sometimes associated with pinocytotic vesicles, was reminiscent of perineurioma ultrastructure. These findings are possibly related to the observations by De Pinieux et al. [Citation[17]], who in a report of HSTGR noted ultrastructural features resembling Schwann cells. The presence of abundant ovoid fibrillary structures, consistent with angulate lysosomes, seen in 25% of cases, was not previously described in this tumor type. In contrast with Bejarano et al. [Citation[18]] or Dobashi et al. [Citation[19]], we did not identify the presence of dense-core granules or actin microfilaments, as evidence of neuroendocrine differentiation or myofibroblastic differentiation, respectively.
Exciting molecular evidence was recently provided by 2 back-to-back reports on the similar genetic abnormalities shared by both FMS and HSTGR [Citation[12], Citation[20]]. Reid et al. [Citation[12]] found a recurrent t(7;16)(q34;p11) translocation in 2 cases each of FMS and HSTGR. The cloning of the t(7;16) fusion gene partners was subsequently carried out by Storlazzi et al. [Citation[20]], showing the fusion of FUS on 16p11 with BBF2H7 at 7q34, a previously uncharacterized gene homologous to DrosophillaBbf − 2 gene. RT-PCR methodology showed that the chimeric fusion transcript is composed of the first 5 exons and part of exon 6 of FUS and part of exon 5 and exons 6–12 of BBH2H7. These results offer the scientific support for a larger scale investigation of the sensitivity and specificity of TLS − BBF2H7 fusion transcript in low-grade fibrosing fibrosarcomas and also might provide a reliable molecular diagnostic test that can support the diagnosis of FMS in difficult and challenging cases.
We would thank Elizabeth Weiss, Kin Kong, and Allyne Manzo for excellent technical assistance. Also we thank Frank Breuer for providing one of the low-grade fibromyxoid sarcoma cases.
REFERENCES
- Angervall L, Kindblom LG, Merck C. Myxofibrosarcoma: a study of 30 cases. Acta Pathol Microbiol Scand [A]. 1977; 85A: 127–140
- Mentzel T, Calonje E, Wadden C, Beham A, Smith MA, Fletcher CDM. Myxofibrosarcoma: clinical analysis of 75 cases with emphasis on the low grade variant. Am J Surg Pathol. 1996; 20: 391–405. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Evans HL. Low-grade fibromyxoid sarcoma: a report of two metastasizing neoplasms having a deceptively bland appearance. Am J Clin Pathol. 1987; 88: 615–619. [PUBMED], [INFOTRIEVE]
- Evans HL. Low-grade fibromyxoid sarcoma: a report of 12 cases. Am J Surg Pathol. 1993; 17: 595–600. [PUBMED], [INFOTRIEVE]
- Folpe AL, Lane KL, Paull G, weiss SW. Low-grade fibromyxoid sarcoma and hyalinizing spindle cell tumor with giant rosettes: a clinicopathologic study of 73 cases supporting their identity and assessing the impact of high-grade areas. Am J Surg Pathol. 2000; 24: 1353–1360. [PUBMED], [INFOTRIEVE], [CSA]
- Lane KL, Shannon RJ, Weiss SW. Hyalinizing spindle cell tumor with giant rosettes: a distinctive tumor closely resembling low grade fibromyxoid sarcoma. Am J Surg Pathol. 1997; 21: 1481–1488. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Woodruff JW, Antonescu CR, Erlandson RA, Boland PJ. Low-grade fibrosarcoma with palisaded granuloma-like bodies (giant rosettes). Am J Surg Pathol. 1999; 23: 1423–1428. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Meis-Kindblom JM, Kindblom L-G, Enzinger FM. Sclerosing epithelioid fibrosarcoma: a variant of fibrosarcoma simulating carcinoma. Am J Surg Pathol. 1995; 19: 979–993. [PUBMED], [INFOTRIEVE], [CSA]
- Antonescu CR, Rosenblum MK, Pereira P, et al, Sclerosing epithelioid fibrosarcoma: a study of 16 cases and confirmation of a clinicopathologically distinct tumor. Am J Surg Pathol. 2001; 25: 699–709. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Weiss SW, Enzinger FM. Myxoid variant of malignant fibrous histiocytoma. Cancer. 1977; 39: 1672–1685. [PUBMED], [INFOTRIEVE]
- Kindblom LG, Merck C, Angervall L. The ultrastructure of myxofibrosarcoma: a study of 11 cases. Virchows Arch A Pathol Anat Histol. 1979; 381: 121–139. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Reid R, de Silva CMV, Paterson L, et al, Low grade fibromyxoid sarcoma and hyalinizing spindle cell tumor with giant rosettes share a common t(7;16)(q34;p11) translocation. Am J Surg Pathol. 2003; 27: 1229–1236. [PUBMED], [INFOTRIEVE], [CSA]
- Merck C, Angervall L, Kindblom LG, et al, Myxofibrosarcoma: a malignant soft tissue tumor of fibroblastic-histiocytic origin: a clinicopathologic and prognostic study of 110 cases using multivariate analysis. Acta Pathol Microbiol Immunol Scand Suppl. 1983; 282: 1–40. [PUBMED], [INFOTRIEVE]
- Huang-Ying H, Lal P, Qin J, Brennan MF, Antonescu CR. Low-grade myxofibrosarcoma: a clinicopathologic analysis of 49 cases treated at a single institution with simultaneous assessment of the efficacy of 3-tier and 4-tier grading systems. Hum Pathol. 2004; 35: 612–621. [CSA], [CROSSREF]
- Nielsen GP, Selig MK, O'Connel JX, et al, Hyalinizing spindle cell tumor with giant rosettes. Am J Surg Pathol. 1999; 23: 1227–1232. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Eyden BP, Manson C, Banerjee SS, Roberts ISD, Harris M. Sclerosing epithelioid fibrosarcoma: a study of five cases emphasizing diagnostic criteria. Histopathology. 1998; 33: 354–360. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Franchi A, Massi D, Santucci M. Hyalinizing spindle cell tumor with giant cell rosettes and low grade fibromyxoid sarcoma: an immunohistochemical and ultrastructural comparative investigation. Ultrastruct Pathol. 2003; 27: 349–355. [PUBMED], [INFOTRIEVE], [CSA]
- De Pinieux G, Anract P, Le Charpentier M, et al, Un cas de tumeur a cellules fusiformes hyalinisante avec rosettes geantes de la region presacree: etude immunohistochimique et ultrastructurale. Ann Pathol. 1998; 18: 488–491. [PUBMED], [INFOTRIEVE]
- Bejarano PA, Padhya TA, Smith R, Blough R, Devitt JJ, Gluckman JL. Hyalinizing spindle cell tumor with giant rosettes—a soft tissue with mesenchymal and neuroendocrine features: an immunohistochemical, ultrastructural, and cytogenetic analysis. Arch Pathol Lab Med. 2000; 124: 1179–1184. [PUBMED], [INFOTRIEVE], [CSA]
- Dobashi Y, Noguchi T, Nasuno S, Jiang S-X, Kameya T. Hyalinizing spindle cell tumor with giant rosettes: report of a case showing remarkable myofibroblastic differentiation. Pathol Res Pract. 2001; 197: 691–697. [PUBMED], [INFOTRIEVE], [CSA]
- Storlazzi CT, Mertens F, Nascimento A, et al, Fusion of the FUS and BBF2H7 genes in low grade fibromyxoid sarcoma. Hum Mol Genet. 2003; 12: 2349–2358. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]