
Abstract
This article provides a third manifestation of a new tradition by which the editors of Comments on Inorganic Chemistry wish to lead by example, whereby we start publishing original research content that, nonetheless, preserves the journal’s identity as a niche for “critical discussion” of contemporary literature in inorganic chemistry. (For the first and second manifestations, see: (1) Otten, B. M.; Melancon, K. M.; Omary, M. A. “All that Glitters is Not Gold: A Computational Study of Covalent vs Metallophilic Bonding in Bimetallic Complexes of d10 Metal Centers: A Tribute to Al Cotton on the 10th Anniversary of His Passing,” Comments Inorg. Chem. 2018, 38, 1–35; (2) Yaseen, W. K.; Sanders, S. F.; Almotawa, R. M.; Otten, B. M.; Bhat, S.; Alamo, D. C.; Marpu, S. B.; Golden, T. D.; Omary, M. A. “Are Metal Complexes ‘Organic’, ‘Inorganic’, ‘Organometallic’, or ‘Metal-Organic’ Materials? A Case Study for the Use of Trinuclear Coinage Metal Complexes as ‘Metal-Organic Coatings’ for Corrosion Suppression on Aluminum Substrates,” Comments Inorg. Chem. 2019, 39, 1–26.) Herein, we contrast the electronic structure of two categories of Pt(II) complexes with mixed imine/thiolato ligands: a new class of [Pt(N^N)2(S^S)] bis(κ1-diimine)dithiolatoplatinum(II) divergent complexes—whereby only one N atom from the “back-to-back” diimine ligand actually coordinates to a Pt(II) center in a non-bridging monodentate manner—vis-à-vis the hitherto well-studied [Pt(N^N)(S^S)] (κ2-diimine)dithiolatoplatinum(II) convergent congeners—whereby both the diimine and dithiolate coordinate as κ2-chelating bidentate. Thus, in pursuit of using luminescent building blocks to generate light-harvesting supramolecular coordination compounds, we report the synthesis, characterization, and reactivity of Pt(pyz)2(mnt), 1, in high yield (pyz = pyrazine, mnt = maleonitriledithiolate). The complex exhibits solvent-dependent exchange of the pyrazine ligands, which can be utilized in the formation of larger complexes containing platinum dithiolate moieties. As proof of concept, 1 has been converted into the more inert Pt(4,4ʹ-bpy)2(mnt), 2, and Pt(4-ap)2(mnt), 3, (4-ap = 4-aminopyridine) complexes. These complexes exhibit redox properties, are strong absorbers of ultraviolet and visible light, and exhibit bright-luminescence at 77 K. Single-crystal X-ray diffraction analysis for 1 and 3 confirms the monotopic coordination of the ligands with intramolecular Pt-S and Pt-N bond lengths being comparable to other complexes of type (κ2-diimine)(dithiolate)Pt(II), whereas significant torsion is exhibited by the two heterocyclic rings due to the lack of tethering to one another. Solvent-dependent stability is uncovered for 1 crystals, whereas the crystal structure of 3 reveals an interesting supramolecular quadrangle resulting from hydrogen bonding between the amine groups of two complexes and two interstitial water molecules. A commentary is presented to contrast the electronic structure of the divergent complexes herein with that of a diimine-dithiolate analogue. Experimentally, the luminescence is believed to be sensitized in the new class of divergent complexes, given that their lack of conjugation of two aromatic rings renders them less-efficient π-acceptors compared to analogous diimines such as 2,2ʹ-bipyridine (2,2ʹ-bpy), hence blue-shifting the absorption toward the blue/near-UV and assisting in keeping the emission well within the visible region, whereby energy gap law considerations would suppress quenching effects in the red/near-infrared regions, especially under experimental conditions conducive for phosphorescence and aggregation effects in the solid state and higher-concentration fluid and/or frozen solutions. Computational studies for monomeric models of the three divergent complexes plus the diimine-dithiolate congener, Pt(2,2ʹ-bpy)(mnt), attain reasonable agreement with experimental structural, spectroscopic, and redox properties and provide excellent insights for the comparison between the two categories of complexes upon which we focus the commentary section. The computed photophysical properties reveal phosphorescence due to higher-lying triplet states, as the T1 is found to luminesce in the near-IR region and entail a ligand-field (dd) transition origin, whereas higher-lying states are shown to be in the visible region, close to experimental phosphorescence energies, and are consistent with the expected charge transfer transition to imine/diimine lowest-unoccupied molecular orbitals (LUMOs) from admixed dithiolate/platinum highest-occupied molecular orbitals (HOMOs). The T1 state is likely the culprit for the vanishing emission intensity at ambient temperature for both classes of complexes. We also find that both the T1 and D1 states for the neutral exciton and radical polaron (anion or cation) species, respectively, are severely distorted in 1–3 vs Pt(2,2ʹ-bpy)(mnt) models. Near-perfect orthogonality of the two heterocyclic rings can be attained for such excited and redox models of Pt(κ1-imine)2(dithiolate), whereas this distortion is hindered in Pt(κ2-diimine)(dithiolate) models due to the steric constraint in their CCNNPt metallacycle.
Graphical Abstract
1. INTRODUCTION
Supramolecular coordination compounds exhibit extensive properties, including light-harvesting capabilities,[Citation1] magnetic behavior,[Citation2] redox functionality,[Citation3] electron transfer,[Citation4] and gas storage and separation for porous metal-organic frameworks (MOFs).[Citation5,Citation6] Further advancements in this field depend upon the creative development and utilization of building blocks possessing both form and function. Traditionally, the geometry and stability of platinum(II) complexes make them commonly-used precursors for supramolecular polygons, polyhedral,[Citation6] and MOFs.[Citation7,Citation8] The addition of photophysical functionality to the square-planar form of platinum(II) is emerging as a new way of generating fluorescent organometallic metallacycles[Citation9,Citation10] or microcrystalline fibers.[Citation11]
The redox and photophysical behavior of platinum(II) dithiolate complexes[Citation12–Citation14] offer an intriguing avenue to engender location-specific functionality to supramolecular coordination complexes. In particular, the photophysical properties of platinum(II) diimine-dithiolate complexes foster applications in light-harvesting devices,[Citation15,Citation16] second-order nonlinear optical materials,[Citation17] and water splitting photocatalysts.[Citation18,Citation19] Supramolecular coordination complexes, however, are typically synthesized using divergent ditopic diimine ligands rather than the terminal chelating diimine ligands employed in platinum(II) diimine-dithiolate compounds. By combining the divergent bridging imine ligands with the cis-directing form and functionality of dithiolatoplatinum(II), a new family of supramolecular building blocks with tunable photophysical and electrochemical properties can be generated. Herein, we report on the rational design of luminescent dithiolatobis(imine)platinum(II) complexes with two divergent imine ligands, both experimentally and computationally. The presence of two κ1-NN ligands in the family of Pt(κ1-arylimine)2(dithiolate) complexes herein allows for greater torsion in the independent arylamine ring planes than the situation in the analogous Pt(κ2-arylimine)(dithiolate) complexes. The extent of this distortion in the neutral/singlet ground, excited/triplet, and the radical cation/doublet and radical anion/doublet electronic states responsible for the photophysical and redox processes is investigated for both classes of complexes using density functional theory (DFT) computations. The role of ligand-field (dd) vs charge transfer transitions in quenching the phosphorescence intensity and altering the redox potentials in 1–3 vs Pt(2,2ʹ-bpy)(mnt) models has been found to be central.
In the typical syntheses of platinum(II) diimine-dithiolate complexes, the dithiolate ligand is added to a Pt(diimine)Cl2 complex.[Citation20] Utilizing this methodology with two monotopically coordinated pyridine ligands results in lower yields and the potential formation of the thermodynamically stable bis(dithiolato)platinate(II) salt.[Citation21,Citation22] We employ an alternative high-yield route to make neutral Pt(κ1-NN)2(mnt) complexes, which minimizes the formation of bis-dithiolate salts. The products are promising candidates for yet further reactivity to form hetero- or homo-metallic supramolecular assemblies, including MOFs and coordination polymers, whereby the divergent complexes herein will be secondary building blocks or linkers with active optoelectronic functionalities. These linkers may be altered to improve their potential use in pertinent devices due to coordination polymer formation that will facilitate electronic communication between such active centers. As such, it is critical to understand the structure and properties of the light-emitting/harvesting excitons and redox-active polarons of such active components, as aspired in this investigation.
2. MATERIALS AND METHODS
Unless otherwise indicated, all operations were performed under a nitrogen atmosphere using standard Schlenk-line techniques with reaction flasks covered with aluminum foil to exclude light. Solvents were purchased from Pharmco-AAPER and reagents were purchased from Aldrich and used as received. [Pt(pyz)4](BF4)2, 4,[Citation23] [Pt(NCMe)4](BF4)2,[Citation24] and (TBA)2[Pt(mnt)2],[Citation25] were synthesized according to previously published work. Electrochemical measurements were conducted using a CHI 620B electrochemical analyzer in a 0.1 M tetra-n-butylammonium hexafluorophosphate (Bu4NPF6) solution using a platinum disk working electrode, a platinum wire counter electrode, and an Ag/AgCl reference electrode with a scan rate of 0.1 V/s. 1H NMR spectra were obtained on a JEOL Eclipse Plus 300 instrument. UV-Vis spectra were obtained using a Hewlett-Packard UV-Vis 8453 diode array spectrophotometer. FTIR spectra measurements were obtained using a Thermo Nicolett 6700 FT-IR spectrometer with FAR-IR range. The photoluminescence measurements were carried out with a PTI Quanta Master Model QM-4 scanning spectrofluorometer.
2.1. Syntheses
2.1.1. Pt(pyz)2(mnt), 1
82.5 mg (0.120 mmol) of [Pt(pyz)4](BF4)2 is dissolved with 322.2 mg (4.02 mmol) pyrazine in 25 mL DI water. A solution of 23.2 mg (0.125 mmol) Na2(mnt) dissolved in 15 mL DI water is added slowly via cannula to the Pt solution while rapidly stirring. The solution is stirred for two hours and the resulting yellow-orange precipitate is filtered through a frit in air and washed with 2 × 5 mL DI water, 2 × 5 mL cold MeOH, and 5 mL Et2O yielding 57.4 mg (97% yield). FT−IR (CsI pressed pellet): (cm−1) 2212, 2200, 2190 (m, C ≡ N), 1594(C = N), 1425, 2256, 122, 1071, 1062, 812, 806, 694, and 474. 1H NMR (d-acetone): 9.00 (dd, Ja = 3.2 Hz, Jb = 1.4 Hz, 4H) and 8.90 (dd, Ja = 3.2 Hz, Jb = 1.4 Hz, 4 H) ppm. ESI-M.S. (MeOH with 1% KBr) 534 [K(Pt(N2C4H4)2)(S2C4N2)]+.
2.1.2. Pt(4,4ʹ-bpy)2(mnt), 2
A solution of Na2mnt (7.5 mg, 0.040 mmol) dissolved in 10 mL of water was slowly added, via addition funnel, to a stirring solution of 4,4ʹbipyridine (164.8 mg, 1.1 mmol) and [Pt(4,4ʹ-bpy)4](BF4)2 (40.2 mg, 0.040 mmol) dissolved in 60 ml of 5:4 methanol: water. After stirring for 15 minutes, the resulting cloudy orange slurry is filtered and resulting orange solid is washed with water and dried in air to give 0.0247 g (95% yield) of an orange solid. FT-IR (CsI pellet): (cm−1) 2203 & 2192 (C ≡ N), 1678, 1647(C = N), 1405, 1352, 1313, 993, 832, 695, 662, 631, and 506. 1H NMR (d-DMSO): 9.10 (dd, Ja = 6.9 Hz, 4 H), 8.76 (dd, Ja = 6.0 Hz, 4 H), 8.08 (dd, Ja = 6.9 Hz, 4 H), and 7.89 (dd, Ja = 6.3 Hz, 4 H) ppm.
2.1.3. Pt(4,4ʹ-bpy)2(mnt), 2, by Pyrazine Replacement
Pt(pyz)2(mnt) (52.2 mg, 0.105 mmol) is dissolved with 4,4ʹ-bipyridine (165.2 mg, 1.06 mmol) in 10 mL degassed MeNO2 and is stirred for one day. The solution is concentrated under reduced pressure to ~5 mL, and the orange product is precipitated with 35 mL degassed Et2O. After isolation by vacuum filtration, the solid is washed with 5 mL cold THF and 2 × 5 mL cold acetone, yielding 57.8 mg (85% yield). FT−IR (CsI pressed pellet): (cm−1) 2203 (m, C ≡ N), 1647(C = N), 1405, 994, 833, 808, 695, 662, and 631. 1H NMR (d-DMSO): 9.10 (dd, Ja = 6.9 Hz, 4 H), 8.76 (dd, Ja = 6.0 Hz, 4 H), 8.08 (dd, Ja = 6.9 Hz, 4 H) and 7.89 (dd, Ja = 6.3 Hz, 4 H) ppm.
2.1.4. Pt(4-ap)2(mnt), 3
Pt(pyz)2(mnt) (53.7 mg, 0.108 mmol) is dissolved with 4-aminopyridine (105 mg, 1.12 mmol) in 10 mL degassed MeNO2 and stirred overnight. The solution is concentrated under reduced pressure to ~5 mL and combined with 30 mL degassed Et2O and filtered. Further product was obtained by condensing the filtrate under reduced pressure to dryness, re-dissolving the solid in 10 mL acetone, then precipitating with 30 mL DI H2O and isolating via frit filtration. Both yellow solids are combined and washed with 2 × 10 mL DI H2O and dried in air to yield 52.8 mg (93% yield). FT−IR (CsI pressed pellet): (cm−1) 3350 (N-H), 2200 (m, C ≡ N), 1641 (C = N), 1518, 1355, 1210, 827 and 525. 1H NMR (d-acetone): 8.12 (dd, Ja = 6.7 Hz, 4 H), 6.66 (dd, Ja = 7.1 Hz, 4 H) and 6.46 (4 H, s) ppm.
2.1.5. [Pt(4,4ʹ-bpy)4](BF4)2, 5
A solution containing 3.87 g (24.8 mmol) of 4,4ʹ-bpy in 30 mL of degassed CH3NO2 is transferred, via cannula, to 0.44 g (0.83 mmol) of [Pt(NCMe)4](BF4)2 to make a peach solution. This solution is mildly heated for two days. The slight orange solution is partially condensed using reduced pressure and then combined with 50 mL of a 1:1 ratio of degassed diethyl ether and THF. The resulting beige precipitate is filtered in air and washed with 15 mL of 1:1 diethyl ether and THF to give 0.414 g (50% yield). FT-IR (CsI): (cm−1), 1678, 1641(C = N), 1621, 1594, 1408, 1353, 1261, 1082 (B-F), 1022, 832, 809, 695, 635, 516, 499, and 395. 1H NMR (d-DMSO): 9.31 (dd, 8 H), 8.76 (dd, 8 H), 8.21 (dd, 8 H) and 7.84 (dd, 8 H) ppm. UV-vis (nm, ε (Lmol−1cm−1)) 240 (1.2 x 105). ESI-M.S. (MeOH) 410 [(Pt(N2C10H8)4)]2+.
2.1.6. [Pt(pyz)4][Pt(mnt)2], 6
A solution of (TBA)2[Pt(mnt)2] (24.7 mg, .026 mmol) in 5 mL degassed acetone is added to a solution of [Pt(pyz)4](BF4)2 (19.6 mg, 0.028 mmol) and pyrazine (154 mg, 1.91 mmol) dissolved in 5 mL degassed acetone and one drop of DMF. The solution is stirred for one hour. The red product is filtered over a frit in air, yielding 16.9 mg (66% yield). 1H NMR (d-DMSO): 9.06(dd) and 8.89(dd) ppm. FT-IR (nujol, CsI plates): (cm−1) 2196 (C ≡ N), 1418, 1151, 1107, 1023.
2.1.7. [Pt(4,4ʹ-bpy)4][Pt(mnt)2], 7
A red solution of 24.9 mg (0.030 mmol) (TBA)2[Pt(mnt)2] in 15 mL of acetone is transferred, via cannula, to a tan solution containing 30.2 mg (0.030 mmol) [Pt(4,4ʹ-bpy)4](BF4)2 in 10 mL of acetone and 5 mL of methanol to immediately form an orange solid. The solid is isolated by filtration and washed with 2 x 5 mL of acetone and 1 mL of Et2O to yield 29.8 mg (85%) of product. 1HNMR (d-DMSO) 9.32 (dd, 8 H), 8.76 (dd, 8 H), 8.20 (dd, 8 H), 7.83(dd, 8 H) ppm. ESI-M.S. (DMSO) 409 [(Pt(N2C10H8)4)]2+, 237 [Pt(S2C4N2)2]2-.
2.2. X-ray Crystallography
Slow evaporation at −20 °C of saturated acetone/xylenes solutions of either 1 or 3 generated orange crystals or orange-yellow crystals, respectively. A hemisphere of data was collected for compounds 1 at 295 K and for 3•C3H6O•2H2O at 291 K. Data were integrated using Bruker SAINT[Citation26] and corrected for absorption effects using SADABS.[Citation27] Space groups were assigned via XPREP.[Citation28] Structures were solved by a charge flipping solution method through the use of the Olex2 program,[Citation29,Citation30] which was also employed to refine all non-hydrogen atoms with anisotropic thermal parameters. Hydrogen atoms were inserted at ideal positions and refined using a riding model with an isotropic thermal parameter. has the crystal data for compounds 1 and 3•C3H6O•2H2O.
Table 1 Crystallographic data for compounds 1 and 3•C3H6O•2H2O
2.3. Computational Studies
All geometry optimizations of 1, 2, 3, and the comparative model Pt(2,2ʹ-bpy)(mnt) were performed using the Gaussian 09 suite of programs.[Citation31] The B3LYP hybrid[Citation32,Citation33] and M06 meta-hybrid[Citation34] functionals were utilized in conjunction with the CEP-31G(d) basis set,[Citation35–Citation37] where d signifies the addition of a polarization function to main group elements besides hydrogen. Geometry optimizations were performed for the ground state (S0), lowest-triplet excited state (T1), and the lowest-doublet radical-cation and radical-anion states (DC1 and DA1, respectively). Adiabatic and diabatic transition and redox energies were calculated. The time-dependent DFT (TD-DFT) method was utilized in order to accurately simulate the absorption spectra, as well as to determine transition types. These results have been used to verify and support the experimental results herein.
3. RESULTS
3.1. Synthetic Studies
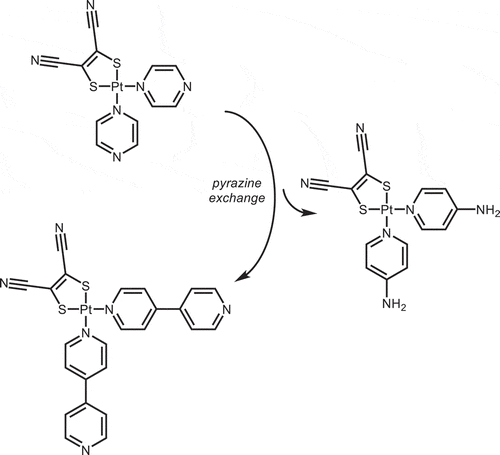
The neutral Pt(κ1-NN)2(mnt), (κ1-NN = pyz, 1, or 4,4ʹ-bpy, 2), complexes were synthesized by reacting either [Pt(pyz)4](BF4)2, 4,[Citation23] or [Pt(4,4ʹ-bpy)4](BF4)2, 5, with Na2mnt in a methanol/water mixture with an excess of the divergent NN ligand ()). The corresponding ion-paired complexes, [Pt(κ1-NN)4][Pt(mnt)2], (κ1-NN = pyz, 6, or 4,4ʹ-bpy, 7), which are coordination isomers of 1 and 2, respectively, were also synthesized and used to confirm the syntheses of Pt(κ1-NN)2(mnt).
Scheme 1 (a) Syntheses of 1 (L = pyz) and 2 (L = 4,4ʹ-bpy) from [Pt(κ1-NN)4]2+ and (b) subsequent exchange of pyz ligands in 1 to form 2 or 3 (L = 4-ap)).
![Scheme 1 (a) Syntheses of 1 (L = pyz) and 2 (L = 4,4ʹ-bpy) from [Pt(κ1-NN)4]2+ and (b) subsequent exchange of pyz ligands in 1 to form 2 or 3 (L = 4-ap)).](/cms/asset/b90fb4ad-1bb5-429d-bacb-7535d05edf1f/gcic_a_1594205_sch0001_b.gif)
In the process of characterizing 1, we observed a solvent-dependent exchange of the κ1-pyrazine (in DMSO or MeNO2). This behavior could be utilized to generate building blocks with larger divergent ligands (such as 4,4ʹ-bpy) or to directly synthesize supramolecular coordination complexes bearing dithiolate corners.[Citation38] As a proof of concept, we exchanged the pyrazine ligands with two less labile κ1-NN ligands in the high-yield syntheses of Pt(4-ap)2(mnt), 3, and an alternative route to form 2 ()).
3.2. 1H NMR Spectroscopy
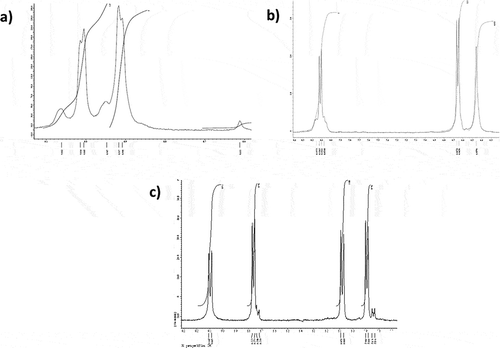
The 1H NMR resonances of compounds 1–7 were consistent with those for the monotopic coordination mode of the pyz, 4,4ʹ-bpy, or 4-ap ligands. The resonances of the aromatic protons in compounds 1 (9.00 and 8.90 ppm) and 3 (8.12, 6.66, and 6.46 ppm) exhibited 1H-195Pt coupling (J= 34 and 25 Hz, respectively) for the downfield resonances associated with the hydrogen atoms nearest the coordinated nitrogen in the ring (see ), which is in range with other pyridyl/pyrazine-containing complexes.[Citation39] The resonances for the coordinated pyridyl group of the (κ1-4,4ʹ-bpy) complexes shift upfield (9.10 and 8.08 ppm for 2 and from 9.31 and 8.21 ppm for 5; see ) upon coordination of the anionic mnt ligand. This is consistent with the larger trans-influence of the chelating dithiolate.[Citation40] The 1H resonances for the ion-paired complexes 6 and 7 are equivalent to the tetrakis(κ1-NN)platinum(II) complexes 4 and 5, respectively.
Figure 1 1H NMR spectra of Pt(pyz)2(mnt), 1 (a), Pt(4-ap)2(mnt), 3 (b) and Pt(4,4ʹ-bpy)2(mnt), 2 (c).
3.3. Structural Studies
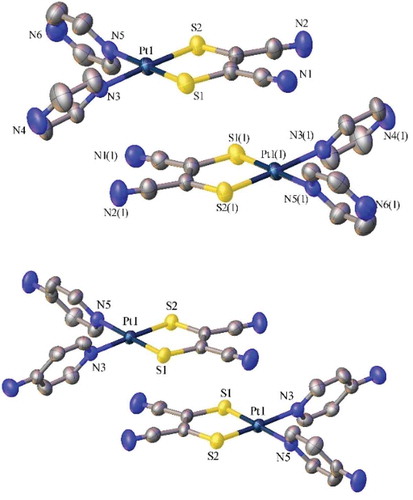
shows the pertinent interatomic distances and bond and torsion angles for 1 and 3 as revealed via single-crystal X-ray diffraction analysis, as well as the computational structural parameters. The structures in confirm the monotopic coordination of the ligands in both 1 and 3, as well as the solvent-dependent stability of 1 (grown in acetone). Intramolecular Pt-S bond lengths are comparable to other complexes of the type (κ2-diimine)(dithiolate)Pt(II).[Citation41–Citation44] The intramolecular Pt-N bond lengths (2.063(5) and 2.049(6) Å) in 3 are within the range of these diimine analogous complexes. The Pt-N distances are longer in the structure of 1, (2.077(3) and 2.078(3) Å), than in 3, which further supports the tendency of 1 to be more exchangeable. These Pt-N bond distances are also longer than those in the tetrakis(pyrazine)platinum(II) (2.012(5) and 2.026(6) Å)[Citation23] or tetrakis(pyridine)platinum(II) (2.016(3) - 2.031 (2) Å),[Citation45] providing further support for the stronger trans-influence of the mnt ligand.[Citation46] The calculated bond lengths are elongated when compared to the crystal structures due to the loss of solvation and packing effects seen in the crystal structures that are absent in the calculated structures (). There is also deviation seen for these reasons for the N-Pt-N and S-Pt-S angles. In comparison to the crystal structure of Pt(2,2ʹ-bpy)(mnt)[Citation47] where the κ2-(2,2ʹ-bpy) is essentially in the same plane as the Pt(mnt), the κ1-NN ligands have a substantial torsion angle from the PtS2C2 plane in both 1 (49.24(12) and 61.33(12)°) and 3 (56.9(2) and 60.8(2)°), which is also reproduced well by the calculations. The loss of planarity of the pyridyl-based ligand is a factor which improves the performance of these types of molecules in light-harvesting materials.[Citation21,Citation22] Both complexes pack in a slipped head-to-tail dimer arrangement (), with plane-to-plane vertical distances of 3.618(2) and 3.607(4) Å, for 1 and 3, respectively. The calculated geometry for 2 is summarized in .
Table 2 Select bond lengths, angles, and torsion angles for Pt(pyz)2(mnt), 1, and Pt(4-ap)2(mnt), 3
Table 3 Select calculated bond lengths, angles and torsion angles for Pt(4,4ʹ-bpy)2(mnt), 2
Figure 2 Crystal structures (ellipsoids at 50%) of the H-bonded quadrangle containing two units of Pt(4-ap)2(mnt), 3, and two water molecules. Other solvent molecules omitted for clarity (top) and Pt(pyz)2(mnt), 1, (bottom).
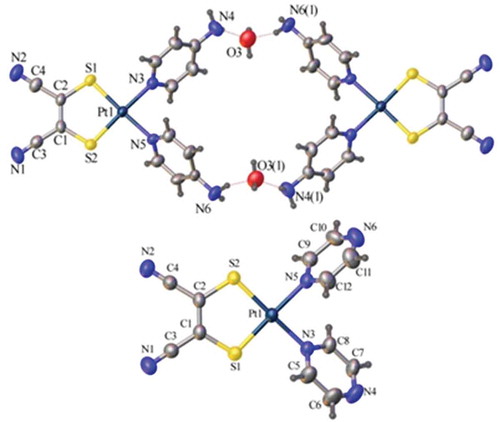
Figure 3 Crystal structures (ellipsoids at 50%) of 1 (top) and 3 (bottom) showing head-to-tail packing. H-atoms and solvent molecules omitted for clarity.
The solid-state packing of 3 reveals both NH2 groups of two distinct complexes are H-bonded with two water molecules (). This generates a supramolecular quadrangular (Pt…Pt distance of 1.2743(4) nm and O…O distance of 0.9624(13) nm), resulting in a cavity (occupied by the remaining solvate molecules; not shown in for clarity). Such H-bonding interactions may be further utilized with customized building blocks to generate supramolecular complexes with specific properties. Investigation of self-assembly reactions to form optoelectronically active supramolecular assemblies, MOFs, and other extended coordination polymer structures whereby such complexes act as building blocks is warranted.
3.4. Electrochemical and Photophysical Results
Compounds 1–3 all exhibit quasi-reversible or irreversible redox properties ( contains experimental data, while compares experimental data versus computational data) consistent with reductions centralized on the pyridyl/pyrazine ligands and oxidations originating from the Pt(mnt) components.[Citation15,Citation16,Citation21,Citation22,Citation48–Citation50] This observation is further supported via orbital analysis of the SOMO and SOMO-1 of the anionic doublet at the optimized ground state structures of 1–3 (). The orbitals of the doublet anion are used in this justification because, in DFT, the orbital shapes and energies are computed based on the electron density. While the HOMO of the ground state should adequately show the nature of the oxidation, the LUMO is not trustworthy to determine the nature of the reduction based on the fact that it has no electron density.[Citation51] Since the HOMO of the ground state is still occupied in this doublet anion, the character of this orbital does not change appreciably. However, the former LUMO is now a SOMO, leading to a more reliable description of this orbital. These orbital contours reveal that the SOMO – 1 (HOMO of the ground state) of each complex is Pt(mnt)-based, supporting the conclusion that the oxidation is based on this moiety, while the SOMOs (LUMO of the ground state) are based on the pyridyl ligands, supporting that the reduction should be pyridyl-based. This orbital analysis is also supported by previous computational work on divergent [Pt(II)(diimine)(dithiolate)] complexes.[Citation21,Citation22] The splitting of the oxidation peaks for 3, observed at 1.06 and 1.21 V ( and ), is indicative of dimerization in solution, analogous to related well-documented phenomena in TTF species,[Citation52–Citation55] and in Pt(tbpy)(benzenedithiolate).[Citation53] The oxidative potential shifts by ~25 mV more negatively for a 10-fold increase in scan rate, confirming this dimerization behavior ().[Citation56] Interestingly, a cathodic peak at the low potential of 0.32 V is also observed for 3 after the thiolate-based oxidation, and the cycle was reproduced at least five times (). This unexpected peak did not appear for 1 or 2, or even more dilute solutions of 3 (), and likely represents a Pt(II)-based reduction that takes place following the thiolate-based oxidation; a control experiment whereby the cyclic voltammetry was limited to 0.0–0.9 V did not show this unexpected peak (). Metal-based redox processes for Pt(II) square-planar complexes aid their association because the pertinent HOMO (σ*dz2) for the oligomer is a formally anti-bonding orbital while the LUMO (σpz) is bonding; this phenomenon is known for simple [Pt(CN)4]2- aqueous solutions, which become redox-active only at high concentration.[Citation57] An alternative hypothesis of a ligand rearrangement to form [Pt(mnt)2]2- is ruled out; although this complex is oxidized at a similar potential, it has a completely reversible oxidation profile (E½ = 0.37 V), which was not observed upon repeated cycles for 3 ().
Table 4 Redox potentials for compounds 1–3 at a scan rate of 0.1 V/s. *Quasi-reversible while others are irreversible. All Ec and Ea values represent peak potentials.
Table 5 Comparison of experimental and calculated reduction/oxidation potentials for 1, 2, 3, and Pt(2,2ʹ-bpy)(mnt). Eabsexp was calculated according to Eabs = Eexp + 4.696 V to convert from a Ag/AgCl reference electrode to vacuum and Eabscalc = ΔGsol/nF where ΔGsol = ΔG°xsol – ΔGredsol. Experimental values for Pt(2,2ʹ-bpy)(mnt) taken from [50]
Figure 4 SOMO–1 (top) and SOMO (bottom) contours for 1 (left), 2 (middle), and 3 (right) (plotted with an isovalue of 0.02), supporting assignment of oxidation and reduction processes.
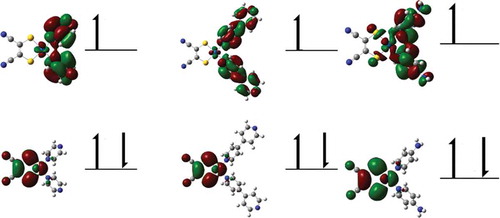
Figure 5 Cyclic voltammagram of 3 with five complete cycles from 0 to 1.5 V (red) and one cycle from 0 to 0.9 V (blue), indicating dependence of cathodic peak on oxidations at higher potential. Performed in acetonitrile using 0.1 M [n-Bu4N][PF6] electrolyte with a scan rate of 0.1 V/s.
![Figure 5 Cyclic voltammagram of 3 with five complete cycles from 0 to 1.5 V (red) and one cycle from 0 to 0.9 V (blue), indicating dependence of cathodic peak on oxidations at higher potential. Performed in acetonitrile using 0.1 M [n-Bu4N][PF6] electrolyte with a scan rate of 0.1 V/s.](/cms/asset/754bdfeb-c524-4e7f-9369-43511f2a07cb/gcic_a_1594205_f0005_oc.jpg)
Figure 6 Cyclic voltammagrams of 3 with increasing scan rate showing a negative shift in the oxidation potential dependent on scan rate.
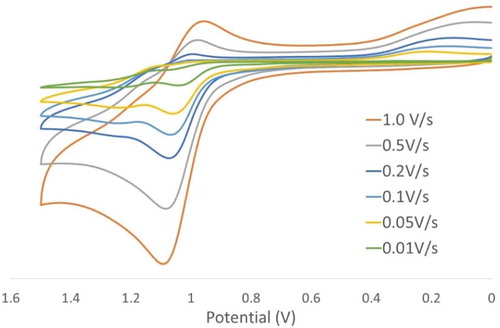
Figure 7 Cyclic voltammagram of 3 with increasing concentrations (0.2 mM, 0.4 mM, and 0.8 mM) indicating concentration dependence of cathodic peak at 0.32 V. Performed in acetonitrile using 0.1 M [n-Bu4N][PF6] electrolyte with a scan rate of 0.1 V/s.
![Figure 7 Cyclic voltammagram of 3 with increasing concentrations (0.2 mM, 0.4 mM, and 0.8 mM) indicating concentration dependence of cathodic peak at 0.32 V. Performed in acetonitrile using 0.1 M [n-Bu4N][PF6] electrolyte with a scan rate of 0.1 V/s.](/cms/asset/3f3cb1d7-e547-4fcf-92da-bdab52046c51/gcic_a_1594205_f0007_oc.jpg)
The electronic absorption spectra of compounds 1–3 () exhibit two types of bands: stronger absorption bands (ε ~104 M−1cm−1) in the mid-UV region attributable to intraligand 1π-π* transitions, and weaker bands (ε ~103 M−1cm−1) in the near-UV extending into the visible region. For the latter bands, we adopt Eisenberg’s assignment of mixed metal-(donor)ligand to (acceptor)ligand charge transfer, 1MMLL’CT (i.e., platinum dithiolate charge transfer to pyrazine or pyridyl-based ligands).[Citation20] This lowest-energy absorption is tunable based on the identity of the κ1-NN ligand, which is manifest in the absorption maxima herein (λmax = 372, 377, and 388 nm for 1, 2 and 3, respectively). These band assignments are not only supported by previous work in the field, but also through a computational analysis of the calculated absorption spectra for 1–3 (). In all computationally studied complexes, the majority of the transitions and bands can be assigned as intraligand charge transfer states (LLCT), while the other bands are made up of MLL`CT states.
Figure 8 Electronic adsorption spectra of compounds 1–3 exhibiting diimine-based π-π* absorptions in the mid-UV region and tunable MMLL’CT in the near-UV extending into the visible region.
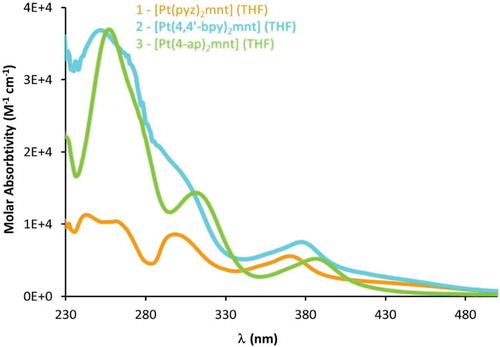
Figure 9 Calculated absorption spectra of 1 (a), 2 (b), and 3 (c) with orbital contours for the transition with the highest oscillator strength.
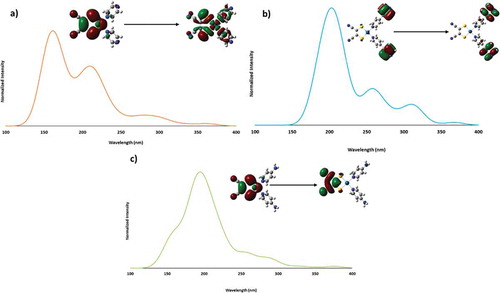
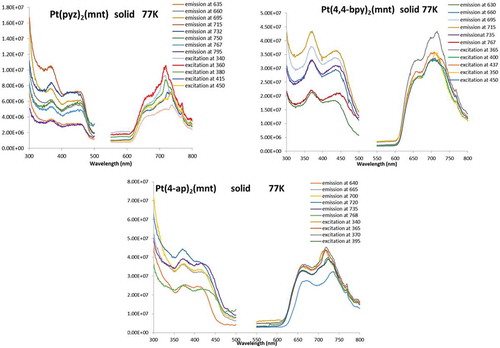
All three complexes exhibit feeble luminescence that can be drastically enhanced upon cooling to cryogenic temperatures. shows the emission and excitation spectra for all three complexes in glassy matrices at 77 K (lifetime data are summarized in ), whereas the analogous spectra in the solid state are shown in (summarized lifetime data in ). The spectral resolution is significantly higher in the frozen glasses than in the solid state, while the spectral energies are rather similar. The lifetimes of the emission bands were consistently and uniformly in the 10 µs regime, clearly suggesting phosphorescence (hence a 3MMLL’CT assignment), as expected for this type of complexes.[Citation20] Typically, it would be assumed that the emissive state would be the lowest triplet, T1. These complexes, however, exhibit T1 states that are too low in energy to be responsible for the emission (i.e., emission energies from the T1 state are in the near-IR). DFT calculations show that the emissive state is from the second-lowest triplet state, T2, which has the expected MMLL`CT character. This emissive state has emission energies in the visible region ().
Table 6 Summary of lifetime data for frozen glassy solutions of 1 (10−3 M), 2 (10−4 M), and 3 (10−3 M) in acetone
Table 7 Summary of lifetime data for solid samples of 1, 2, and 3 at 77 K
Table 8 Comparison of calculated emission energies from the T1 and T2 states and excitation energy from S0 → T1
Figure 10 Photoluminescence excitation (left/thinner curves) and emission (right/thicker curves) spectra for frozen glassy solutions of 1, 2, and 3 in acetone.
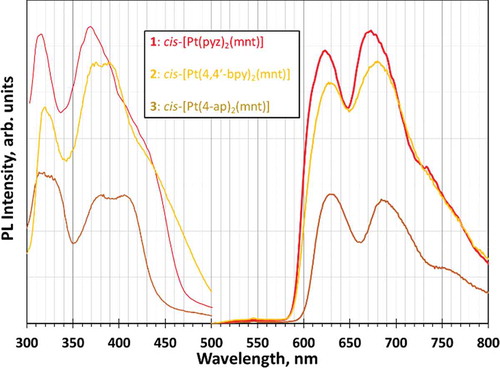
Figure 11 Photoluminescence excitation (left/thinner curves) and emission (right/thicker curves) spectra for solid samples of 1, 2, and 3 at 77 k.
The excitation bands correspond to the spin-forbidden direct absorption transition to the 3MMLL’CT triplet state of the same 1MMLL’CT lower-energy absorption bands (based on band energy). There is a red shift in the absorption or PL excitation bands, which is reproduced computationally () and which is consistent with the trend of the κ1-NN ligand’s ability to act as a π-acceptance (trend increases from 4-ap (3) < pyz (1) < 4,4ʹ-bpy (2)), which is consistent with previous work.[Citation21,Citation22] The presence of only one N-heterocyclic nitrogen atom in the aromatic ring, and an electron-donating NH2 group in 3, renders weaker π-acceptance and blue-shifted absorption/excitation bands. The addition of a second N-heterocyclic aromatic ring in 4,4ʹ-bpy of 2 expands the π conjugation beyond that seen in 1, leading to 2 having better π-accepting behavior. Interestingly, however, the emission bands are essentially indifferent to the κ1-NN ligand identity; i.e., they do not follow the same trend seen in the corresponding absorption/excitation bands. This is a qualitatively different result from that known for Pt(diimine)(dithiolate) complexes, whose emission and absorption energies correlate with one another. We hypothesize that the steric factor is more significant than the electronic factor for the excited state distortion in 1–3, given that two κ1-NN ligands rearrange more easily than one κ2-NN ligand bonded as one unit to the Pt(II) center in these Pt(II)(diimine)(dithiolate) complexes.
3.5. Commentary on Divergent vs Convergent Pt(II)-Diimine Complexes
While this study has focused on divergent Pt(II)-diimine complexes, there are plenty of already known convergent complexes. Computationally, it is easier to achieve more accurate structures for these convergent complexes, given the rigid nature of their structure (). Due to the rigid nature of this structure, the bond lengths and angles are much closer than those calculated for the divergent ligands. In addition to this rigidity, the N-Pt-N angle is smaller for the convergent complex than the divergent complexes (88.3° and 86.6° for 1 and 3, respectively, vs 79.9°). The bond lengths are similar in the convergent complex to the divergent complexes, with only slight decreases in the Pt-S and Pt-N bond lengths. Additionally, the convergent complexes are completely planar, with the dihedral angle between the imine rings being 0.0° and in the same plane as the mnt ligand.
Table 9 Select bond lengths, angles, and torsion angles for Pt(2,2ʹ-bpy)(mnt). Experimental values for Pt(2,2ʹ-bpy)(mnt) taken from [47Citation47]
The electron density of the SOMO-1 is localized on the dithiolate ligand for the divergent pyridyl ligands, while, for the convergent complex, Pt(2,2ʹ-bpy)(mnt), the SOMO-1 is not localized on the dithiolate and has some electron density on the κ2-2,2-bpy ligand (). The SOMO for the doublet anion for the convergent complex possesses a SOMO that is comparable to that of 3, where there is some electron density on the mnt ligand, but it is mostly localized on the pyridyl ligand. These similarities in the orbital analysis lead to oxidation and reduction potentials that are comparable to those seen for 1, 2, and 3 ().
Figure 12 SOMO and SOMO-1 for the convergent Pt(2,2ʹ-bpy)(mnt) complex along with experimental oxidation and reduction values (in DMF) taken from [50] and calculated oxidation and reduction potentials.
![Figure 12 SOMO and SOMO-1 for the convergent Pt(2,2ʹ-bpy)(mnt) complex along with experimental oxidation and reduction values (in DMF) taken from [50] and calculated oxidation and reduction potentials.](/cms/asset/0f872000-cbb6-4a2d-b8c8-6100dba0ea3a/gcic_a_1594205_f0012_oc.jpg)
Similar to the divergent complexes, the convergent complex exhibits two types of electron transitions in the calculated absorption spectrum. A detailed orbital analysis revealed there to be intraligand charge transfer (π-to-π* transitions that are pyridyl based) as well as MMLL`CT states that involve the transfer of electron density from the Pt(mnt) moiety to the diimine ligand (both types of transitions are depicted in ). These transition types are consistent with previous literature,[Citation44] as well as with the characteristics seen for the divergent complexes in both character and the energy range in which the transitions occur.
Figure 13 Calculated absorption spectra of Pt(2,2ʹ-bpy)(mnt) with orbital contours for the transition with the highest oscillator strength.
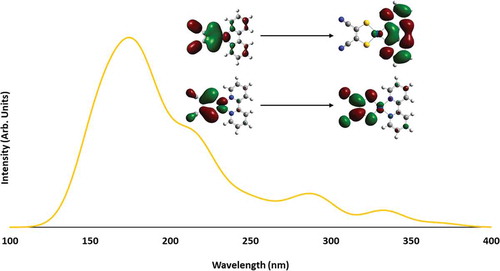
Finally, the convergent complex has an emissive state that is similar to the divergent complexes. The convergent complex's emissive state is shown to have the same character of MMLL`CT. The emissive state for the convergent complex is also the T2 instead of the T1. The excitation energy, however, is significantly red-shifted from the divergent complexes (749 nm vs around 550 nm for the divergent complexes). The T2 emission energy falls in the range of the divergent complex emission energies, being blue-shifted from 1 and 2 (644 vs 659 and 712, respectively) but red-shifted from 3 (587 nm).
4. CONCLUSIONS
A series of neutral Pt(κ1-NN)2(mnt) and ion-paired [Pt(κ1-NN)4][Pt(mnt)2] complexes have been synthesized and characterized for their molecular and electronic structures. Representatives of the former Pt(κ1-NN)2(mnt) complexes have been found to exhibit luminescence and redox properties akin to those that are characteristic of the diverse platinum(II) diimine-dithiolate family. This is the case despite having two non-chelating imine ligands as opposed to a chelating diimine, such as 2,2ʹ-bpy. The new complexes also exhibit the potential to extend the coordination core by shifting the coordination mode from a κ1-monotopic to a μ-bridging mode. As such, these new complexes hold promise as building blocks for supramolecular compounds, MOFs, and other coordination polymers with photophysical and/or electrochemical functions.
Acknowledgments
We are grateful to Dr. Vladimir Nesterov and Prof. Guido Verbeck from the University of North Texas for helpful discussions on X-ray crystallography and MS measurements at the Laboratory of Imaging Mass Spectrometry, respectively.
This work was supported by the Welch Foundation (AD-0007, BWS) and (B-1542, MAO) and the National Science Foundation (CHE-1413641 for research support and CHE-1726652 for equipment support) (MAO). The SMART-1000 diffractometer was provided by generous donations from Bruker and Austin College. Calculations were completed on a supercomputer at the University of North Texas supported by the National Science Foundation (CHE-1531468).
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Balzani, V.; Bergamini, G.; Marchioni, F.; Ceroni, P. Ru(II)-bipyridine Complexes in Supramolecular Systems, Devices and Machines. Coord. Chem. Rev. 2006, 250, 1254–1266. DOI: 10.1016/j.ccr.2005.11.013.
- Schelter, E. J.; Karadas, F.; Avendano, C.; Prosvirin, A. V.; Wernsdorfer, W.; Dunbar, K. R. A Family of Mixed-Metal Cyanide Cubes with Alternating Octahedral and Tetrahedral Corners Exhibiting A Variety of Magnetic Behaviors Including Single Molecule Magnetism. J. Am. Chem. Soc. 2007, 129(26), 8139–8149. DOI: 10.1021/ja0683281.
- Klosterman, J. K.; Iwamura, M.; Tahara, T.; Fujita, M. Energy Transfer in a Mechanically Trapped Exciplex. J. Am. Chem. Soc. 2009, 131(27), 9478–9479. DOI: 10.1021/ja901674g.
- Sautter, A.; Kaletaş, B. K.; Schmid, D. G.; Dobrawa, R.; Zimine, M.; Jung, G.; van Stokkum, I. H. M.; De Cola, L.; Williams, R. M.; Würthner, F. Ultrafast Energy-Electron Transfer Cascade in a Multichromophoric Light-Harvesting Molecular Square. J. Am. Chem. Soc. 2005, 127(18), 6719–6729. DOI: 10.1021/ja0448216.
- Corma, A.; García, H.; Llabrés I Xamena, F. X. Engineering Metal Organic Frameworks for Heterogeneous Catalysis. Chem. Rev. 2010, 110(8), 4606–4655. DOI: 10.1021/cr9003924.
- Cook, T. R.; Zheng, Y.-R.; Stang, P. J. Metal–Organic Frameworks and Self-Assembled Supramolecular Coordination Complexes: Comparing and Contrasting the Design, Synthesis, and Functionality of Metal–Organic Materials. Chem. Rev. 2013, 113(1), 734–777. DOI: 10.1021/cr3002824.
- Guo, Z.; Kobayashi, T.; Wang, -L.-L.; Goh, T. W.; Xiao, C.; Caporini, M. A.; Rosay, M.; Johnson, D. D.; Pruski, M.; Huang, W. Selective Host–Guest Interaction between Metal Ions and Metal–Organic Frameworks Using Dynamic Nuclear Polarization Enhanced Solid-State NMR Spectroscopy. Chem. - Eur. J. 2014, 20(49), 16308–16313. DOI: 10.1002/chem.v20.49.
- Cui, J.; Xu, Z. An Electroactive Porous Network from Covalent Metal–Dithiolene Links. Chem. Commun. 2014, 50(30), 3986–3988. DOI: 10.1039/C4CC00408F.
- Ghosh, S.; Mukherjee, P. S. Self-Assembly of a Nanoscopic Prism via a New Organometallic Pt3 Acceptor and Its Fluorescent Detection of Nitroaromatics. Organometallics. 2008, 27(3), 316–319. DOI: 10.1021/om701082y.
- Flynn, D. C.; Ramakrishna, G.; Yang, H.-B.; Northrop, B. H.; Stang, P. J.; Goodson, T. Ultrafast Optical Excitations in Supramolecular Metallacycles with Charge Transfer Properties. J. Am. Chem. Soc. 2010, 132(4), 1348–1358. DOI: 10.1021/ja9082655.
- Mauro, M.; Aliprandi, A.; Cebrián, C.; Wang, D.; Kübel, C.; De Cola, L. Self-Assembly of a Neutral Platinum(Ii) Complex into Highly Emitting Microcrystalline Fibers through Metallophilic Interactions. Chem. Commun. 2014, 50(55), 7269–7272. DOI: 10.1039/C4CC01045K.
- Eisenberg, R.; Gray, H. B. Noninnocence in Metal Complexes: A Dithiolene Dawn. Inorg. Chem. 2011, 50(20), 9741–9751. DOI: 10.1021/ic2011748.
- Espa, D.; Pilia, L.; Makedonas, C.; Marchiò, L.; Mercuri, M. L.; Serpe, A.; Barsella, A.; Fort, A.; Mitsopoulou, C. A.; Deplano, P. Role of the Acceptor in Tuning the Properties of Metal [M(II) = Ni, Pd, Pt] Dithiolato/Dithione (Donor/Acceptor) Second-Order Nonlinear Chromophores: Combined Experimental and Theoretical Studies. Inorg. Chem. 2014, 53(2), 1170–1183. DOI: 10.1021/ic402738b.
- Smucker, B. W.; Hudson, J. M.; Omary, M. A.; Dunbar, K. R. Structural, Magnetic, and Optoelectronic Properties of Diiminedithiolatoplatinum(II) and -Palladium(II) Complexes and Their Charge-Transfer Adducts with Nitrile Acceptors. Inorg. Chem. 2003, 42(15), 4714–4723. DOI: 10.1021/ic0259585.
- Islam, A.; Sugihara, H.; Hara, K.; Singh, L. P.; Katoh, R.; Yanagida, M.; Takahashi, Y.; Murata, S.; Arakawa, H.; Fujihashi, G. Dye Sensitization of Nanocrystalline Titanium Dioxide with Square Planar Platinum(II) Diimine Dithiolate Complexes. Inorg. Chem. 2001, 40(21), 5371–5380.
- Browning, C.; Hudson, J. M.; Reinheimer, E. W.; Kuo, F.-L.; McDougald, R. N.; Rabaâ, H.; Pan, H.; Bacsa, J.; Wang, X.; Dunbar, K. R.; et al. Synthesis, Spectroscopic Properties, and Photoconductivity of Black Absorbers Consisting of Pt(Bipyridine)(Dithiolate) Charge Transfer Complexes in the Presence and Absence of Nitrofluorenone Acceptors. J. Am. Chem. Soc. 2014, 136(46), 16185–16200. DOI: 10.1021/ja506583k.
- Pintus, A.; Aragoni, M. C.; Bellec, N.; Devillanova, F. A.; Lorcy, D.; Isaia, F.; Lippolis, V.; Randall, R. A. M.; Roisnel, T.; Slawin, A. M. Z.; et al. Structure–Property Relationships in PtII Diimine-Dithiolate Nonlinear Optical Chromophores Based on Arylethylene-1,2-Dithiolate and 2-Thioxothiazoline-4,5-Dithiolate. Eur. J. Inorg. Chem. 2012, 2012(22), 3577–3594. DOI: 10.1002/ejic.201200346.
- Zhang, J.; Du, P.; Schneider, J.; Jarosz, P.; Eisenberg, R. Photogeneration of Hydrogen from Water Using an Integrated System Based on TiO2 and Platinum(II) Diimine Dithiolate Sensitizers. J. Am. Chem. Soc. 2007, 129(25), 7726–7727. DOI: 10.1021/ja071789h.
- Zheng, B.; Sabatini, R. P.; Fu, W.-F.; Eum, M.-S.; Brennessel, W. W.; Wang, L.; McCamant, D. W.; Eisenberg, R. Light-Driven Generation of Hydrogen: New Chromophore Dyads for Increased Activity Based on Bodipy Dye and Pt(Diimine)(Dithiolate) Complexes. Proc. Natl. Acad. Sci. 2015, 112(30), E3987–E3996. DOI: 10.1073/pnas.1509310112.
- Cummings, S. D.; Eisenberg, R. Tuning the Excited-State Properties of Platinum(II) Diimine Dithiolate Complexes. J. Am. Chem. Soc. 1996, 118(8), 1949–1960. DOI: 10.1021/ja951345y.
- Moorcraft, L. P.; Jack, L. A.; Jennings, J. R.; Peter, L. M.; Yellowlees, L. J.; Robertson, N. Synthesis and Properties of [Pt(4-Co2ch3-Py)2(Dmit)] and [Pt(4-No2-Py)2(Mnt)]: Exploring Tunable Pt Dyes. Polyhedron. 2009, 28(18), 4084–4090. DOI: 10.1016/j.poly.2009.09.017.
- Moorcraft, L. P.; Morandeira, A.; Durrant, J. R.; Jennings, J. R.; Peter, L. M.; Parsons, S.; Turner, A.; Yellowlees, L. J.; Robertson, N. Synthesis and Properties of [Pt(4-Co2ch3-Py)2(Mnt)]: Comparison of Pyridyl and Bipyridyl-Based Dyes for Solar Cells. Dalton Trans. 2008, (48), 6940–6947. DOI: 10.1039/b811943k.
- Derry, P. J.; Wang, X.; Smucker, B. W. Tetra-pyrazine-platinum(II) bis-(tetra-fluoro-borate) acetonitrile hemisolvate . Acta Crystallogr. Sect. E Struct. Rep. Online 2008, 64(Pt 11), m1449. DOI: 10.1107/S1600536808033679.
- Wendt, F. O.; Kaiser, N.-F. K.; Elding, L. I. Acetonitrile and Propionitrile Exchange at palladium(II) and platinum(II) †. J. Chem. Soc. Dalton Trans. 1997, (24), 4733–4738. DOI: 10.1039/a705048h.
- Davison, A.; Holm, R. H.; Benson, R. E.; Mahler, W. Metal Complexes Derived from Cis-1,2-Dicyano-1,2-Ethylenedithiolate and Bis(Trifluoromethyl)-1,2-dithiete. Inorg. Synth. 1967, 10, 8–26. Muetterties, E. L., Mc-GrawHill Inc.
- Bruker. Saint; Bruker AXS Inc: Madison, Wisconsin, USA, 2007.
- Bruker. SADABS; Bruker AXS Inc: Madison, Wisconsin, USA, 2007.
- Bruker. XPREP; Bruker AXS Inc: Madison, Wisconsin, USA, 2007.
- Bourhis, L. J.; Dolomanov, O. V.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. The Anatomy of a Comprehensive Constrained, Restrained Refinement Program for the Modern Computing Environment - Olex2 Dissected. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71(1), 59–75. DOI: 10.1107/S2053273314022207.
- Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crsytallogr. 2009, 42(2), 339–341. DOI: 10.1107/S0021889808042726.
- Frisch, M. J.; W. T, G.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; et al. 09 Revision E01, Wallingford, CT, USA, 2016.
- Becke, A. D.;. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A. 1988, 38(6), 3098–3100. DOI: 10.1103/PhysRevA.38.3098.
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results Obtained with the Correlation Energy Density Functionals of Becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157(3), 200–206. DOI: 10.1016/0009-2614(89)87234-3.
- Zhao, Y.; Truhlar, D. G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120(1), 215–241.
- Cundari, T. R.; Stevens, W. J. Effective Core Potential Methods for the Lanthanides. J. Chem. Phys. 1993, 98(7), 5555–5565. DOI: 10.1063/1.464902.
- Stevens, W. J.; Basch, H.; Krauss, M. Compact Effective Potentials and Efficient Shared‐Exponent Basis Sets for the First‐ and Second‐Row Atoms. J. Chem. Phys. 1984, 81(12), 6026–6033. DOI: 10.1063/1.447604.
- Stevens, W. J.; Krauss, M.; Basch, H.; Jasien, P. G. Relativistic Compact Effective Potentials and Efficient, Shared-Exponent Basis Sets for the Third-, Fourth-, and Fifth-Row Atoms. Can. J. Chem. 1992, 70(2), 612–630. DOI: 10.1139/v92-085.
- Zheng, Y.-R.; Stang, P. J. Direct and Quantitative Characterization of Dynamic Ligand Exchange between Coordination-Driven Self-Assembled Supramolecular Polygons. J. Am. Chem. Soc. 2009, 131(10), 3487–3489. DOI: 10.1021/ja809788x.
- Willermann, M.; Mulcahy, C.; Sigel, R. K. O.; Cerdà, M. M.; Freisinger, E.; Sanz Miguel, P. J.; Roitzsch, M.; Lippert, B. Pyrazine as a Building Block for Molecular Architectures with PtII. Inorg. Chem. 2006, 45(5), 2093–2099. DOI: 10.1021/ic051966k.
- Melnik, M.; Holloway, C. E. Stereochemistry of Platinum Coordination Compounds. Coord. Chem. Rev. 2006, 250(17), 2261–2270. DOI: 10.1016/j.ccr.2006.02.020.
- Adams, C. J.; Fey, N.; Parfitt, M.; Pope, S. J. A.; Weinstein, J. A. Synthesis, Structures and Properties of a New Series of Platinum–Diimine–Dithiolate Complexes. Dalton Trans. 2007, (39), 4446–4456. DOI: 10.1039/b709252k.
- Makedonas, C.; Mitsopoulou, C. A.; Lahoz, F. J.; Balana, A. I. Synthesis, Characterization, and Crystal Structure of the Pd(Phen)(Bdt) Complex. A DFT and TDDFT Study of Its Ground Electronic and Excited States Compared to Those of Analogous Complexes. Inorg. Chem. 2003, 42(26), 8853–8865. DOI: 10.1021/ic034978j.
- Connick, W. B.; Gray, H. B. Photooxidation of Platinum(II) Diimine Dithiolates. J. Am. Chem. Soc. 1997, 119(48), 11620–11627. DOI: 10.1021/ja9723803.
- Bevilacqua, J. M.; Eisenberg, R. Synthesis and Characterization of Luminescent Square-Planar Platinum(II) Complexes Containing Dithiolate or Dithiocarbamate Ligands. Inorg. Chem. 1994, 33(13), 2913–2923. DOI: 10.1021/ic00091a034.
- Wei, C. H.; Hingerty, B. E.; Busing, W. R. Structure of tetrakis(pyridine)platinum(II) Chloride Trihydrate: Unconstrained Anisotropic Least-Squares Refinement of Hydrogen and Non-Hydrogen Atoms from Combined X-Ray-Neutron Diffraction Data. Acta Crystallogr., Sect. C: Struct. Chem. 1989, 45(1), 26–30. DOI: 10.1107/S0108270188009515.
- Geary, E. A. M.; Yellowlees, L. J.; Jack, L. A.; Oswald, I. D. H.; Parsons, S.; Hirata, N.; Durrant, J. R.; Robertson, N. Synthesis, Structure, and Properties of [Pt(Ii)(Diimine)(Dithiolate)] Dyes with 3,3‘-, 4,4‘-, and 5,5‘-Disubstituted Bipyridyl: Applications in Dye-Sensitized Solar Cells. Inorg. Chem. 2005, 44(2), 242–250. DOI: 10.1021/ic048799t.
- Nesterov, V. N.; Reinheimer, E. W.; Smucker, B. W. (2,2`-Bipyridine)(1,2-Dicyanoethene-1,2-Dithiolato)Platinum(Ii). IUCrData. 2019, 4(x190158). DOI: 10.1107/S2414314619001585.
- Lazarides, T.; McCormick, T. M.; Wilson, K. C.; Lee, S.; McCamant, D. W.; Eisenberg, R. Sensitizing the Sensitizer: The Synthesis and Photophysical Study of Bodipy−Pt(II)(diimine)(dithiolate) Conjugates. J. Am. Chem. Soc. 2011, 133(2), 350–364. DOI: 10.1021/ja1070366.
- Zuleta, J. A.; Bevilacqua, J. M.; Proserpio, D. M.; Harvey, P. D.; Eisenberg, R. Spectroscopic and Theoretical Studies on the Excited State in Diimine Dithiolate Complexes of platinum(II). Inorg. Chem. 1992, 31(12), 2396–2404. DOI: 10.1021/ic00038a019.
- Zuleta, J. A.; Burberry, M. S.; Eisenberg, R. Platinum(II) Diimine Dithiolates. New Solution Luminescent Complexes. Coord. Chem. Rev. 1990, 97(47). DOI: 10.1016/0010-8545(90)80079-9.
- Koopmans, T.;. Über die Zuordnung von Wellenfunktionen und Eigenwerten zu den Einzelnen Elektronen Eines Atoms. Physica. 1934, 1(1), 104–113. DOI: 10.1016/S0031-8914(34)90011-2.
- Guasch, J.; Grisanti, L.; Souto, M.; Lloveras, V.; Vidal-Gancedo, J.; Ratera, I.; Painelli, A.; Rovira, C.; Veciana, J. Intra- and Intermolecular Charge Transfer in Aggregates of Tetrathiafulvalene-Triphenylmethyl Radical Derivatives in Solution. J. Am. Chem. Soc. 2013, 135(18), 6958–6967. DOI: 10.1021/ja400281b.
- Pap, J. S.; Benedito, F. L.; Bothe, E.; Bill, E.; DeBeer George, S.; Weyhermüller, T.; Wieghardt, K. Dimerization Processes of Square Planar [Ptii(Tbpy)(Dithiolato•)]+ Radicals. Inorg. Chem. 2007, 46(10), 4187–4196. DOI: 10.1021/ic070130+.
- Spanggaard, H.; Prehn, J.; Nielsen, M. B.; Levillain, E.; Allain, M.; Becher, J. Multiple-Bridged Bis-Tetrathiafulvalenes: New Synthetic Protocols and Spectroelectrochemical Investigations. J. Am. Chem. Soc. 2000, 122(39), 9486–9494. DOI: 10.1021/ja000537c.
- Spruell, J. M.; Coskun, A.; Friedman, D. C.; Forgan, R. S.; Sarjeant, A. A.; Trabolsi, A.; Fahrenbach, A. C.; Barin, G.; Paxton, W. F.; Dey, S. K.; et al. Highly Stable Tetrathiafulvalene Radical Dimers in [3]Catenanes. Nat. Chem. 2010, 2(10). DOI: 10.1038/nchem.749.
- Zanello, P.;. Inorganic Electrochemistry, The Royal Society of Chemistry: Cambridge, UK, 2003. DOI: 0.1039/9781847551146.
- Schindler, J. W.; Adamson, A. W. Emission Quenching and Photochemistry of Aqueous Tetracyanoplatinate(2-). Inorg. Chem. 1982, 21(12), 4236–4240. DOI: 10.1021/ic00142a025.