
Abstract
As the tenth year that follows the premature passing of the inorganic chemistry legend F. Albert (Al) Cotton comes to a close, we provide some soul food that we believe Al would have enjoyed as he looks down upon two generations of disciples, a grandchild and two great grandchildren, in his scientific family tree. This article also represents a new tradition by which the editors of Comments on Inorganic Chemistry wish to lead by example, whereby we start publishing original research content that nonetheless preserves the journal’s identity as a niche for “critical discussion of the current literature” of inorganic chemistry. Thus, we show novel demonstrations of a posteriori validation and a priori prediction of possible counterintuitive covalent M-M’ bonds—after and before synthesis, respectively—contrasted with non-covalent M···M interactions of the metallophilic type in bridge-dimeric systems of d10 metal centers. Mixing of (n + 1)s0/p0 orbitals with (n)d10 orbitals in the former heterobimetallics case—and lack thereof in the latter homobimetallics case—is hypothesized to be responsible for the presence or absence of covalency, respectively. However, we also demonstrate that the consequent d-s’ and/or d-p’ orbital mixing/hybridization can occur even in homobimetallic complexes that contain asymmetric bridging ligands (e.g., with -S^C- donor atoms that lead to orbital interactions between M-S and M-C fragments). Hence, the “control” metallophilic case for the hypothesized d10-d10 polar-covalent bonding herein is best manifest by M2(P^P)2 homobimetallics vis-à-vis M2(S^C)2 embodiments. Finally, we also offer a point/counterpoint commentary to the readers to contrast factors that argue for covalency vs metallophilicity and forewarn against exaggerating the extent of covalent bonding in such d10-d10 species. Thus, scrutinizing the various theoretical parameters has pointed to M-M’ and M-M partially bonded S^C-bridge dinuclear d10 complexes vis-à-vis the classic Cotton-type multiple metal-metal bond description in d0<n<10 species.
GRAPHICAL ABSTRACT
1. INTRODUCTION
Metal-metal bonds have captured the hearts and minds of inorganic chemists since Cotton’s paramount discovery of quadruply bonded metal centers in 1964.[Citation1] His work inspired many to further investigate the underlying chemistry and applications of metal-metal bonds that continue to persist today, even as we enter the second half of the century after their original discovery.[Citation2,Citation3] Additional significant findings include the research of James P. Collman and colleagues regarding multiply bonded, porphyrin-stabilized heterodinuclear transition metal complexes.[Citation4–Citation10] Other notable work includes Roald Hoffmann’s[Citation11–Citation14] and Pekka Pyykko’s[Citation15–Citation17] contributions concerning strong closed-shell metal-metal interactions, as well as extended studies on covalent radii for singly and multiply bonded atoms.[Citation16,Citation18–Citation21] Harry B. Gray’s contributions to the field concerning the photophysical properties of multiply bonded metal atoms and binuclear d10-d10 complexes are also of great significance.[Citation22–Citation26]
Distinct from bona fide covalent M-M bonds has been the discovery of metallophilic interactions, a type of non-covalent M···M interaction that is usually attributed to correlation and/or hybridization effects of closed-shell metal centers.[Citation11–Citation15,Citation17] Due to their counterintuitive nature, the existence of M-M bonds in such systems has been both supported and refuted at various levels of theory.[Citation17] While bonds are expected between open-shell metal centers due to the unequal occupation of bonding and antibonding orbitals, this is not the expectation for closed-shell metal centers (e.g., linear or trigonal d10, or square-planar d8 with strong ligand fields). The existence of M-M bonds between closed-shell metal centers relies on more than merely the nature of the filled orbitals; it is dictated by the availability of low-lying metal orbitals—i.e., the (n + 1)s/p orbitals—to mix with the (n)d orbitals to give significant ds/dp orbital overlap. The mixing of these low-lying, higher-shell metal orbitals with the formally anti-bonding (dz2-dz2)σ* leads to an orbital that is more non-bonding than antibonding in character,[Citation27] which, when occupied, can result in partial bond orders.[Citation28–Citation30] While there have been many cases of covalent bonding between open-shell metals,[Citation21,Citation31–Citation38] fewer examples exist for covalent bonding between closed-shell and pseudo-closed shell metals.[Citation39–Citation41] To our knowledge, only one example exists for a genuine covalent bond between two closed-shell metals, which was published in 2017 by Omary and co-workers for a hexanuclear CuI2AuI4 azolate/imidazolate cluster.[Citation42] In that system, the authors scrutinized crystallographic, spectroscopic, and electronic structure considerations that pointed to the existence of two Cu(I)-Au(I) covalent bonds from multi-faceted experimental and computational results that cross-correlated to one another to arrive at that conclusion.
This investigation encompasses computational findings that distinguish the differences in bonding character in bridged homo- and hetero-bimetallic complexes of AuI/AgI/CuI, where the bridging ligand is MTP(Ph)2 = diphenylmethylenethiophosphinate, whereas the truncated analogue thereof, MTP(H)2, is used as a simplified theoretical model susceptible to higher levels of theory; see and . We also include the Pt2(PCP)2 model (PCP = H2PCH2PH2; ) for comparison and a prelude to other ongoing investigations of the same bonding phenomena herein for related dimers of zero-valent group 10 metal centers with diphosphines.
Figure 1 Structures of AuAg[MTP(R)]2 (A) and AuCu[MTP(R)]2 (B) models (R = Ph or H) and the Pt2(PCP)2 model (C).
![Figure 1 Structures of AuAg[MTP(R)]2 (A) and AuCu[MTP(R)]2 (B) models (R = Ph or H) and the Pt2(PCP)2 model (C).](/cms/asset/306280cb-5800-4d89-beba-0111c43e5d4c/gcic_a_1467315_f0001_b.gif)
2. COMPUTATIONAL METHODOLOGIES
To deduce differences in structural characteristics and bonding trends, all complexes were modeled using the M06 functional[Citation43–Citation45] combined with the CEP-31G(d) basis set.[Citation46–Citation48] M06 was selected based on its previous performance on modeling and detection of weak intermolecular forces, among other features that have led Truhlar to tout it as a “desert island functional.”[Citation45] The M06 functional has previously been employed by our group to study metallophilic interactions between closed-shell coinage metal complexes,[Citation49] and, like other DFT methods, M06 can describe covalent interactions. This led us to the conclusion that the M06 functional can be used to differentiate between metallophilic interactions and true covalent interactions. We realize that other functionals have been developed and used to treat dispersion interactions.[Citation43,Citation50–Citation59] Calculations at a higher level of theory were performed on a model complex where the MTP(Ph) ligand was changed to MTP(H), whereby the phenyl rings were exchanged with hydrogen atoms, using MP2 as the method[Citation60–Citation64] and cc-pVTZ as the basis set,[Citation65] with pseudopotentials added for the Ag and Au metals.[Citation66]
To analyze these systems, ground-state geometries (1S0) were calculated and compared against previously published crystal structure data when available. As a preliminary approach to identifying potential bonding character, the Kohn-Sham orbitals were modeled to identify the bonding and antibonding orbitals as well as non-bonding orbitals, when relevant, with isodensity values of 0.03 a.u. (default in Gaussian) or 0.02 a.u. (varied to magnify the weaker bonding interactions). The total density plots for these complexes were also generated, to determine bonding character through variation of the isodensity value from 0.01 to 0.1 a.u., where higher isodensity values represent stronger interactions while lower values are used to illustrate weaker interactions. Time-dependent DFT (TD-DFT) calculations were completed to identify optical transitions in both the homo- and heterobimetallic complexes. All resulting data were compared to surmise bonding trends and differences in the strength of metal-metal bonding interactions in these homo- and heterobimetallic complexes.
3. RESULTS AND DISCUSSION
3.1. “Control” Cases of M···M Metallophilic Bonding
We have assessed two types of posssible “control” models for the hypothesized d10-d10 polar-covalent bonding herein that would presumably exhibit metallophilic bonding. The results, however, have demonstrated that homobimetallic M2(S^C)2 models of monovalent group 11 coinage metals do not serve as good metallophilic controls as do M2(P^P)2 models of neutral group 10 metals, as detailed in the rest of this section.
Calculated structures for the ground state of the homometallic Ag2[MTP(Ph)]2 and Au2[MTP(Ph)]2 complexes were compared against previously reported crystal structures ().[Citation67–Citation69] For both complexes, the calculated metal-metal distances were within 0.1 Å of the crystallographic distances, with the metal-metal distance in the AuAu and AgAg complexes calculated to be 3.15 Å and 3.04 Å, respectively. These distances fall significantly outside the range of the summation of the covalent radii for two metal centers, with a greater departure of >0.3 Å for gold (2.72 Å) than for silver (~0.1 Å departure from 2.90 Å),[Citation70] which would argue to categorize the interactions as “metallophilic.” The CuCu system, yet to be synthesized, is analogous to the other homobimetallic congeners with differences arising in geometrical aspects. The C-Cu-S bond angles are not equivalent (), whereas in the AuAu and AgAg complexes the analogous C-M-S angles are identical (). One bond angle deviates from linearity by 3° while the other bond angle deviates from linearity by 8°. The metal-metal distance for this CuCu complex (2.84 Å) resembles that of the AuAu complex, with the distance falling more significantly outside the sum of the covalent radii (by ~0.2 Å vs 2.64 Å).[Citation70]
Table 1 Selected bond lengths (Å) and angles (°) for the coordination geometry in the Ag2[MTP(Ph)]2 and Au2[MTP(Ph)]2 complexes
Figure 2 Calculated geometry of the Cu2[MTP(Ph)]2 complex with the C-Cu-S angles (°) shown.
![Figure 2 Calculated geometry of the Cu2[MTP(Ph)]2 complex with the C-Cu-S angles (°) shown.](/cms/asset/eb863193-7b54-4b78-9154-c130d55abdca/gcic_a_1467315_f0002_oc.jpg)
Based on the errors in the calculated metal-metal distances seen for the two known homobimetallic complexes, which stem from known limitations of DFT methods, one may argue that the crystallographic distances could be within the sum of the most liberal interpretation of the covalent radii, whereby one adds the uncertainty (esd) to each r value in ref [Citation70]. This begs an interesting question regarding these homobimetallic complexes: If the metal-metal distances are within, or close to, the covalent radii, could metal-metal covalent bonding be taking place? Or are the metal centers close to one another merely due to the small bite size of the bridging ligands? This situation prompted us to analyze the molecular orbitals and total density plots for these complexes. What is expected for homobimetallic complexes with closed-shell metals is the generation of five bonding and five antibonding orbitals, whereas d-s’ or d-p’ hybridization due to mixing from the (n + 1)s/p vacant shell may alter the situation and lead to covalent bonding. Cotton and co-workers have refuted this hybridization in their experimental/computational study of M2(form)2 systems (M = Cu or Ag; form = formamidinate).[Citation71] In a total density plot, it would be expected to see the electron density between the metal centers drop off at low isodensity values. The isodensity values used to analyze these complexes range from 0.01 to 0.1 and, while these loosely represent van der Waals interactions and covalent interactions, respectively, these are not hard and fast guidelines.[Citation72,Citation73] In general, for these complexes, stronger interactions will persist at larger isodensity values.
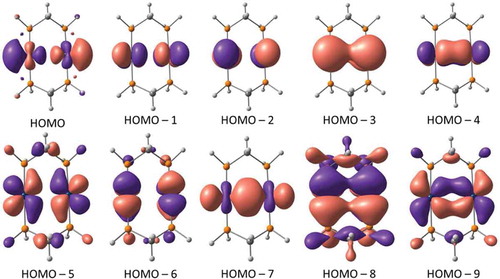
In the case of the AuAu complex, one can deduce the generation of at least five bonding orbitals and five antibonding orbitals among the primarily metal-based frontier orbitals (). A few other orbitals are difficult to assign as bonding or anti-bonding without altering the isodensity value. Given that both metal centers have a formally d10 electronic configuration, this leads to equal occupation of the bonding and the antibonding orbitals, thus leading to a formal bond order of zero; however, we notice that all five bonding orbitals are strongly bonding while three out of the five antibonding orbitals are weakly anti-bonding and exhibit strong ligand contribution. The ordering of all such metal-based orbitals is not intuitive in terms of not being sequentially ordered between HOMO to HOMO-4 for the five anti-bonding and HOMO-5 to HOMO-9 for the five bonding orbitals, as one would predict from Cotton’s classical scheme.[Citation1] Additional insights are provided in the discussion of similar orbital descriptions for unsubstituted models (vide infra).
Figure 3 Molecular orbital contours of the Au2[MTP(Ph)]2 complex (isodensity = 0.03 a.u.). Orbital notation is given to describe relative energy (e.g., HOMO–6 = the orbital that lies sixth below the highest occupied molecular orbital).
![Figure 3 Molecular orbital contours of the Au2[MTP(Ph)]2 complex (isodensity = 0.03 a.u.). Orbital notation is given to describe relative energy (e.g., HOMO–6 = the orbital that lies sixth below the highest occupied molecular orbital).](/cms/asset/45977e14-ef9f-43e6-ae50-2793689ae7f8/gcic_a_1467315_f0003_oc.jpg)
We hypothesize that the degree of mixing of the (n + 1)s/p with the (n)d orbitals is responsible for the deviation of M-based frontier orbitals’ relative energies in M2[MTP]2 from the classical metal-metal bonding scheme. We have addressed this issue upon careful analysis of orbital composition in M2[MTP(H)]2 unsubstituted model complexes (–).
Table 2 Kohn-Sham orbital contribution analysis for [Au(MTP(H))]2
Table 3 Kohn-Sham orbital analysis for [Ag(MTP(H))]2
Table 4 Kohn-Sham orbital analysis for [Cu(MTP(H))]2
In this homometallic case, the Kohn-Sham HOMO of the M = Au complex entails significant mixing with the 6s orbital of each metal center (28% 5d-character and 46% 6s-character). The Ag and Cu complexes give rise to 12 occupied frontier molecular orbitals with strong d-character according to analysis of the Kohn-Sham orbitals. Such orbitals of these complexes also show significant mixing of the (n)d and (n + 1)s orbitals (32% 4d-character 24% 5s-character for the Ag complex and 24% 3d-character with 28% 4s-character for the Cu complex). This mixing is hypothesized to be a result of the energy gap between the d and s orbitals of each metal center. This is demonstrated in , where the energy level gap between the 1S0 state of the coinage metal ions and the lower 3D3 state is shown.[Citation74]
Table 5 Atomic energy level splitting for Au(I), Ag(I), and Cu(I) reported in cm−1 units for the singlet and triplet excited states, including the weighted average of the spin-orbit microstates of the latter
The energy gaps are smaller for the Au(I) and Cu(I) ions, and this is reflected in the contributions of the Au and Cu homometallic complexes, which are higher than that seen for the Ag homometallic complex (46% and 28% vs 24%). While this analysis is quantified based upon Kohn-Sham orbitals (–), a similar conclusion is reached upon performing natural bond order (NBO) analysis[Citation75,Citation76] of these complexes (). NBO analysis reveals significant mixing of the nd and (n + 1)s orbitals for all three complexes where all of the d orbitals are denoted as lone pair orbitals, and each metal center has five of these orbitals. Within these orbitals, the first four orbitals are all pure d orbitals, with no contribution from the (n + 1)s/p orbitals for all three complexes. In the fifth d orbital, mixing is evident at both M centers in the M2[MTP(H)]2 models, with the Au complex showing the most mixing (85% d-character and 15% s-character) and the Cu and Ag being about the same (95%/94% d-character and 5%/6% s-character, respectively). The role and contribution of (n + 1)p as acceptor orbitals to the (n)d lone pairs, based on the NBO analysis, is discussed in a subsequent section. – show contribution results from the Kohn-Sham orbital analyses for the M2[MTP(H)]2 models. It can be concluded that metal-metal interactions persist even in these homobimetallic cases, based on the contributions of the metal orbitals to the frontier molecular orbitals of these complexes. While this is true, the contributions from the ligands to this orbital space cannot be ignored. The asymmetry of the MTP ligand leads to mixing of the ligand and metal orbitals, which results in the manifestation of more than the expected 10 orbitals with metal contributions.
Table 6 Natural bond order orbital (NBO) analysis for M2[MTP(H)]2 complexes
Figure 4 Molecular orbital contours of the Ag2[MTP(H)]2 complex (isodensity = 0.03 a.u.).
![Figure 4 Molecular orbital contours of the Ag2[MTP(H)]2 complex (isodensity = 0.03 a.u.).](/cms/asset/b06b7ab8-e0db-400e-ac4a-7e3db9149b19/gcic_a_1467315_f0004_oc.jpg)
Figure 5 Molecular orbital contours of the Cu2[MTP(H)]2 complex (isodensity = 0.03 a.u.).
![Figure 5 Molecular orbital contours of the Cu2[MTP(H)]2 complex (isodensity = 0.03 a.u.).](/cms/asset/4583e3a3-6359-4a2b-ada9-e0389a7ba4b4/gcic_a_1467315_f0005_oc.jpg)
Regarding the total density plots, there is a strong interaction at an isodensity value of 0.01, which drops off at 0.03 (). This indicates an interaction that is stronger than a van der Waals interaction, but not as strong as a covalent interaction. Even when the focus is shifted to the AgAg and CuCu complexes, similar results are observed. Looking in to the total density plots admits similar results as those seen for the AuAu complex. Although there is an interaction between the metal centers, it is arguable whether it is strong enough to be classified as a covalent bond. On the one hand, the distances between the metal centers are short enough that a covalent bond could be possible, especially when substantiated by the mixing of the nd and (n + 1)s/p orbitals, enhanced by the asymmetry of the MTP ligand, based on Kohn-Sham and NBO orbital composition analyses. On the other hand, the total density plots are inconsistent with a fully covalent bond and there is lack of abundant clarity that an excess of M-M bonding vs antibonding occupied molecular orbitals is obtained from the metal-based frontier orbital contours (without manipulating the isodensity values to exaggerate the electron density). Additional insights are discussed in the point/counterpoint commentary in Section 4 for both the homoatomic and heteroatomic species herein.
Figure 6 Selected total density contours for the Au2[MTP(Ph)]2 complex (isodensity value labeled in each as “Isovalue” in a.u. units).
![Figure 6 Selected total density contours for the Au2[MTP(Ph)]2 complex (isodensity value labeled in each as “Isovalue” in a.u. units).](/cms/asset/75880b8d-e92c-4af0-89cd-2aef74e7b6d8/gcic_a_1467315_f0006_oc.jpg)
The results for the truncated models, M2[MTP(H)]2, were analyzed to determine if these model complexes can accurately represent the phenomena seen in the full M2[MTP(Ph)]2 complexes. To address this concern, the structure of the Au2[MTP(H)]2 complex was compared against the previously mentioned structure of the Au2[MTP(Ph)]2 complex at the same M06/CEP-31G(d) level of theory (). The Au-Au bond distance remains essentially the same at ~3.15 Å (indeed, if anything it is—rather surprisingly—elongated, albeit slightly by a mere 0.006 Å, despite the lesser steric effect). There is also reasonable reproduction of the C-Au-S bond angles as well as the R-P-R angles, despite the smaller substituents. This structural phenomenon is seen with the other homobimetallic complexes as well, as all truncated models exhibit bond lengths that exhibit small variations within 0.00–0.06 Å vs the full model, so as to deem the truncated models sufficient for higher levels of theory and/or further oligomerization to model the solid state in subsequent studies. Presumably, the placement of phenyl groups on P- instead of S- or C-donor atoms, and their positioning away from the coordination geometry, are factors that act to ameliorate their steric or electronic influence on the coordination geometry and intramolecular or intermolecular metal-metal bonding.
Table 7 Selected bond lengths (Å) and angles (°) for Au2 complexes at M06/CEP-31g(d)
In addition to the aforementioned structural analysis, we also compare the electronic properties of these complexes. We find that the HOMO of the Au2[MTP(Ph)]2 and the Au2[MTP(H)]2 models resemble one another, corresponding to an antibonding σ* orbital with a major contribution from the Au 6s orbitals, which offsets its contribution in the σ orbital upon mixing with the 5dz2 (in the HOMO–21 of the Au2[MTP(Ph)]2 model; see ). Meanwhile, the engagement of the 6p orbitals is limited to the virtual space in both Au2[MTP(Ph)]2 and Au2[MTP(H)]2 models. While the LUMO of the MTP(Ph) dinuclear complex model is mostly phenyl-based, with little electron density on the Au2 cage, the LUMO for the analogous MTP(H)-containing model is mostly Au2 cage-based that manifests the σ bonding from primarily 6pz-6pz orbital interaction.
On the other hand, such Au2 cage contributions from 6pz-6pz orbital interactions in the Au2[MTP(Ph)]2 model are manifest in higher-lying orbitals than the LUMO; due to Koopmans’ Theorem considerations,[Citation77] aside from presence vs absence of Ph rings, one should accept such differences, as there is little credence merited for the relative energy of virtual orbitals. All in all, orbital mixing from the n = 6 shell of gold in Au2[MTP(Ph)]2 or Au2[MTP(H)]2 models is limited to 6s/5dz2 (d-s’ hybrid) σ-bonding and σ*-antibonding orbitals, whereas the 6p orbitals act as Lewis acid recipients of the lone pairs in the Lewis-basic d-s’ hybrid orbitals. The qualitative description of the bonding and anti-bonding metal-based orbitals is complicated by lack of abundant clarity that an excess of M-M bonding vs antibonding occupied molecular orbitals is obtained from the metal-based frontier orbital contours, as stated earlier. The relative energy of such metal-based orbitals with respect to one another, or with respect to other frontier orbitals that have strong ligand contribution, is different from the classical bonding scheme, at least in part due to strong asymmetric interaction with two different ligand donor atoms, S and C(H2).
Natural bond orbital (NBO) analysis of the homobimetallic complexes indicates that there are significant donor/acceptor interactions that involve the metal atoms, leading to stabilization of the M2 species. Each complex exhibits two such interactions, with one arising from the donation of electron density from the M nd orbital to the M’ (n + 1)p orbital (). These interactions are illustrated in .
Table 8 Natural bond order analysis for MM[MTP(Ph)]2
Figure 7 Illustration of the donor-acceptor interaction in Au2[MTP(H)]2 by superimposing the Au 5d orbital with the Au’ 6p orbital from NBO population analysis (isodensity = 0.03 a.u.).
![Figure 7 Illustration of the donor-acceptor interaction in Au2[MTP(H)]2 by superimposing the Au 5d orbital with the Au’ 6p orbital from NBO population analysis (isodensity = 0.03 a.u.).](/cms/asset/e3875fcb-dd34-4d5c-884f-be025876e913/gcic_a_1467315_f0007_oc.jpg)
A more intuitive situation, however, has been considered as a better “control” case—for the Pt2(PCP)2 model shown in . The PCP ligand is symmetrical, hence both M-L fragments are identical and symmetrical (P-Pt···Pt-P). Thus, shows that all frontier orbitals from the HOMO to the HOMO–9 are sequentially representative of the five bonding orbitals (σ, 2δ and 2π) and five antibonding orbitals (σ*, 2δ* and 2π*) that bear out Cotton’s classical bonding scheme of Pt(0)···Pt(0) bonding. The electron density is largely localized on the metal atoms in the Pt2 cage with negligible ligand contribution so as to impart a formal bond order of zero more genuinely with greater abundant clarity than the situation in the Au2[MTP]2 models. Little orbital mixing from the primarily σ-donating PCP phosphine (with only weak π-acceptance in absence of substituents) is responsible for this situation. In contrast, the MTP ligand entails at least strong π-donation from the S-donor atom on one fragment to shift its orbital energies from those of the AuCH2 fragment. That situation in MTP models necessitated the involvement of as low as HOMO-21 orbitals and non-sequential ordering of the Au-based bonding and anti-bonding orbitals in . There is also little to no mixing of the 6s/6p orbitals with the 5d orbitals of the Pt in Pt2(PCP)2 model, which results in orbitals that have only d-character. This leads to this Pt2 complex being a good benchmark for d10-d10 metallophilic bonding when there is no participation of the higher-lying metallic orbitals.
Figure 8 Molecular orbital contours of the Pt2(PCP)2 model (isodensity = 0.03 a.u.).
We also would like to remark that method/basis set alteration changes the relative energies of the 10 bonding and anti-bonding Pt-based orbitals in (e.g., some combinations led to the more intuitive sequencing with the σ(z2) bonding orbital being the lowest-energy as in Cotton’s classical scheme), but they remain ordered from HOMO (highest energy) to HOMO-9 (lowest energy), where all 10 orbitals are energetically close to one another and are found as a continuous group. Further description of the electronic structure, including investigations into total electron density, energetics, and geometries of M2(PCP)2 and MM’(PCP)2 models of zero-valent group 10 dimers (including those with larger R groups on the P-donor atoms), will be described elsewhere when the ongoing study concludes, as here we only wish to describe the limiting case for the Pt2(PCP)2 model as a more genuine “control” that better emulates homometallic d10-d10 bonding in Cotton’s classical scheme (with non-covalent metallophilic interactions and zero formal bond order) than does the Au2[MTP]2 model.
3.2. “Discovery” Cases of Covalent M-M’ Bonding
More intriguing are the cases for heterobimetallic complexes. These complexes consist of AuAg[MTP(H)]2 and AuCu[MTP(H)]2 illustrated in and , for which the present investigation suggests that they exhibit genuine covalent M-M’ covalent bonding. This situation entails a potential a posteriori validation of the potential existence of an Ag(I)-Au(I) polar-covalent bond for AuAg[MTP]2 species, vis-à-vis the argento-aurophilic interaction concluded by Fackler and co-workers in 2002 for the Ph-substituted complex, AuAg[MTP(Ph)]2.[Citation67] The findings herein for AuCu[MTP(H)]2, on the other hand, entail a potential a priori prediction of the potential existence of a Cu(I)-Au(I) polar-covalent bond for the yet-to-be-synthesized AuCu[MTP]2 species, vis-à-vis cupro-aurophilic interaction for future R-substituted complexes, AuCu[MTP(R)]2. We are in the process of targeted syntheses of such complexes, as guided by the predictive computational results herein and expansion thereof, whereby we shall attempt to alter the R group identities to maximize the fragment mixing and corresponding formation of d-s’ and d-p’ Au-Cu or Cu-Au interatomic hybrid orbitals.
3.2.1. The Case of a Posteriori Validation of Covalent Cu(I)-Au(I) Bonding
Given the coordination preference of organometallic complexation to gold experimentally (e.g., see ref. [Citation67] and references cited therein), we restricted the models to those with Au-C coordinate-covalent bonding in the isomers shown in - and did not investigate the CuAg[MTP(H)]2 variant. These complexes are structurally analogous to their homobimetallic counterparts, as shown in and . The metal-metal distance for the AuAg complex is 2.83 Å, which lies within the crystallographic experimental error for the sum of the covalent radii (2.81 Å and a high end of 2.92 Å if one considers the esd values), based on a recent survey of such radii.[Citation70] The MP2-computed Ag-Au distance in the AuAg[MTP(H)]2 model is shorter than the experimental crystallographic distance of AuAg[MTP(Ph)]2, which in turn is shorter than the DFT-computed distance for AuAg[MTP(Ph)]2. This result is expected, given that MP2 is a higher level of theory than DFT and that the geometry optimizations are performed for gas-phase models for a single molecule at 0 K, whereas the experimental structure is determined for packed complexes in a single crystal at 110 K.[Citation67] The metal-containing bond angles (C-Au-C’ and S-Ag-S’) deviate from linearity, with a much greater deviation of ~15° for the S-Ag-S’ angle being 165.8° vs almost nearly negligible deviation for the C-Au-C’ angle of 178.5° (). This suggests that the Au center does not experience significant attraction to the Ag center, whereas the Ag center experiences the major attraction to the Au center, as was found experimentally.[Citation67] This finding also substantiates the conclusions from our recent work that such external bond angle under-deviations from linearity represent a major figure of merit in describing whether covalent bonding is manifested by the short d10-d10 metal-metal distance (i.e., when the external L-M-L’ bond angle bends away from the M-M’ bond axis, it manifests stronger covalency, and vice versa when it bends toward the two metals).[Citation42] The calculated metal-metal distance and the metal-containing bond angles suggest that this complex has the potential to exhibit covalent character from a structural viewpoint. Section 3.2.3 provides complementary evidence from an electronic structure standpoint to substantiate the existence of the Ag(I)-Au(I) bond hypothesized herein.
Table 9 Selected bond lengths (Å) and angles (°) for AuAg[MTP(R)]2
Table 10 Selected bond lengths (Å) and angles (°) for AuCu[MTP(R)]2
3.2.2. The Case of a Priori Prediction of Potential Covalent Cu(I)-Au(I) Bonding
The same bonding trend seen for AuAg is also uncovered in the AuCu complex, for which the Cu-Au distance is calculated to be 2.73 Å, which is also tangentially inside the sum of the covalent radii of Au and Cu within experimental error (2.68–2.78 Å with the esd upper limit). This distance is remarkably short, even shorter than: (a) the analogous Cu-Au distance of 2.88 Å in the hetrobimetallic hexanuclear Au4Cu2 cluster for which two quanta of polar-covalent Cu-Au bonds have been claimed to hold two Au2Cu cyclotrimer molecules together, as has been substantiated in 2017 both experimentally and computationally by Galassi et al.; and (b) the Ag-Au bond distance of 2.84 Å in the AuAg[MTP(H)]2 model per MP2 computations herein or 2.91 Å experimentally for the crystallographic distance in AuAg[MTP(Ph)]2. The C-Au-C’ bond angle is still close to linear (179°), while there is a remarkably-large attractive distortion of the S-Cu-S bond angle to give an external bond angle of 158°. Similar to the situation in the AuAg complex, this indicates that the Au center does not experience significant attraction to the Cu center, given the linearity of the C-Au-C’ bond. However, the Cu center experiences significant attraction to the Au center, indicated by the significant deviation from linearity of the S-Cu-S’ bond angle. This deviation of 22° from linearity is much larger than that in either aforementioned (a) and (b) analogous contrasts vs the N-Cu-N’ bond angle in the Au4Cu2 cluster (167° or 13° deviation from linearity) or S-Ag-S’ bond angle deviations in AuAg[MTP(R)]2 (165.8° or ~14° deviation for the R = H model and 171.7° or ~8° deviation for the R = Ph crystal structure). Both structural figures of merit validate the strength of the hypothesized Cu(I)-Au(I) bonding, namely the Cu-Au distance and the S-Cu-S’ bond angle deviation from linearity (), are underdescribed by 0.09 Å and 0.9°, respectively, via the DFT treatment of M06/CEP-31G(d) vs the more rigorous ab initio treatment of MP2/cc-pVTZ, according to , as discussed in the previous section. The subsequent Section 3.2.3 provides complementary evidence from an electronic structure standpoint to substantiate the existence of the Cu(I)-Au(I) bond hypothesized herein.
Figure 9 Calculated geometry of AuCu[MTP(Ph)]2 complex with the C-Cu-S angles (°) shown.
![Figure 9 Calculated geometry of AuCu[MTP(Ph)]2 complex with the C-Cu-S angles (°) shown.](/cms/asset/65a84cf2-df08-409a-81cb-0e6d2db586e8/gcic_a_1467315_f0009_oc.jpg)
Figure 10 Calculated geometry of AuCu[MTP(Ph)]2 complex with the C-Cu-S angles (°) shown.
![Figure 10 Calculated geometry of AuCu[MTP(Ph)]2 complex with the C-Cu-S angles (°) shown.](/cms/asset/0fe69a30-01d4-4aba-ad7d-55dca8509cb6/gcic_a_1467315_f0010_oc.jpg)
3.2.3. Molecular Orbital Formalisms and Total Electron Density Profiles of Covalent M-M’ vs Metallophilic M···M Bonding
As was alluded to earlier, the metal-metal distance is not enough to unequivocally determine the bonding character between two metal centers. Since the bonding character of interest is covalent, orbital contributions must be considered. In the case of these heterobimetallic complexes, there is an energy mismatch between the (n)d orbitals of M vs M’, which simultaneously improves the d-s’/d-p’ interatomic hybridization between the (n)d orbitals on M and (n + 1)s/p orbitals on M’, the extent of which depends on the (M, M’) and (ML, M’L’) fragment in the pertinent system. In the case of the AuAg complex, the mixing of the 4d10(Ag)/5d10(Au) and 5s05p0(Ag)/6s06p0(Au) orbitals leads to six strongly bonding orbitals (HOMO-16, HOMO-14, HOMO-13, HOMO-12, HOMO-10, and HOMO-5) and four strongly antibonding orbitals (HOMO, HOMO-6, HOMO-8, and HOMO-9), along with several non-bonding orbitals (). With the use of a more liberal isodensity threshold, the number of Ag-Au bonding orbitals increases at least to eight to exaggerate the bonding character in the HOMO-22 and HOMO-2 orbitals, although the former is countered by a corresponding anti-bonding HOMO-18 partner orbital. The situation is likewise for analogous Kohn-Sham molecular orbitals in the R = Ph model from DFT calculations, which—indeed more clearly than the R = H model—attains eight bonding vs four anti-bonding orbitals, even at the default isodensity threshold. Nevertheless, we caution that ligand contribution is not trivial and that various bond order approximations and electrostatic vs orbital overlap decomposition analyses we are pursuing via separate collaborative efforts for both the bridged-dinuclear systems herein and our recently reported Au4Cu2 hexanuclear species so far suggest less than multiple or even single integral M-M’ formal bond order but, rather, a partially bonded character; see the concluding section for additional “counterpoint” arguments.
Figure 11 Selected Hartree-Fock molecular orbital contours of [AuAg(MTP)2] from MP2 calculations (isodensity = 0.03 a.u.).
![Figure 11 Selected Hartree-Fock molecular orbital contours of [AuAg(MTP)2] from MP2 calculations (isodensity = 0.03 a.u.).](/cms/asset/eb9db5ba-a9e8-4018-b293-466e1388634a/gcic_a_1467315_f0011_oc.jpg)
When comparing the nature of the occupied frontier molecular orbital (OFMO) space for the AuAu and AgAg versus the AuAg complex, all three complexes exhibit OFMOs that are both antibonding and bonding in nature, nearly equally from the mixing of the (n)d and the (n + 1)s’ orbitals for the two homometallic species (as discussed in Section 1; vide supra), whereas both this d-s’ and its analogous d-p’ interatomic orbital mixing have greater contributions in the bonding orbitals for the AgAu heterobimetallic species. This is demonstrated through a population analysis that revealed there to be significant mixing of the (n + 1)s orbitals with the d-orbitals in the HOMO–5 orbital as well as HOMO. In the HOMO–5, the orbital was determined to have contributions from the Au 6s (33%) as well as the Ag 5s (10%), whereas the HOMO exhibits contributions from the Au 6s (38%). The mixing of these orbitals with the d-orbitals is what leads to the difference between this AuAg heterobimetallic complex and the Pt2(PCP)2 “control” complex, where there is a distinct lack of interatomic d-s’ hybridization. This analysis is further supported by an NBO analysis, which shows there to be significant mixing of the Au 6s orbital (18%) with the 5d orbital (82%). There is still mixing seen on the Ag center, but it is much smaller (97% d-character and 3% s-character).
On the other hand, analysis of total density plots somewhat quenches one’s excitement regarding the extent of the Ag-Au bonding in the dinuclear MTP complexes herein. The heterobimetallic complex becomes virtually indistinguishable from its two analogous homobimetallic complexes at either extreme of conventional thresholds used to designate covalent bonding vs van der Waals/dispersion interactions. There are strong interactions in all three complexes at the 0.01 a.u. isodensity value, which is a common threshold for the detection of dispersion or van-der-Waals-level interactions. The interaction drops off at an isodensity value of 0.03 a.u. for the homobimetallic complexes, whereas this is not true for the AuAg complex (). This suggests that the amount of electron density between the two metal centers in the two homobimetallic complexes is virtually the same, whereas there is some modest, yet significant, increase in electron density between the metal centers of the AuAg complex. This suggests that the degree of bonding between these metal centers is not equivalent, leading to the conclusion that the AuAg complex exhibits slightly stronger bonding between the Au and Ag metal centers than those in the corresponding homobimetallic complexes. We caution, however, that the 0.03 a.u. is too liberal a threshold to distinguish covalent bonding from dispersion, van-der-Waals-level interactions compared to traditional threshold of ~0.10 a.u., at which the AuAg bonding does not “survive,” leading to the conclusion that this bonding is in the weakly covalent/upper metallophilic range to counter the other arguments based on molecular structure and interatomic d-s’/d-p’ hybrid orbital formation in the previous sections.
Figure 12 Total density contours for Au2[MTP(H)]2, AuAg[MTP(H)]2, and Ag2[MTP(H)]2 (isodensity value labeled in each as “Isovalue” in a.u. units).
![Figure 12 Total density contours for Au2[MTP(H)]2, AuAg[MTP(H)]2, and Ag2[MTP(H)]2 (isodensity value labeled in each as “Isovalue” in a.u. units).](/cms/asset/323dce8b-2cd3-436b-b0af-fb8372730dd7/gcic_a_1467315_f0012_oc.jpg)
There is significant deviation from what is expected of the homobimetallic complexes also for the AuCu heterobimetallic complex as that was seen for the AuAg complex. The AuCu[MTP(H)]2 complex exhibits a greater number of bonding than antibonding orbitals, which results in a non-zero bond order (), in contrast to the situation in the analogous Au2 and Cu2 homobimetallic complexes. Other arguments used earlier regarding the bonding character of the OFMO space and the associated d-s’ interatomic hybrid orbitals in AuAg[MTP]2 are also valid for AuCu[MTP]2 vs analogous Au2 and Cu2 counterparts; hence we do not repeat them here for the analysis of contour plots for the R = H complexes from MP2 calculations and analogous Kohn-Sham contour plots for the R = Ph complexes from DFT calculations. The percent mixing values based on the Kohn-Sham orbitals (as well as the NBO analysis) for this complex are similar to those seen in the AuAg complex.
Figure 13 Selected Hartree-Fock molecular orbital contours of AuCu[MTP(H)]2 from MP2 calculations (isodensity = 0.03 a.u.).
![Figure 13 Selected Hartree-Fock molecular orbital contours of AuCu[MTP(H)]2 from MP2 calculations (isodensity = 0.03 a.u.).](/cms/asset/c7c06feb-d8b1-4de2-9f14-55bd1c9a4b1f/gcic_a_1467315_f0013_oc.jpg)
Figure 14 Total density contours for Au2[MTP(H)]2, AuCu[MTP(H)]2, and Cu2[MTP(H)]2 (isodensity value labeled in each as “Isovalue” in a.u. units).
![Figure 14 Total density contours for Au2[MTP(H)]2, AuCu[MTP(H)]2, and Cu2[MTP(H)]2 (isodensity value labeled in each as “Isovalue” in a.u. units).](/cms/asset/a8f705ef-a7bf-4bc8-8f9b-c869d41a71c9/gcic_a_1467315_f0014_oc.jpg)
Dissimilarities were also observed in the total density plots for this complex versus the homobimetallic complexes. For all three complexes (AuAu, AuCu, CuCu), there is a strong interaction at the 0.01 isodensity value, which is to be expected, as these complexes exhibit at least van der Waals interactions. The electron density between the metal centers still drops off to create a hole or density vacancy between the two metals at 0.03 for the homobimetallic complexes but not for the AuCu complex (). This leads to the conclusion that the bonding character between the Au and Cu metal centers is slightly different (i.e., stronger) than that seen in the homobimetallic complexes, such as that seen for the AuAg heterobimetallic structure. This observation leads to the question of why the bonding interaction is stronger in the AuCu and AuAg complexes. The bonding and antibonding orbitals for these complexes result from the combination of the (n)d orbitals on the metal centers, as well as the extent of (n + 1)s/p orbital mixing/hybridization. The energy difference between the Cu and Au (n)d orbitals will be greater than the difference in the Au and Ag (n)d orbitals, causing a greater degree of (n + 1)s/p interatomic orbital mixing that attains d-s’ and d-p’ hybrid orbitals to a greater extent in the AuCu species. The asymmetry of the MTP ligand also plays a significant role in increasing the probability to form strong d-s’ and d-p’ interatomic hybrid orbitals because the bonding of the S-donor π-donor moiety to Ag and Cu raises the energy of their (n)d valence orbitals, hence increasing the likelihood for their interaction with the 6s/6p orbitals of gold.
The claim of mixing of the Au 5d orbitals with (n + 1)p orbitals on the Cu and Ag in the heterobimetallic cases of these complexes is supported by the NBO analysis, as quantified in and illustrated in . Similar to the results from the Au2[MTP(Ph)]2 case, there are two donor/acceptor interactions for each complex that significantly contribute to the stabilization of the complex. In both cases, there is donation from the Au 5d orbital to the (n + 1)p orbital for the Cu and Ag. It is also true that there is “back-donation” seen from the (n)d orbitals to the 6p orbital, but this donation is much weaker. In this case, that means Au is acting as a Lewis base and the Ag/Cu centers are acting as Lewis acids. This is in agreement with previous studies for Au in Lewis acid/base pairs containing transition metals. In one previous study by Mebs and co-workers in 2012,[Citation78] the Au(I) center donates electrons to the Bi(III) center, with little back donation from the Bi(III) center to the Au(I). In that study, the two metal centers were also held together through the use of a bridging ligand.[Citation78] However, another study has shown that Pt(II) is capable of donating electrons to form a coordinate-covalent bond between Pt and Au.[Citation39] In this case, the two metal centers were not bridged by a ligand and the stabilization energy derived from the interaction was weaker than the interaction energy between Au and C in the same complex.
Table 11 Natural bond order analysis for AuAg[MTP(Ph)]2 and AuCu[MTP(Ph)]2
Figure 15 Illustration of the donor-acceptor interaction in AuCu[MTP(H)]2 from the Au 5d donor orbital (left) to the Cu 6p orbital (middle) to form the superimposed NBO orbital (right) (isodensity = 0.03 a.u.).
![Figure 15 Illustration of the donor-acceptor interaction in AuCu[MTP(H)]2 from the Au 5d donor orbital (left) to the Cu 6p orbital (middle) to form the superimposed NBO orbital (right) (isodensity = 0.03 a.u.).](/cms/asset/c3747747-39d4-48eb-8c1d-2ca2f3312328/gcic_a_1467315_f0015_oc.jpg)
The difference between the homo- and hetero-bimetallic cases lies in the magnitude of the stabilization energy. While both interactions for the homometallic case have the same stabilization energy, this is not the case with the heterobimetallic complexes. The donation seen from the Au 5d orbitals to the (n + 1)p orbitals of the other metal center have much higher stabilization energies, which quantifies the extent of the unrequited “crush” that Ag and Cu feel towards Au, as seen crystallographically for the former and computationally for both in the optimized models herein (based on greater departure from linearity of Ag(I) and Cu(I) centers to approach the Au(I) center).
3.3. Photophysical Properties
While these complexes are structurally interesting, they also exhibit interesting optical properties. When conducting TD-DFT analysis, the full MTP(Ph) models were used based on metal-to-ligand charge transfer states facilitated by the phenyl rings. For the homobimetallic complexes, the spectra are calculated (with M06/CEP-31G(d)) to be dominated by metal-to-ligand charge transfer (MLCT) states. The calculated spectra for the AuAu and AgAg complexes are in good agreement with the experimental spectra,[Citation67] with the λmax falling around 280 nm. While there is no experimental data for the CuCu complex, the calculations suggest the absorption peak aligns with that of the AuAu complex (). The results here bear out the trend of excited state energies of the free M(I) ions known from Moore’s tables ().
Figure 16 Calculated normalized absorption spectra of Au2[MTP(Ph)]2, Cu2[MTP(Ph)]2, and Ag2[MTP(Ph)]2 with the highest orbital contribution from Au2[MTP(Ph)]2 (isodensity = 0.03 a.u. for the orbital contours depicted in the inset for the major transition).
![Figure 16 Calculated normalized absorption spectra of Au2[MTP(Ph)]2, Cu2[MTP(Ph)]2, and Ag2[MTP(Ph)]2 with the highest orbital contribution from Au2[MTP(Ph)]2 (isodensity = 0.03 a.u. for the orbital contours depicted in the inset for the major transition).](/cms/asset/05e414f8-7710-4b6f-8353-50a76d9ffb71/gcic_a_1467315_f0016_oc.jpg)
When looking at the heterobimetallic complexes, the original question pertains to the λmax placement. Experimentally, the AuAg band was shown to be slightly blue-shifted from its position in the AuAu and AgAg complexes, if not identical.[Citation67] This is a trend that is reproduced by the present calculations. The orbital analysis for this complex also reveals that MLCT states are responsible for the AuAg absorption spectrum ().
Figure 17 Calculated normalized absorption spectra of Au2[MTP(Ph)]2, Ag2[MTP(Ph)]2 and AuAg[MTP(Ph)]2 (isodensity = 0.03 a.u. for the orbital contours depicted in the inset for the major transition in the AuAg complex).
![Figure 17 Calculated normalized absorption spectra of Au2[MTP(Ph)]2, Ag2[MTP(Ph)]2 and AuAg[MTP(Ph)]2 (isodensity = 0.03 a.u. for the orbital contours depicted in the inset for the major transition in the AuAg complex).](/cms/asset/e42ff6da-178a-4474-ad3f-6c39a3330b14/gcic_a_1467315_f0017_oc.jpg)
The case becomes more interesting for the AuCu complex. For this complex, the calculations show that the absorption band is red-shifted versus its position for the homobimetallic cases, unlike the AuAg case, where the band is slightly blue-shifted. Another interesting point is that while the orbital analysis reveals MLCT states for the AuAg analogue, more significantly, there is the emergence of Au-to-Cu intermetallic charge transfer states (MM’CT). The composition of the unoccupied orbitals reveals electron density being transferred into an orbital that resembles a 4s/p orbital (). When comparing this against the AuAg complex, the appearance of this s/p-type orbital for the AuCu case and not for the AuAg case lies in the difference between the 3d and 4d metals. The energy gap between the (n)d and (n + 1)s/p orbitals increases on going from Cu(I) to Ag(I); see . So, of all the complexes, the Cu 4s orbitals should be the lowest in energy and would be expected to appear and interact with the ligand or Au orbitals before the appearance of the Ag or Au analogous 5s and 6s orbital interactions with the other metal and/or the ligand.
3.4. Point/Counterpoint Commentary
3.4.1. Point: The Case for Genuine Polar-Covalent M-M’ and (S)M-M(C) Bonds
To make the case for genuine polar covalency, we distinguish M-M’ bonds the corresponding author had hypothesized at the outset of this work as an extension to the work in ref. [Citation42] (i.e., the Omary bonds) from the (S)M-M(C) bonds that the student authors have discovered upon completion of additional computational analysis throughout this investigation (i.e., the Otten-Melançon bonds).
The M-M’ systems herein are akin to other systems shown in the literature with varying degrees of metal-metal distances and deviations in geometry—one in particular showing rather strong Pt(II)-Tl(III) polar-covalent bonding in [(NC)5Pt-Tl(CN)n]n- complexes discovered by Glaser and co-workers that possess short intramolecular M-M’ distances; subsequent computational studies completed by Pyykkö and Patzschke supported the polar-covalent bond argument.[Citation79,Citation80] In Glaser’s complexes, the Pt(II)-Tl(III) centers exhibit a difference in electronegativity with corresponding orbital energy mismatch, leading to a significant shortening of M-M’ distances. For the closed-shell metal center complexes herein, on the other hand, d10 metal-metal interactions lead to an energy mismatch of the d-orbitals of the metal centers due to both metal and ligand asymmetry, where one fragment is (S)-M’-(S), and the other is (CH2)-M-(CH2) in the hypothesized Omary bonds, where M = Au and M’ = Ag or Cu with the M-C and M’-S coordination corresponding to the crystal structure of the AgAu(MTP)2 complex. Thus, both the synthesized AuAg(MTP)2 complex and the yet-to-be-synthesized AuCu(MTP)2 complex exhibit attractive deviations from linearity for the (S)-M’-(S) bond angle, suggesting the affinity of the lighter metal center to the Au(I) organometallic center, leading to short intramolecular M-M’ distances. For the (S)M-M(C) Otten-Melançon bonds, on the other hand, the energy mismatch is hypothesized to be caused by the two π-donating S-donor moieties (situated diagonally in the trans-geometric isomers modeled herein or their cis-analogues) to raise the energy of the (n)d orbitals, which then facilitates the ds’ or dp’ mixing with the (n + 1)s’/p’ orbitals on the metal centers stabilized by ligand-field effects of the C-donor moieties.
Our investigations into natural bond order (NBO) analysis provide further insights into the stabilization of M-M’ and (S)M-M(C) interactions. The primary donor/acceptor interactions observed are shown in and , respectively. The heterobimetallic interactions are composed of two unequal interactions, representing the 2-electron donation from the Au 5d orbital to the (n + 1)p orbital of the Cu and Ag and the back donation from the Cu or Ag (n)d orbitals to the Au 6p (). Although this result could be indicative of a dative interaction, the “back-donation” might sway support to a theory that it is not purely dative but possibly polar-covalent. Recent literature by Heaven and coworkers[Citation81] has examined the extent of dative bonding between two nominally closed-shell main group atoms by second-order perturbation theory analysis. The results indicate two purely dative interactions from the F 2s/2pz hybrid orbital into a Be 2s/2pz orbital that is 87% 2pz as the primary source of attraction in a BeF- with a smaller yet significant contribution from the F 2px/2py to the Be 2px/2py orbitals.[Citation81] There is similar, if not stronger, lone pair donation from the ds’ filled hybrid orbitals to the p’ acceptor orbital in our heterobimetallic complexes than the situation described for BeF-, which exhibits closed-shell dative bonding.
The metal-metal interactions observed in the homobimetallic complexes, MM(MTP)2, shown in –, , and , were investigated further in terms of the underlying extent of orbital mixing and possible covalency. The asymmetry of the MTP ligands, (S)-M-(CH2), leads to the mixing of the ligand and metal orbitals to increase the mixing of (n)d with (n + 1)s’/p’ orbitals to generate stronger bonding than anti-bonding interactions, as supported by Kohn-Sham orbital and NBO analyses. The results obtained from the NBO analysis show two identical donor-acceptor interactions, one from Au 5d to the Au’ 6p and the other Au’ 5d to the Au 6p, with equivalent stabilization energies where the Au-Au complex exhibits the highest value, Ag-Ag the second highest, and the lowest stabilization energy is shown for the Cu-Cu complex. In all NBO analyses, however, the population vs donor-acceptor models point to different origins of orbital mixing, as the former suggests mixing of nd and (n + 1)s’ orbitals (), while the latter entails mixing of nd and (n + 1)p’ orbitals (). The discrepancy is explainable by insights from theoretical colleagues we have consulted with (vide infra; see Acknowledgments), who suggested that canonical molecular orbitals one typically plots are not unique; they merely represent one among an infinite number of possible solutions to the Schrödinger (or Kohn-Sham) equation. Additionally, the NBO method is an attempt to fit the best Lewis structure (a valence bond concept) to a molecular orbital-based total density concept. Population methods require an assumption to dissect the total electron density (which is an experimental observable, akin to what is measured in X-ray diffraction) into chemically meaningful partitions. There is no unique way, however, to partition the total electron density, and so any population scheme—be it Mulliken, Löwdin, NBO, Mayer, etc.—has an underlying assumption that it has to make in order to do the partition. Since the partitions are not unique, no one is more theoretically rigorous than another, and the hope is that one is at least more chemically intuitive (e.g., one can localize the eight electrons in methane to be either t2 and a1 levels in one approach, or four equivalent sp3 hybrid orbitals in another). Nevertheless, the NBO approach is advantageous to the default Mulliken population analysis used in Gaussian in that its dependence on the basis set size is minimal, among other known advantages for NBO methods.[Citation82]
With the qualifications noted earlier (and some to be noted later) considered regarding population analyses, the case for covalent Au-Au and Cu-Cu bonds in MTP complexes herein is further manifested in and , respectively. complements by showing four additional molecular orbitals—besides the five shown in —whereby decreasing the isodensity value to 0.02 a.u. has imparted a more magnified bonding character between the two Au(I) centers, whereas the default 0.03 a.u. value merely showed in-phase orientation of the component atomic orbitals localized on each. Doing likewise for the Cu2 analogue also shows an excess of bonding molecular orbitals, shown in , unlike the mere five attained in the “control” systems and classical Cotton formalism for d10 dimers. Caution, however, must be drawn to the multiple caveats discussed in the subsequent counterpoint section, in particular those that pertain to limitations of population analysis methods.
Figure 18 Calculated normalized absorption spectra of Au2[MTP(Ph)]2, Cu2[MTP(Ph)]2, and AuCu[MTP(Ph)]2 with the major orbital contribution for the AuCu[MTP(Ph)]2 complex (isodensity = 0.03 a.u. for the orbital contours depicted in the inset for the major transition).
![Figure 18 Calculated normalized absorption spectra of Au2[MTP(Ph)]2, Cu2[MTP(Ph)]2, and AuCu[MTP(Ph)]2 with the major orbital contribution for the AuCu[MTP(Ph)]2 complex (isodensity = 0.03 a.u. for the orbital contours depicted in the inset for the major transition).](/cms/asset/bf428fab-4d58-4e93-9ec6-8fdf643a1535/gcic_a_1467315_f0018_oc.jpg)
Figure 19 Selected molecular orbital contours of the Au2[MTP(Ph)]2 complex with isodensity values varied from 0.02 a.u. (top) to magnify the bonding interaction vs the 0.03 a.u. values used in and other figures.
![Figure 19 Selected molecular orbital contours of the Au2[MTP(Ph)]2 complex with isodensity values varied from 0.02 a.u. (top) to magnify the bonding interaction vs the 0.03 a.u. values used in Figure 3 and other figures.](/cms/asset/7f3752bb-5079-429e-afc5-56e730655300/gcic_a_1467315_f0019_oc.jpg)
Figure 20 Molecular orbital contours of the Cu2[MTP(Ph)]2 complex that manifest a Cu-Cu bonding character upon using 0.02 a.u. isodensity value.
![Figure 20 Molecular orbital contours of the Cu2[MTP(Ph)]2 complex that manifest a Cu-Cu bonding character upon using 0.02 a.u. isodensity value.](/cms/asset/559f51c9-625b-460f-bb6b-0f3a55154f1b/gcic_a_1467315_f0020_oc.jpg)
3.4.2. Counterpoint: The Case for Strong-Metallophilic-Yet-Non-Covalent Bonds
As a counterpoint, we wish to discuss a few items that further caution the reader (and ourselves as authors) from exaggerating the CuAu and AgAu bonding in these bridged-dinuclear MTP systems:
Total density plots: While this counterpoint argument is already discussed earlier, we wish to further elaborate that we have investigated plots for the simple Hg2 ground-state singlet dimer vs its *Hg2 triplet excimer, which clearly distinguished the metallophilic nature in the former and covalent nature in the latter with the traditional 0.01 a.u. and 0.10 a.u. isodensity values, respectively.
Summed covalent radii: When one employs the more conservative sum of the covalent radii for Ag-Au and Cu-Au by Schmidbaur et al.,[Citation83] and Omary/Cundari/Dias et al.,[Citation84] than that used in the previous sections based on crystal structure surveys by Alvarez et al.,[Citation70] the computed interatomic separations noted earlier become well above those other more strict limits of 2.36–2.38 Å for Cu-Au and 2.58–2.59 Å for Ag-Au.
Formal bond order calculations: Partial bond orders of no more than 0.15–0.52 have been obtained so far via various conventional approximations, such as the Mayer[Citation85] and “fuzzy” (delocalization index)[Citation86] approaches in an ongoing collaboration with the Grochala group for this type of M-M’ bonds, albeit for the precedent Au4Cu2 mixed-azolate hexanuclear cluster reported in ref. [Citation42] instead of the bridged-dinuclear MTP systems herein.
Bonding decomposition: It may be too naïve for the systems herein or, indeed, any other ligand-containing metal complexes to ascribe their bonding to a single type such as metal-metal bonds. Indeed, we have refrained in this article from invoking bond energy calculations because we have found that too high dissociation energies are obtained to be ascribed only to M-M or M-M’ single (or even multiple) covalent bonds, which are trumped by the strong electrostatic interactions between the formally +2 charge from the two monovalent coinage metals on the one hand and -2 charge from the two monoanionic MTP ligands. Thus, we have started a collaboration with Bagus et al. that seeks to decompose the bonding forces via projection operators, corresponding orbitals, and succeeding steps of the constrained space orbital variation (CSOV) method.[Citation87]
Orbital and population analysis, and theoretical level’s dependence thereof: While both canonical and NBO orbitals are included in this analysis, it is important to keep in mind the differences and similarities between the two. The canonical (Kohn-Sham) orbitals are non-unique solutions to the Kohn-Sham equations used by DFT. This means that these orbitals will be dependent upon the functional and basis set one uses, so the results will vary in this regard. Using orbitals to determine the bond order for this type of system can be misleading. NBO analysis is another method to generate orbitals whereby the electron density is partitioned and localized based on the investigated geometry. While this is true for other population analysis methods, most of these rely on properties of the structure, including the form of the wavefunction. In contrast, the NBO analysis does not suffer this pitfall, as it is primarily concerned with the eigen properties of the first-order reduced density matrix, which can be attributed to the lack of dependence on the basis set.[Citation82] The partition of the electron density is dependent upon the type of population analysis (see #3 and analogous arguments in Section 3.4.1), so some chemical intuition is necessary to determine which partition is the most chemically sound. Extended basis sets that one uses to obtain better properties can also cloud the orbital description vs intuitive concepts, in that they are often quite diffuse and can have significant orbital character away from the nucleus they are associated with. In general, the “extra” basis sets—diffuse functions, polarizations, the outer part of triple zeta, etc.—tend to pollute the bottom of the conduction band rather than the more chemically meaningful orbitals that one would attain in a minimal basis set computation. Additionally, for such large metal complexes as those treated herein, it is not likely to saturate the basis set at all angular momenta, which enables other orbitals to fill in, as dictated by the variational principle to attain a lower energy, whether the underlying situation corresponds to the simple chemical intuition such as that of Cotton’s M-M bonding scheme or not. To address these issues, multi-configurational self-consistent field (MC-SCF) calculations can be pursued, as well as other techniques that seek to project out the “extra” basis functions and mimic what the virtual orbitals would look like if done in a minimal basis set. Another aspect of this counterpoint related to population analysis is in relation to the orbital contours. While the iso-surfaces hold appeal due to the fact they give attractive illustrations to the orbital coefficients to help assign chemical meaning, there is a pitfall to using small isodensities (such as the 0.02 a.u. plots in –). This issue arises from the long-range asymptotic behavior of the wavefunction for the occupied orbitals. This can distort the relative size depicted for orbitals plotted with the same isodensity.[Citation88] For example, some of the orbital depictions in – include bonding and anti-bonding regions simultaneously in the same orbitals, whereas others include bonding due to contribution from (n-1)d and (n)s’/p’ sub-valence atomic orbirals, instead of being based merely upon the (n)d and (n + 1)s’/p’ valence orbirals.
We shall describe efforts to guard against the possible multi-faceted pitfalls in these counterpoints in future reports. We also invite constructive criticism as both/either “point” and/or “counterpoint” future articles in Comments on Inorganic Chemistry on what we believe to be stimulating and thought-provoking issues invoked in this article with regard to M-M’ covalent bonding and the associated d-s’/d-p’ interatomic hybridization in formally d10 metal cluster species. There remains work to be done in this investigation to thoroughly elucidate the bonding character within these bimetallic systems as well as other multinuclear systems. In going back to the drawing board, answers can be found by looking into these metal-metal bonded systems and performing a more complete analysis on these structures, to create guidelines and methodology to determine bonding where there is expected to be none (intuitively) and justify these stronger-than-expected interactions.
4. CONCLUSIONS AND FUTURE WORK
Metal-metal bonding has been a topic of hot debate in recent years and is one of the forefronts of inorganic chemistry.[Citation2,Citation3,Citation42] The complexes discussed herein demonstrate the difference in bonding character between selected homobimetallic and heterobimetallic complexes of group 11 monovalent cations with an MTP (methylene-thio-phosphinate) -S^C - bridging ligand. Although not conclusive to the extreme claim of strong covalency in these complexes—certainly not the multiple metal-metal bonding in Cotton’s classical systems—by employing orbital analysis and total density plots, we have shown that the interactions between the metal centers in the AuCu complex can be concluded to be stronger than those present in the homobimetallic complexes as well as the other heterobimetallic complex discussed. The strong interactions in the lower covalent/upper metallophilic range arise from the energy difference in (n)d orbitals with the (n + 1)s’/p’ orbitals, caused by different metal (M/M’) centers in heterobimetallic complexes and asymmetric –S^C– ligand in both homobimetallic and heterobimetallic complexes, so as to generate d-s’ and d-p’ interatomic hybrid orbitals. This proposition is evident from the orbital contributions, whereby the strongly antibonding orbitals are outnumbered by the strongly bonding orbitals in the occupied frontier molecular orbital space. This difference in bonding character also leads to a difference in the optical properties, with the AuCu case showing Au-to-Cu intermetallic charge transfer states with only a few MLCT states.
ACKNOWLEDGMENTS
Herein we wish to acknowledge other people and entities besides the major dedication subject, the late and great F.A.C., who has inspired the career of all authors since the onset of their respective doctoral graduate careers. Thus, B.M.O., K.M.M., and M.A.O. would like to acknowledge Profs. Tom Cundari, Andrés Cisneros, and John Berry for their helpful insights and unwavering guidance in substantiating and “decoding” these closed-shell bonding systems. M.A.O. also wishes to acknowledge stimulating discussions he has had with Profs. Harry Gray, Roald Hoffmann, Pekka Pyykko, and Alan Balch, as well as his collaborators on this project’s expansion—both computationally with Profs. Paul Bagus (and his co-worker Dr. David Hrovat) and Wojciech Grochala (and his postdoc, Dr. Pawel Szarek), and experimentally with Profs. Manal Rawashdeh-Omary and Rossana Galassi. The authors also wish to thank the three anonymous reviewers who—together with the handling editor, Prof. Berry—have offered excellent insights that have enriched the manuscript upon revision. Lastly, we thank Ms. Kaleigh Sunday (the managing editor of this journal), whose English language expertise has helped in the revisions, including those related to the use of a priori/a posteriori to both satisfy the concern expressed by one reviewer and sustain the use of that verbiage in honor of the late Prof. P. L. Goodfriend, who habitually used the verbiage during his humor-laden quantum mechanics classes at the University of Maine during Omary’s Ph.D. studies in the 1990s.
Additional information
Funding
REFERENCES
- Cotton, F. A.; Curtis, N. F.; Harris, C. B.; Johnson, B. F. G.; Lippard, S. J.; Mague, J. T.; Robinson, W. R.; Wood, J. S. Science. 1964, 145, 1305–1307. DOI: 10.1126/science.145.3638.1305.
- Murillo, C. A.;. Comments Inorg. Chem. 2015, 35, 39–58. DOI: 10.1080/02603594.2014.1002033.
- Berry, J. F.; Lu, C. Comments Inorg. Chem.. 2017, 56, 7577–7581. DOI: 10.1021/acs.inorgchem.7b01330.
- Collman, J. P.; Arnold, H. J. Acc. Chem. Res. 1993, 26, 586–592. DOI: 10.1021/ar00035a004.
- Collman, J. P.; Barnes, C. E.; Woo, L. K. Proc. Natl. Acad. Sci. U. S. A. 1983, 80, 7684–7688. DOI: 10.1073/pnas.80.24.7684.
- Collman, J. P.; Boulatov, R. Angew. Chem., Int. Ed. 2002, 41, 3948–3961. DOI: 10.1002/1521-3773(20021104)41:21<3948::AID-ANIE3948>3.0.CO;2-K.
- Collman, J. P.; Harford, S. T.; Franzen, S.; Shreve, A. P.; Woodruff, W. H. Inorg. Chem. 1999, 38, 2093–2097. DOI: 10.1021/ic9810337.
- Collman, J. P.; Harford, S. T.; Maldivi, P.; Marchon, J. C. J. Am. Chem. Soc. 1998, 120, 7999–8000. DOI: 10.1021/ja980233e.
- Tait, C. D.; Garner, J. M.; Collman, J. P.; Sattelberger, A. P.; Woodruff, W. H. J. Am. Chem. Soc. 1989, 111, 7806–7811. DOI: 10.1021/ja00202a022.
- Werner, H.;. Angew. Chem., Int. Ed. 1988, 100, 1145–1146. DOI: 10.1002/ange.19881000838.
- Dedieu, A.; Hoffmann, R. J. Am. Chem. Soc. 1978, 100, 2074–2079. DOI: 10.1021/ja00475a017.
- Jiang, Y.; Alvarez, S.; Hoffmann, R. Inorg. Chem. 1985, 24, 749–757. DOI: 10.1021/ic00199a023.
- Mehrotra, P. K.; Hoffmann, R. Inorg. Chem. 1978, 17, 2187–2189. DOI: 10.1021/ic50186a032.
- Merz, K. M.; Hoffmann, R. Inorg. Chem. 1988, 27, 2120–2127. DOI: 10.1021/ic00285a022.
- Pyykkö, P.; Li, J.; Runeberg, N. Chem. Phys. Lett. 1994, 218, 133–138. DOI: 10.1016/0009-2614(93)E1447-O.
- Pyykkö, P.; Mendizabal, F. Chem. Eur. J. 1997, 3, 1458–1465. DOI: 10.1002/chem.19970030912.
- Pyykkö, P.;. Chem. Rev. 1997, 97, 597–636. DOI: 10.1021/cr940396v.
- Pyykkö, P.;. J. Phys. Chem. A. 2015, 119, 2326–2337. DOI: 10.1021/jp5065819.
- Pyykkö, P.; Runeberg, N.; Mendizabal, F. Chem. Eur. J. 1997, 3, 1451–1457. DOI: 10.1002/chem.19970030911.
- Pyykko, P.; Xiong, X.-G.; Li, J. Faraday Discuss. 2011, 152, 169–178. DOI: 10.1039/c1fd00018g.
- Xiong, X.-G.; Pyykko, P. Chem. Commun. 2013, 49, 2103–2105. DOI: 10.1039/c2cc37875b.
- Trogler, W. C.; Gray, H. B. Acc. Chem. Res. 1978, 11, 232–239. DOI: 10.1021/ar50126a002.
- Winkler, J. R.; Gray, H. B. Molecular Electronic Structures of Transition Metal Complexes I; Springer Berlin Heidelberg: Berlin, Heidelberg, 2012. DOI: 10.1007/978-3-642-27370-4.
- Levenson, R. A.; Gray, H. B. J. Am. Chem. Soc. 1975, 97, 6042–6047. DOI: 10.1021/ja00854a015.
- Miskowski, V. M.; Gray, H. B. Understanding Molecular Properties: A Symposium in Honour of Professor Carl Johan Ballhausen, Held at the Royal Danish Academy of Sciences and Letters; April 4-5, 1986. Springer Netherlands: Dordrecht, 1987.
- Harvey, P. D.; Gray, H. B. J. Am. Chem. Soc. 1988, 110, 2145–2147. DOI: 10.1021/ja00215a023.
- Bercaw, J. E.; Durrell, A. C.; Gray, H. B.; Green, J. C.; Hazari, N.; Labinger, J. A.; Winkler, J. R. Inorg. Chem. 2010, 49, 1801–1810. DOI: 10.1021/ic902189g.
- Dahl, L. F.; Connelly, N. G. J. Am. Chem. Soc. 1970, 92, 7472–7474. DOI: 10.1021/ja00728a042.
- Kornecki, K. P.; Berry, J. F.; Powers, D. C.; Ritter, T. Progress in Inorganic Chemistry; Volume 58. John Wiley & Sons, Inc.: Hoboken, New Jersey, 2014. DOI: 10.1002/9781118792797.ch04.
- Berry, J. F.;. Acc. Chem. Res. 2016, 49, 27–34. DOI: 10.1021/acs.accounts.5b00517.
- Irwin, M. D.; Abdou, H. E.; Mohamed, A. A.; Fackler, J. J. P. Chem. Commun. 2003, 2882–2883. DOI: 10.1039/B309724M.
- Nguyen, T.; Sutton, A. D.; Brynda, M.; Fettinger, J. C.; Long, G. J.; Power, P. P. Science. 2005, 310, 844–847. DOI: 10.1126/science.1116789.
- Kreisel, K. A.; Yap, G. P. A.; Dmitrenko, O.; Landis, C. R.; Theopold, K. H. J. Am. Chem. Soc. 2007, 129, 14162–14163. DOI: 10.1021/ja076356t.
- Li Manni, G.; Dzubak, A. L.; Mulla, A.; Brogden, D. W.; Berry, J. F.; Gagliardi, L. Chem. Eur. J. 2012, 18, 1737–1749. DOI: 10.1002/chem.201103096.
- Rudd, P. A.; Liu, S.; Planas, N.; Bill, E.; Gagliardi, L.; Lu, C. C. Angew. Chem. Int. Ed. 2013, 52, 4449–4452. DOI: 10.1002/anie.201208686.
- Brogden, D. W.; Turov, Y.; Nippe, M.; Li Manni, G.; Hillard, E. A.; Clérac, R.; Gagliardi, L.; Berry, J. F. Inorg. Chem. 2014, 53, 4777–4790. DOI: 10.1021/ic5007204.
- Sunderland, T. L.; Berry, J. F. Chem. Eur. J. 2016, 22, 18564–18571. DOI: 10.1002/chem.201604007.
- Wu, B.; Wilding, M. J. T.; Kuppuswamy, S.; Bezpalko, M. W.; Foxman, B. M.; Thomas, C. M. Inorg. Chem. 2016, 55, 12137–12148. DOI: 10.1021/acs.inorgchem.6b01543.
- Baya, M.; Belío, Ú.; Fernández, I.; Fuertes, S.; Martín, A. Angew. Chem. Int. Ed. 2016, 55, 6978–6982. DOI: 10.1002/anie.201602081.
- Hlina, J. A.; Pankhurst, J. R.; Kaltsoyannis, N.; Arnold, P. L. J. Am. Chem. Soc. 2016, 138, 3333–3345. DOI: 10.1021/jacs.5b10698.
- Oeschger, R. J.; Chen, P. J. Am. Chem. Soc. 2017, 139, 1069–1072. DOI: 10.1021/jacs.6b12152.
- Galassi, R.; Ghimire, M. M.; Otten, B. M.; Ricci, S.; McDougald, R. N.; Almotawa, R. M.; Alhmoud, D.; Ivy, J. F.; Rawashdeh, A.-M.-M.; Nesterov, V. N.; Reinheimer, E. W.; Daniels, L. M.; Burini, A.; Omary, M. A. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, E5042–E5051. DOI: 10.1073/pnas.1700890114.
- Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. DOI: 10.1007/s00214-007-0310-x.
- Zhao, Y.; Truhlar, D. G. Acc. Chem. Res. 2008, 41, 157–167. DOI: 10.1021/ar700111a.
- Truhlar, D. G.; Ernest R. Davidson Lecture; University of North Texas: Denton, Texas, 2009.
- Cundari, T. R.; Stevens, W. J. J. Chem. Phys. 1993, 98, 5555–5565. DOI: 10.1063/1.464902.
- Stevens, W. J.; Basch, H.; Krauss, M. J. Chem. Phys. 1984, 81, 6026–6033. DOI: 10.1063/1.447604.
- Stevens, W. J.; Krauss, M.; Basch, H.; Jasien, P. G. Can. J. Chem. 1992, 70, 612–630. DOI: 10.1139/v92-085.
- McDougald, R. N.; Chilukuri, B.; Jia, H.; Perez, M. R.; Rabaâ, H.; Wang, X.; Nesterov, V. N.; Cundari, T. R.; Gnade, B. E.; Omary, M. A. Inorg. Chem. 2014, 53, 7485–7499. DOI: 10.1021/ic500808q.
- Determan, J. J.; Sinha, P.; Wilson, A. K.; Omary, M. A. J. Phys. Chem. C. 2015, 119, 2015–2028. DOI: 10.1021/jp5034189.
- Schwabe, T.; Grimme, S. Phys. Chem. Chem. Phys. 2006, 8, 4398–4401. DOI: 10.1039/b608478h.
- Møller, C.; Plesset, M. S. Phys. Rev. 1934, 46, 618–622. DOI: 10.1103/PhysRev.46.618.
- Tozer, D. J.; Andrews, J. S.; Amos, R. D.; Handy, N. C. Chem. Phys. Lett. 1992, 199, 229–236. DOI: 10.1016/0009-2614(92)80111-N.
- Becke, A. D.;. J. Chem. Phys. 1997, 107, 8554–8560. DOI: 10.1063/1.475007.
- Schmider, H. L.; Becke, A. D. J. Chem. Phys. 1998, 108, 9624–9631. DOI: 10.1063/1.476438.
- Grimme, S.;. J. Comp. Chem. 2006, 27, 1787–1799. DOI: 10.1002/jcc.20495.
- Rabaâ, H.; Omary, M. A.; Taubert, S.; Sundholm, D. Inorg. Chem. 2017, 57, 718–730. DOI: 10.1021/acs.inorgchem.7b02593.
- Tao, J.; Perdew, J. P.; Staroverov, V. N.; Scuseria, G. E. Phys. Rev. Lett. 2003, 91, 146401. DOI: 10.1103/PhysRevLett.91.146401.
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. J. Chem. Phys. 2010, 132, 154104. DOI: 10.1063/1.3382344.
- Frisch, M. J.; Head-Gordon, M.; Pople, J. A. Chem. Phys. Lett. 1990, 166, 275–280. DOI: 10.1016/0009-2614(90)80029-D.
- Frisch, M. J.; Head-Gordon, M.; Pople, J. A. Chem. Phys. Lett. 1990, 166, 281–289. DOI: 10.1016/0009-2614(90)80030-H.
- Head-Gordon, M.; Pople, J. A.; Frisch, M. J. Chem. Phys. Lett. 1988, 153, 503–506. DOI: 10.1016/0009-2614(88)85250-3.
- Sæbø, S.; Almlöf, J. Chem. Phys. Lett. 1989, 154, 83–89. DOI: 10.1016/0009-2614(89)87442-1.
- Head-Gordon, M.; Head-Gordon, T. Chem. Phys. Lett. 1994, 220, 122–128. DOI: 10.1016/0009-2614(94)00116-2.
- Peterson, K. A.; Puzzarini, C. Theor. Chem. Acc. 2005, 114, 283–296. DOI: 10.1007/s00214-005-0681-9.
- Figgen, D.; Rauhut, G.; Dolg, M.; Stoll, H. Chem. Phys. 2005, 311, 227–244. DOI: 10.1016/j.chemphys.2004.10.005.
- Rawashdeh-Omary, M. A.; Omary, M. A.; Fackler, J. J. Inorg. Chim. Acta. 2002, 334, 376–384. DOI: 10.1016/S0020-1693(02)00864-2.
- Wang, S.; Fackler, J. P.; Carlson, T. F. Organometallics. 1990, 9, 1973–1975. DOI: 10.1021/om00156a045.
- Mazany, A. M.; Fackler, J. P. J. Am. Chem. Soc. 1984, 106, 801–802. DOI: 10.1021/ja00315a062.
- Cordero, B.; Gomez, V.; Platero-Prats, A. E.; Reves, M.; Echeverria, J.; Cremades, E.; Barragan, F.; Alvarez, S. Dalton Trans. 2008, 2832–2838. DOI: 10.1039/b801115j.
- Cotton, F. A.; Feng, X.; Matusz, M.; Poli, R. J. Am. Chem. Soc. 1988, 110, 7077–7083. DOI: 10.1021/ja00229a021.
- Janotti, A.; Van de Walle, C. G. Nat. Mater. 2006, 6, 44. DOI: 10.1038/nmat1795.
- Autschbach, J.;. Comments Inorg. Chem. 2016, 36, 215–244. DOI: 10.1080/02603594.2015.1121874.
- Moore, C. E.;. National Bureau of Standard Circular 467; Government Printing Office: WashingtonD.C., 1958.
- Glendening, E. D.; Landis, C. R.; Weinhold, F. Wiley Interdiscip. Reviews: Computational Molecular Science. 2012, 2, 1–42. DOI: 10.1002/wcms.51.
- Glendening, E. D.; Landis, C. R.; Weinhold, F. J. Comp. Chem. 2013, 34, 1429–1437. DOI: 10.1002/jcc.23266.
- Phillips, J. C.;. Phys. Rev. 1961, 123, 420–424. DOI: 10.1103/PhysRev.123.420.
- Tschersich, C.; Limberg, C.; Roggan, S.; Herwig, C.; Ernsting, N.; Kovalenko, S.; Mebs, S. Angew. Chem., Int. Ed. 2012, 51, 4989–4992. DOI: 10.1002/anie.201200848.
- Jalilehvand, F.; Maliarik, M.; Sandström, M.; Mink, J.; Persson, I.; Persson, P.; Tóth, I.; Glaser, J. Inorg. Chem. 2001, 40, 3889–3899. DOI: 10.1021/ic010055h.
- Pyykko, P.; Patzschke, M. Faraday Discuss. 2003, 124, 41–51. DOI: 10.1039/B211364C.
- Green, M. L.; Jean, P.; Heaven, M. C. J. Phys. Chem. Lett. 2018, 1999–2002. DOI: 10.1021/acs.jpclett.8b00784.
- Weinhold, F.; Landis, C. R.; Glendening, E. D. Int. Rev. Phys. Chem. 2016, 35, 399–440. DOI: 10.1080/0144235X.2016.1192262.
- Bayler, A.; Schier, A.; Bowmaker, G. A.; Schmidbaur, H. J. Am. Chem. Soc. 1996, 118, 7006–7007. DOI: 10.1021/ja961363v.
- Omary, M. A.; Rawashdeh-Omary, M. A.; Gonser, M. W. A.; Elbjeirami, O.; Grimes, T.; Cundari, T. R.; Diyabalanage, H. V. K.; Gamage, C. S. P.; Dias, H. V. R. Inorg. Chem. 2005, 44, 8200–8210. DOI: 10.1021/ic0508730.
- Mayer, I.;. Chem. Phys. Lett. 1983, 97, 270–274. DOI: 10.1016/0009-2614(83)80005-0.
- Mayer, I.; Salvador, P. Chem. Phys. Lett. 2004, 383, 368–375. DOI: 10.1016/j.cplett.2003.11.048.
- Bagus, P. S.; Illas, F. J. Chem. Phys. 1992, 96, 8962–8970. DOI: 10.1063/1.462875.
- Haranczyk, M.; Gutowski, M. J. Chem. Theory Comput. 2008, 4, 689–693. DOI: 10.1021/ct800043a.