
Abstract
Chimeric antigen receptor (CAR) T-cells are redirected T-cells that can recognize cancer antigens in a major histocompatibility complex (MHC)-independent fashion. A typical CAR is comprised of two main functional domains: an extracellular antigen recognition domain, called a single-chain variable fragment (scFv), and an intracellular signaling domain. Based on the number of intracellular signaling molecules, CARs are categorized into four generations. CAR T-cell therapy has become a promising treatment for hematologic malignancies. However, results of its clinical trials on solid tumors have not been encouraging. Here, we described the structure of CARs and summarized the clinical trials of CD19-targeted CAR T-cells. The side effects, safety management, challenges, and future prospects of CAR T-cells for the treatment of cancer, particularly for solid tumors, were also discussed.
1. Introduction
Cancer is the world’s second leading cause of death after heart attack. Despite advances in standard therapies, including surgery, radiation, and chemotherapy, the overall survival of cancer patients has not significantly improved. Over the last decade, immunotherapy has emerged as a very promising treatment, with adoptive T-cell therapy (ACT) at the forefront. One ACT approach is the infusion of autologous lymphocytes with antitumor properties. These lymphocytes are derived from genetically engineered T-cells that recognize tumor antigens by either T-cell receptors (TCRs) or chimeric antigen receptors (CARs) (Morgan et al., Citation2006). Although both TCR- and CAR-based T-cell therapies have had success in recent years, CAR T-cells have several advantages over TCR-modified T-cells for cancer therapy: (1) CAR T-cells can be used to treat cancer patients regardless of human leukocyte antigen (HLA) type because they recognize antigens in a major histocompatibility complex (MHC)-independent fashion; (2) CAR T-cells can resist some tumor escape mechanisms because cancer cells with downregulated expression of class I MHC molecules are readily killed; (3) any protein, carbohydrate, or glycolipid expressed on the tumor cell can be targeted, so CAR T-cells have a broader range of potential targets; and (4) CARs can be modified with additional co-stimulatory domains (CD28, 4-1BB, or OX40) to improve the anti-tumor function and persistence of CAR T-cells in vivo (Shaffer, Zhou, & Gottschalk, Citation2014). Here, we described the architecture of CARs, the clinical application of CAR T-cells for cancer treatment, the side effects and challenges of the possible solutions, and the future directions of CAR T-cells for cancer treatment.
2. CAR anatomy
CARs are engineered receptors on T-cells that can be activated upon antigen recognition. A typical CAR is usually comprised of an extracellular antigen recognition region, a spacer, a transmembrane domain, and an intracellular signaling region (Figure ) (Kowolik et al., Citation2006; Loskog et al., Citation2006).
Figure 1. The general structure of CARs of different generations.
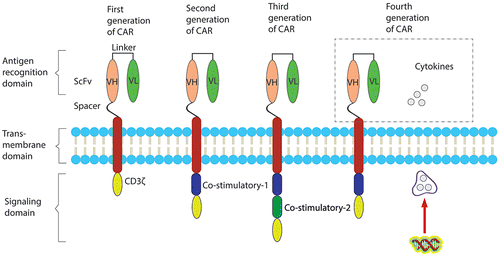
The most common type of extracellular antigen recognition region is the single-chain variable fragment (scFv), which consists of a variable heavy chain (VH), a flexible linker, and a variable light chain (VL) region (Hegde et al., Citation2013). Other types of extracellular antigen recognition moieties include ligands (Altenschmidt et al., Citation1996; Muniappan, Banapour, Lebkowski, & Talib, Citation2000), peptides (Pameijer et al., Citation2007), chimeric ligands (Davies et al., Citation2012), receptor derivatives (Scholler et al., Citation2012; Zhang, Wu, & Sentman, Citation2012), single domain antibodies (Sharifzadeh et al., Citation2013), and Fab fragments (Hegde et al., Citation2013). The origin of the scFv is very important, as it can affect the tumor specificity and other functions of the CAR. The most common source of scFvs is murine antibodies (Lamers et al., Citation2011). However, the murine-derived scFv can stimulate the human body to generate the antibodies against it, resulting in the blockade of the interaction between CAR and the target tumor antigen. This problem has been solved using humanized scFvs or scFvs derived from human monoclonal antibodies. In addition, the application of CAR T-cell treatment is often limited by the non-availability of a scFv for the binding of the target antigens due to its configuration. This issue can be solved using an antibody in its natural configuration, such a CAR is called a dual chain CAR (dcCAR) (Faitschuk, Nagy, Hombach, & Abken, Citation2016).
The spacer/hinge that connects the extracellular antigen recognition region to the transmembrane domain should be flexible enough to move in different directions to facilitate antigen recognition (Moritz & Groner, Citation1995). The sequences of the spacer are commonly derived from IgG subclasses (IgG1 or IgG4), IgD, or CD8 (Hombach, Hombach, & Abken, Citation2010). The spacer is followed by the transmembrane domain, which is derived from CD28, and less commonly from CD4, CD8, OX40, and H2-Kb. The main function of the transmembrane domain is to provide stability to the CAR (Harris & Kranz, Citation2015).
The intracellular signaling region is another important component of the CAR. Currently, CARs have been categorized into four ‘generations’ according to the number of intracellular signaling molecules (Figure ). First-generation CAR contains a single intracellular signaling domain of CD3ζ, which has a low potential for the activation and proliferation of T-cells (Irving & Weiss, Citation1991). Second-generation CAR was developed by combining a CD3ζ signaling motif with an intracellular signaling motif from a co-stimulatory molecule, most commonly, CD28. Other potential signaling co-stimulatory molecules include ICOS, OX40 (CD134) (Brentjens et al., Citation2007; Finney, Akbar, & Lawson, Citation2004), 4-1BB (CD137) (Finney et al., Citation2004; Imai et al., Citation2004), CD27 (Song et al., Citation2012), DAP10 (Brentjens et al., Citation2007), and 2B4 (CD244) (Altvater et al., Citation2009). Due to the additional co-stimulatory molecule, second-generation CAR produces more cytokines compared to first-generation CAR. Multiple experimental studies have shown that the CD28-CD3ζ combination is the strongest signal for cytokine production (Brentjens et al., Citation2007; Kofler et al., Citation2011; Sadelain, Brentjens, & Rivière, Citation2013; Sato et al., Citation2007; Wu, Roybal, Puchner, Onuffer, & Lim, Citation2015); however, this combination also increases the production of the inhibitory cytokine, IL-10. In contrast, the co-stimulatory molecule, 4-1BB, produces IL-2 in addition to IFN-γ and ultimately boosts the responses of CD8 + T-cells. Previous studies have suggested that 4-1BB co-stimulation is considered more effective than CD28 co-stimulation because 4-1BB signals also play an essential role in the persistence of memory CD8 + T-cells (Campana, Schwarz, & Imai, Citation2014). Yet currently, the CD28-CD3ζ combination is still preferred because CD28 signaling potentially circumvents CTLA-4 inhibition (Dubinski et al., Citation2015). In third-generation CAR, CD3ζ signaling moiety is fused with two co-stimulatory molecules, which are derived from p56 lck + CD28 (Geiger, Nguyen, Leitenberg, & Flavell, Citation2001), OX40 + CD28 (Hombach, Heiders, Foppe, Chmielewski, & Abken, Citation2012; Pule et al., Citation2005; Wilkie et al., Citation2008), or 4-1BB + CD28 (Carpenito et al., Citation2009; Wang et al., Citation2007; Wilkie et al., Citation2008; Zhong, Matsushita, Plotkin, Riviere, & Sadelain, Citation2010). In addition to their relatively high potency, third-generation CAR can reduce the undesirable anti-inflammatory effect of IL-10 due to the participation of a signaling domain from OX40 (Hombach et al., Citation2012) or 4-1BB (Milone et al., Citation2009). The disadvantages of third-generation CAR are their risk of signal leakage and their excessive cytokine production. Recently, to optimize the tumoricidal effect of CAR T-cells, fourth-generation CAR has also been engineered by combining second-generation CARs with a cytokine expression cassette, which is called T-cells redirected for universal cytokine-mediated killing (TRUCKs) (Chmielewski, Citation2015). As an example of TRUCKs, tumor-targeted T-cells modified to secrete IL-12 have been shown to be able to safely eradicate tumors without the need for prior conditioning (Pegram et al., Citation2012).
3. Application of CAR T-cells
The birth of CAR technology occurred in 1989, when Eshhar and his colleagues engineered the first functional CAR T-cells for cancer treatment (Gross, Waks, & Eshhar, Citation1989). Over the last few years, CAR T-cell therapy has become a game changer in the field of cancer treatment. CAR T-cell therapy has been considered a promising treatment for B-cell malignancies (hematologic malignancies). Clinical trials using CAR T-cells have also been conducted against solid tumors, for example, ovarian cancer, neuroblastoma, carcinoma, colorectal cancer, glioblastoma, sarcoma, and so on. However, the success rates of CAR T-cell treatments against solid tumors are much lower than those against hematological malignancies due to various challenges, which will be discussed in more detail later in this review.
For CAR T-cell therapy against B-cell malignancies, including chronic lymphocytic leukemia (CLL), B-cell acute lymphoblastic leukemia (B-ALL), and many Non-Hodgkin lymphomas (NHLs), CD19 has proved itself an ideal target due to the expression of CD19 in almost all B-cell malignancies. Normal tissues, except for those originating from the B-cell lineage, do not express CD19 (Depoil et al., Citation2008). Moreover, CD19 is not expressed on hematopoietic stem cells. Therefore, the elimination of all CD19 + B-cells from the body is manageable (Scheuermann & Racila, Citation1995). However, CD19, as a surface expression target, is not essential for maintaining the tumorigenic phenotype, and its escape variants have also been noted (Evans et al., Citation2015; Grupp et al., Citation2013).
In the beginning, CD19-directed CAR T-cell trials were conducted on small numbers of patients. After seeing complete remission (CR) in patients, studies were expanded to include a large number of patients (Brentjens et al., Citation2011; Grupp et al., Citation2013; Kalos et al., Citation2011; Porter, Levine, Kalos, Bagg, & June, Citation2011). CARs used for these studies are mostly involved in either CD28- or 4-1BB-based second-generation CD19-specific CARs
3.1. CD19-targeted CAR T-cells in acute lymphoblastic leukemia (ALL)
Many groups have published large-scale studies of CD19 CAR T-cell therapy in patients with B-cell ALL, which showed striking CR rates (Davila et al., Citation2014; Lee et al., Citation2015; Maude et al., Citation2014). In 2011, a group at Memorial Sloan Kettering Cancer Center (MSKCC) announced that a patient with ALL was treated with CD19 CAR T-cells for the first time (Brentjens et al., Citation2011). In 2013, the same researchers reported that five patients with ALL had achieved CR (Brentjens et al., Citation2013). Yet subsequently, they found one case of relapse, which was thought to be due to the use of high-dose steroids for treating cytokine release syndrome (CRS) (Brentjens et al., Citation2013). According to their latest update, CD19 CAR T-cell treatment led to a CR rate of approximately 88% in a series of 16 ALL patients. To achieve a better therapeutic effect, most of the patients in this series also underwent an allogeneic hematopoietic stem cell transplant (HSCT) (Davila et al., Citation2014). According to data from the 2015 annual conference of the American Society of Clinical Oncology (ASCO), MSKCC has treated 33 patients in a phase I trial to date, with 29 patients achieving CR (Park et al., Citation2015). MSKCC researchers also reported that the CR rate was associated with disease burden; patients with lower disease burden were more likely to achieve CR compared to patients with greater disease burden at the time of CAR T-cell infusion (Park et al., Citation2015). At the same time, similar studies were conducted at the University of Pennsylvania (UPenn). According to an early report from UPenn researchers, two children with relapsed and highly refractory ALL achieved CR after treatment with CD19 CAR T-cells. Afterwards, one patient had ongoing remission, and the other patient relapsed after two months due to blast cells that no longer expressed CD19 (Grupp et al., Citation2013). Later, a more comprehensive report from the same group showed a high CR rate (27/30) in ALL patients treated with CD19 CAR T-cells (Maude et al., Citation2014). With a follow-up period of 2 to 24 months, sustained remissions were observed in 19 patients. Three patients with relapsed disease were characterized by the emergence of CD19 null disease due to the intense selective pressure applied by the CAR T-cells (Maude et al., Citation2014). A further update from the 2014 annual meeting of the American Society of Hematology reported that a 92% CR rate was attained in 39 pediatric patients with ALL at UPenn. In contrast, a study by Lee et al. at the National Cancer Institute (NCI) reported that the CD19 CAR T-cell approach had resulted in CR in only 14/20 children and young adults with ALL (Lee et al., Citation2015).
All reported response rates are unprecedented for a novel agent that has been clinically trialed for the first time in humans with an untreatable malignancy. The response rates from the three centers were different, possibly due to the differences in the disease burden, prior treatment history, conditioning regimen or CAR construct. The key difference of CAR construct used for treating B-cell ALL between the three centers is that MSKCC and NCI used CD28 as the co-stimulatory molecule, while UPenn used 4-1BB instead (Maher, Citation2014). It has been noted that CD19 CAR T-cells with 4-1BB as the co-stimulatory domain show longer persistence up to two years (Maude, Teachey, Porter, & Grupp, Citation2015), while CAR T-cells therapy with CD28 showed only 1–3 months persistence post-infusion. Overall, relapse after CD19-directed T-cell therapy remains a challenge.
3.2. CD19-targeted CAR T-cells in CLL
CAR T-cells targeting CD19 have shown impressive clinical responses in 40 patients with advanced chemotherapy-refractory and high-risk CLL at the NCI (Kochenderfer et al., Citation2013; Kochenderfer et al., Citation2012; Kochenderfer et al., Citation2015), MSKCC (Brentjens et al., Citation2011), Baylor College of Medicine (Cruz et al., Citation2013), and the Abramson Family Cancer Research Institute at UPenn (Kalos et al., Citation2011; Porter et al., Citation2015; Porter et al., Citation2011). Notably, of these 40 patients, 10 achieved CR, 10 achieved partial remission (PR), and five achieved stable disease. In one of these studies, 14 patients who showed relapse and refractory CLL after treatment with CD19 CAR T-cells were again infused with CD19 CAR T-cells, with an overall response rate of 8/14 including four CR and four PR (Porter et al., Citation2015). Some groups have demonstrated that CAR T-cells targeting CD19 containing the CD3ζ signaling domain and the 4-1BB co-stimulatory domain show dramatic proliferation in vivo, remission in the patients with high disease burden, and persistence with ongoing functional activity beyond three years (Kalos et al., Citation2011; Porter et al., Citation2011). In addition, other groups have also reported clinically significant responses in patients who received CD28 co-stimulatory domain containing CAR T-cells in CLL and in some other types of cancer. (Ahmed et al., Citation2015; Eshhar, Waks, Bendavid, & Schindler, Citation2001; Porter et al., Citation2015; Singh & Paterson, Citation2007; Zhou & Levitsky, Citation2012).
3.3. CD19-targeted CAR T-cells in Non-Hodgkin lymphomas (NHLs)
NHL is a diverse group of lymphoid malignancies that includes all types of lymphoma other than Hodgkin’s lymphomas. Every year, nearly 70,000 new cases of NHL are reported in the United States. According to early reports, the first-generation CD19-directed CAR T-cells containing only the CD3ζ intracellular signaling domain were used to treat two patients with follicular lymphoma (FL) in 2010; however, CAR T-cells were not detected in the peripheral blood of the patients a few days after infusion due to very poor persistence (Jensen et al., Citation2010).
At the same time, NCI started a major clinical trial to treat CD19-positive lymphomas with CD19-redirected CAR T-cells. One of their patients with advanced FL was treated with CAR T-cells after lymph depletion, followed by intravenous administration of IL-2 (Kochenderfer et al., Citation2010). CAR T-cells were detected for up to 27 weeks after transfusion in this patient, who showed PR up to 8 months. Later, the patient was retreated with the same CAR T-cells and had PR for another 26 months (Kochenderfer & Rosenberg, Citation2011). In this trial, all eight patients were treated with CD19 CAR T-cells; however, the treatment was only effective in 6 patients, 4 of which were B-cell NHL (three FL and one splenic marginal zone lymphoma). Due to the presence of severe, cytokine release-related toxicity (Kochenderfer et al., Citation2012), amended protocols were developed in a subsequent trial for different types of NHL, for example, utilizing a lower dose of CD19-redirected CAR T-cells and without IL-2 expression. Overall, 12/13 patients showed a partial or complete response to this CD19-specific CAR T-cell therapy (Kochenderfer et al., Citation2015). According to the ASCO 2015 report, the response rate was 67% for the phase IIa trial against NHL and was 100% in 6 patients with FL (Schuster et al., Citation2015).
3.4. Challenges for CD19 CAR T-cell therapy
CD19 CAR T-cell therapy has been considered a promising treatment for hematologic malignancies. However, relapse after CAR T-cell therapy remains a challenge. The approaches for preventing the shortcomings of CD19 CAR T-cells therapy include the optimization of CAR design or manufacture and the improvement of T-cell persistence. The latter is considered a more promising approach. A difference in CAR T-cell persistence has been observed between cells with CARs carrying CD28 and cells with CARs carrying 4-1BB as the co-stimulatory molecule. As we have mentioned above, the CD28-CD3ζ signaling combination is the strongest signal for cytokine production (Brentjens et al., Citation2007; Kofler et al., Citation2011; Sadelain et al., Citation2013; Sato et al., Citation2007; Wu et al., Citation2015), but this combination also increases the production of the inhibitory cytokine, IL-10. In addition, studies have indicated that cells having second-generation CAR containing the CD28 co-stimulatory molecule underwent more rapid in vivo expansion and decline (Van der Stegen, Hamieh, & Sadelain, Citation2015). Conversely, preclinical in vivo studies have suggested that 4-1BB-CAR produces better proliferation and/or persistence than CD28 CAR (Campana et al., Citation2014; Kalos et al., Citation2013; Milone et al., Citation2009; Van der Stegen et al., Citation2015). CAR T-cells with 4-1BB-CAR are expected to persist for more than 6 months and can persist for up to 2 years in many ALL patients treated with CTL019 T-cells (i.e. cells with CD19-redirected CARs containing the CD3ζ signaling domain and the 4-1BB co-stimulatory domain) (Lee et al., Citation2015). In addition, CTL019 cells have been shown to eliminate large volumes of tumors (Kalos et al., Citation2013). This excellent response of tumors to 4-1BB-engineered CARs is due to their ability to release multifunctional cytokines (such as IL-2, TNFα, and IFN-γ) and their stronger signaling for sustained T-cell survival (Carpenito et al., Citation2009). To avoid escape variants due to single target (CD19) therapy, CAR T-cells targeting B-cell-associated antigens such as CD20 (Wang et al., Citation2004), CD22 (Haso et al., Citation2013), CD38 (Mihara et al., Citation2009, 2010), ROR1 (Hudecek et al., Citation2010) and k light chain (Vera et al., Citation2006) also have been developed for treating CD19-negative relapse in combination with CD19-directed CAR T-cells.
Recently, Novartis announced that CTL019 (tisagenlecleucel), an investigational CAR T-cell therapy for the treatment of patients with B-ALL, has been approved by the US Food and Drug Administration (FDA) and the Oncologic Drugs Advisory Committee (ODAC).
3.5. CAR T-cell therapy in solid tumors against different targets
In this section, we summarized therapeutic targets in treating solid tumors (Table ) and focused the molecular basic and biological function of the most common targets used for CAR T-cell therapy of solid tumors in multi-research institutes.
Table 1. Therapeutic targets in treating solid tumors.
3.5.1. HER2
Human epidermal growth factor receptor 2 (HER2) is a 185 KDa transmembrane glycoprotein belonging to the epidermal growth factor receptor (EGFR) family (Cho et al., Citation2003; Yarden & Sliwkowski, Citation2001). Overexpression of HER2 is associated with cellular transformation, carcinogenesis, and poor clinical outcome (Serrano-Olvera, Duenas-Gonzalez, Gallardo-Rincon, Candelaria, & De la Garza-Salazar, Citation2006; Thompson, Sullivan, Davies, & Ruszkiewicz, Citation2011). In addition, it also plays an important role in the biological behavior and pathogenesis of some human cancers (Hudziak, Schlessinger, & Ullrich, Citation1987). HER2 is one of the most commonly studied antigens for cancer immunotherapy because it is overexpressed in many tumors, such as in 80% of glioblastomas (Zhang et al., Citation2008), 60% of osteosarcomas (Gorlick et al., Citation1999), 40% of medulloblastomas (Ahmed et al., Citation2007), and 25–30% of breast and ovarian cancers (Slamon et al., Citation1989). Overexpression of HER2 is an ideal target for CAR T-cell therapy of ovarian cancer, breast cancer, glioblastoma, colon cancer, osteosarcoma and medulloblastoma (Shengnan et al., Citation2017).
3.5.2. EGFRvIII
The epidermal growth factor receptor variant III (EGFRvIII) is a 170 KDa transmembrane receptor and a member of the ErbB oncogene family (Hynes & MacDonald, Citation2009). EGFRvIII is the most common oncogenic EGFR mutant found in glioblastoma (Heimberger, Suki, Yang, Shi, & Aldape, Citation2005). EGFRvIII is not expressed in normal tissue (Maus, Citation2015) and only expressed in several cancer types, including glioblastoma, breast cancer, and non-small cell lung cancer (NSCLC). EGFRvIII plays a role in promoting tumor cell growth, invasion, migration, and therapeutic resistance (Luo et al., Citation2013). EGFRvIII, being a neoantigen, is the best target for CAR T-cell therapy of glioblastoma (Shengnan et al., Citation2017).
3.5.3. Mesothelin
Mesothelin is a 40 KDa cell surface tumor antigen with unknown biological function (Hassan, Bera, & Pastan, Citation2004). Mesothelin is a tumor-associated antigen that is overexpressed in many types of cancer, such as mesothelioma, pancreatic cancers, and ovarian cancers (Beatty et al., Citation2014). Mesothelin is an attractive target for CAR T-cell therapy of mesothelioma, pancreatic cancers, and ovarian cancers (Shengnan et al., Citation2017).
3.5.4. GD2
GD2 is a disialoganglioside with a molecular weight of 105–115 KDa. GD2 is expressed on the plasma membranes of various types of malignant cells and is involved in transducing the cell death signal in tumor cells (Doronin et al., Citation2014). GD2 is one of the most promising antigens for cancer immunotherapy and is thought to be one of the safest targets for immunotherapy (Krug et al., Citation2015). GD2 is a promising target for CAR T-cell therapy of neuroblastoma, melanoma, and glioma (Shengnan et al., Citation2017).
3.5.5. CEA
Carcinoembryonic antigen (CEA) is a 180 KDa glycoprotein involved in cell adhesion. CEA is one of best tumor markers, as it is overexpressed in many epithelial cancers, especially in colorectal adenocarcinoma and pancreatic tumors (Chmielewski, Rappl, Hombach, & Abken, Citation2012). CEA is an ideal target for CAR T-cell therapy of pancreatic adenocarcinoma, breast cancer and colorectal carcinoma (Shengnan et al., Citation2017).
3.5.6. EphA2
Ephrin type-A receptor 2 (EphA2), a member of the ephrin receptor subfamily of the protein-tyrosine kinase family (Davis et al., Citation1994; Kinch & Carles-Kinch, Citation2003), is an attractive emerging target for immunotherapy of glioblastoma due to its overexpression in glioma. EphA2-redirected CAR T-cell therapy has been proven to result in the regression of glioma xenografts in a mouse model (Chow et al., Citation2013).
Published clinical studies on CAR T-cell therapy for both hematological malignancies and for solid tumors are summarized in Tables and , respectively.
Table 2. Published clinical studies on CAR T-cells specific for hematological malignant antigens.
Table 3. Published clinical studies on CAR T-cells specific to solid tumor antigens.
4. Side effects of CAR T-cell therapy
4.1. On-target/off-tumor adverse reactions
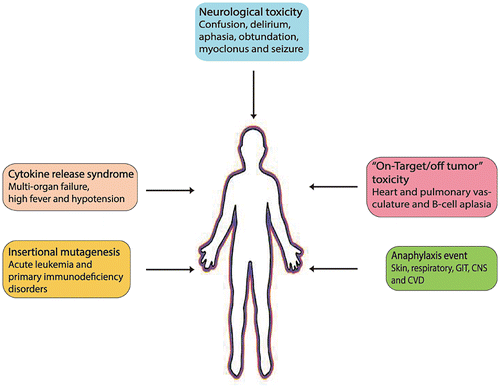
The major safety concern of CAR T-cell therapy is its off-tumor adverse reactions, which are usually caused by the toxicity of CAR T-cells in normal tissues. On-target/off-tumor toxicities following CAR T-cell infusion have been observed in many clinical trials (Wu et al., Citation2015). These reactions can either be minor and manageable or life-threatening if they take place in vital organs like heart, liver, and lungs (Morgan et al., Citation2010). The earliest observation of this kind of adverse reaction was cholestasis after the infusion of CAR T-cells targeting carbonic anhydrase IX for renal cell carcinoma, which was possibly due to the expression of carbonic anhydrase IX in the bile duct epithelium (Lamers et al., Citation2013; Lamers et al., Citation2006). The most severe acute toxicities were reported in a patient infused with third-generation CAR T-cells specific for HER2/neu, who developed rapid respiratory failure due to the reactivity of CAR T-cells against the pulmonary tissue. In contrast, CD19-specific CAR T-cells caused B-cell aplasia, an adverse reaction that was reversed successfully by the infusion of gamma globulin (Morgan et al., Citation2010). Fortunately, many treatments, including CD171-, GD2-, CEA-, and IL13Rα2-redirected CAR T-cell therapy, have shown no major toxicities (Brown et al., Citation2015; Katz et al., Citation2015; Kershaw et al., Citation2006; Pule et al., Citation2008).
4.2. Cytokine-related syndrome
Cytokine-related syndrome (CRS) is a kind of on-target side effect usually caused by overproduction of cytokines from activated CAR T-cells, resulting in cytokine release syndrome, tumor lysis, and macrophage activation syndrome (MAS) (Maus & Levine, Citation2016). CRS is associated with elevated circulating cytokines, including IL-6, IFN-γ, and TNF-α. CRS can lead to high fever, multi-organ failure, hypotension, and hypoxia. However, a majority of the patients with hematologic malignancies showed only mild CRS symptoms during CAR T-cell therapy, for example, a fever. Production of C-reactive protein in the liver can be a predictive biomarker for CRS (Davila et al., Citation2014; Grupp et al., Citation2013).
4.3. Neurologic toxicities
To date, CD19 CAR T-cell therapy has been reported to cause self-reversible neurological side effects, including confusion, delirium, expressive aphasia, obtundation, myoclonus, and seizures in almost all patients (Buie, Pecoraro, Horvat, & Daley, Citation2015; Topp et al., Citation2015). The exact mechanisms of these neurologic side effects are still unknown, although it is believed that elevated levels of cytokines might be the inducing factor.
4.4. Anaphylaxis
Anaphylaxis is an acute allergic reaction to an antigen and may cause death (Tintinalli, Citation2010). The major symptoms of anaphylaxis involve the skin, respiratory tract, cardiovascular system, gastrointestinal system, and central nervous system (Simons, Citation2009). It has been noted that the non-human origin of the scFv can increase the chances of fatal anaphylaxis after multiple infusions of CAR T-cells. In a clinical trial of four patients undergoing mesothelin-specific CAR T-cell therapy, one of the patients treated with multiple infusions of CAR T-cells was found to have cardiorespiratory failure during the third infusion (Maus et al., Citation2013). Further study revealed the presence of human anti-mouse antibodies against foreign proteins derived from CAR T-cells and elevated trypsin in the patient’s serum, suggesting an IgE-mediated anaphylactic reaction (Maus et al., Citation2013).
4.5. Expected risks of insertional mutagenesis
CAR T-cells are usually generated using viral vectors, including retrovirus and lentivirus. Insertional mutagenesis caused by the insertion of viral vectors into sites proximate to proto-oncogenes could potentially result in transactivation of proto-oncogenes and malignancy, but could also trigger acute leukemia and primary immunodeficiency disorders, including X-linked severe combined immunodeficiency, chronic granulomatous disease, and Wiskott–Aldrich syndrome (Hacein-Bey-Abina et al., Citation2008; Hacein-Bey-Abina et al., Citation2003). However, to date, no case of insertional mutagenesis has been reported in patients undergoing CAR T-cell treatment (Bonifant, Jackson, Brentjens, & Curran, Citation2016). All potential side effects of CAR T-cells are summarized in Figure .
Figure 2. Expected side effects of CAR T-cells.
5. Safety management of CAR T-cell therapy in cancer
5.1. Safety management by supportive treatment
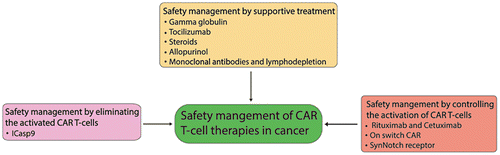
Toxicities generated during CAR T-cell therapy can be controlled through supportive treatment. On-target toxicities such as uveitis and colitis are often managed with topical or systemic steroids. Hypogammaglobulinemia can be controlled with gamma globulin replacement. A cytokine-blocking drug like tocilizumab, a novel IL-6R inhibitor, can be used to overcome CRS/MAS without affecting CAR T-cell efficacy (Zhou et al., Citation2014). Steroids are a safe and effective treatment option for CRS patients (Davila et al., Citation2014; Grupp et al., Citation2013; Lee et al., Citation2015). However, persistent use of corticosteroids decreases the overall efficacy of CAR T-cell therapy (Davila et al., Citation2014). Allopurinol is mostly used for the prevention of tumor lysis syndrome during all types of cancer treatment (Jeha, Citation2001). In addition, other immunosuppression therapy, such as with monoclonal antibodies and lymphodepletion chemotherapy, have also been suggested to prevent the toxicities caused by CAR T-cell therapy. Recently, CRS has been defined by clinical parameters, which helps to properly manage this side effect without decreasing the anti-leukemic activity of CAR T-cells (Davila et al., Citation2014).
5.2. Safety management by eliminating the activated CAR T-cells
CAR T-cells play a key role in causing on-target/off-tumor side effects during CAR T-cell therapy. Therefore, safety management could be achieved by eliminating activated CAR T-cells. The strategy for eradicating activated CAR T-cells is to induce their apoptosis when their toxic effects are out of control, which could be achieved by inserting ‘suicide genes’ into CAR T-cells to eliminate the infused T-cells (Ciceri et al., Citation2009). This strategy could be carried out by fusing an inducible caspase9 (ICasp9) protein to a human FK506 binding protein (FKBP), which allows conditional dimerization of the fusion protein in the presence of a small molecule. This dimerization subsequently activates downstream effector caspases and ultimately induces apoptosis (Tey, Dotti, Rooney, Heslop, & Brenner, Citation2007; Thomis et al., Citation2001). The action of the small dimerizing agent (AP1903) is so rapid and efficient that a single dose can eliminate more than 90% of the ICasp9-modified CAR T-cells within 30 min. This rapid and unstoppable action can reverse graft-versus-host disease (GVHD) without recurrence (Di Stasi et al., Citation2011; Zhou et al., Citation2014). The ICasp9 strategy has the drawbacks caused by immunogenicity (Berger, Flowers, Warren, & Riddell, Citation2006; Marktel et al., Citation2003; Traversari et al., Citation2007) and there can be a delayed response because the suicide switch process takes time, and it cannot control early toxicities of CAR T-cells during infusion (Bonifant et al., Citation2016).
5.3. Safety management by controlling the activation of CAR T-cells
Controlling CAR T-cell activation is another strategy for safety management, which could be achieved by different methods. The first method is to block the activation of CAR T-cells by masking the overexpressed cell surface antigenic markers of tumor cells, such as CD20 or EGFR, via corresponding monoclonal antibodies. Chimeric monoclonal antibodies (rituximab for CD20 and cetuximab for EGFR) have been used for this purpose (Liu et al., Citation2015; Philip et al., Citation2014; Wang et al., Citation2011). These monoclonal antibodies could protect healthy cells from CAR T-cells’ attack. However, this attractive strategy cannot fulfill the actual mission of safety management because these antigens are also present on the surface of some normal cells of the body. Moreover, cytotoxicity of these antibodies has also been reported (Bonifant et al., Citation2016).
The second method for controlling the activation of CAR T-cells is achieved by ‘on-switch’ CAR T-cells, which need both an antigen and a small molecule for their activation (Wu et al., Citation2015). Like natural receptors, on-switch CARs consist of two parts that are assembled only in the presence of a small molecule like rapalog. One part of this receptor is the scFv for extracellular antigen recognition, and the other part is the immunoreceptor tyrosine-based activation motifs (ITAMs) from the T-cell receptor CD3ζ subunit for intracellular signaling (Weiss & Littman, Citation1994). The small molecule acts like a priming factor to control CAR activities. The most important advantage of this strategy is that it can control the initiation of CAR T-cell activation, thus can control the early toxicities occurring immediately after infusion. Another important advantage of the strategy is the positive regulation of CAR T-cells without the waste of an expensive drug (Wu et al., Citation2015).
In addition to the methods discussed above, the activation of CAR T-cells could also be controlled by a synthetic Notch (synNotch) receptor. In this method, T-cells expressing the synNotch receptor recognize the first antigen (e.g. the scFv or tumor antigen), then the orthogonal transcription factor of the synNotch receptor (e.g. TetR-VP64 or Gal4- VP64) is cleaved from the cytoplasmic tail and enters the nucleus, where it induces the expression of the CAR for targeting the second tumor antigen (Roybal et al., Citation2016). Theoretically, CAR T-cells can only be activated when two tumor antigens are simultaneously expressed in the tumor. Therefore, this strategy of modifying T-cells with the synNotch receptor could effectively reduce the off-tumor toxicities of CAR T-cells. Safety management of CAR T-cell therapy is summarized in Figure .
Figure 3. Safety management of CAR T-cell therapy in cancer.
6. Challenges in CAR T-cell therapy for solid tumors
Although CAR T-cell therapy has been considered a promising treatment for hematologic malignancies, the results from clinical trials on solid tumors have not been encouraging due to various challenges. To improve the therapeutic effects of CAR T-cell therapy on solid tumors and the anti-tumor spectrum of CAR T-cells, the following limitations need to be overcome.
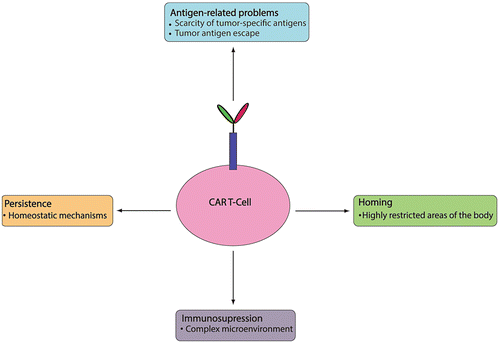
6.1. The limitations of tumor target antigens
An ideal antigen target for CAR T-cell therapy would be one that is exclusively expressed on tumor cells but not on normal cells. However, it is rather infeasible to find such an antigen. One possible alternative would be to find functionally dispensable antigens that are only expressed on a single cell lineage or antigens that are differentially expressed on tumor cells compared to normal cells. CD19 is the most attractive target for CAR T-cell-mediated therapy of hematologic malignancies because it is only present on the surface of normal and malignant B-cells (Li, Hayakawa, & Hardy, Citation1993; Li, Wasserman, Hayakawa, & Hardy, Citation1996). Many targets for hematologic malignancies other than CD19 have also been tested for their potential as therapeutic targets, including: (1) CD20 (Wang et al., Citation2004), CD22 (Haso et al., Citation2013), CD38 (Mihara et al., Citation2009, 2010), ROR1 (Hudecek et al., Citation2010), and k light chain (Vera et al., Citation2006) for nearly all B-cell malignancies, including CLL, ALL and many NHLs; (2) Lewis-Y (Ritchie et al., Citation2013), CD33 (Wang et al., Citation2015), and CD123 (Mardiros, Forman, & Budde, Citation2015) for myeloid malignances; and (3) CD30 for Hodgkin’s lymphoma (Savoldo et al., Citation2007). The updated clinical trials of CAR T-cell therapy using CD19 and CD20, CD33 and Lewis-Y as targets for hematologic malignancies are summarized in Table .
The selection of antigens is more challenging for solid tumors than hematological malignancies for the following two reasons. First, not many proteins are exclusively expressed on the surface of solid tumor cells; hence, it is difficult for CAR T-cells to discriminate between target cells and normal cells. To overcome this limitation, a few selectively expressed proteins have been identified in some solid tumors, e.g. mesothelin, which is expressed in mesothelioma, and cancers of the pancreas, brain, ovaries, and breast. Mesothelin-directed CAR T-cell therapy has been used in trials for many types of cancers (NCT01583686, NCT02159716, and NCT02465983). GD2 protein, commonly expressed in melanoma, neuroblastoma, and some sarcomas, has also been used in trials (e.g. NCT02107963). Neoantigens, short peptides of 8 to 12 amino acids, arise as a consequence of tumor-specific mutations. Neoantigens are only found on oncogenic cells. EGFRvIII is a well-known example of a neoantigen not expressed in normal tissues but widely expressed in glioblastoma, and it has been used as a cancer therapy target for neoplasms (Fritsch, Hacohen, & Wu, Citation2014; Sampson et al., Citation2010). The recently developed synNotch receptor is another option for CAR T-cell treatment, as it can discriminate between tumor and non-tumor cells (Roybal et al., Citation2016). The second hurdle in the way of CAR T-cell therapy, especially for solid tumors, is antigen escape due to the outgrowth of antigen null tumor cells (Hegde et al., Citation2013). This phenomenon has been observed in clinical studies of CD19-targeted CAR T-cells for hematologic malignancies (Grupp et al., Citation2013; Maude et al., Citation2014) as well as CAR T-cell therapy in solid tumors. In light of this issue, CAR T-cells targeting the B-cell antigen CD22 have been developed to overcome antigen escape (Wang et al., Citation2004). Strategies for overcoming antigen escape in solid tumors include: (1) combining CAR T-cells targeting separate antigens such as HER2 and IL13Ra (Hegde et al., Citation2013); (2) targeting two antigens with T-cells expressing two separate CARs (Hegde et al., Citation2013); and (3) applying TanCAR, a single CAR having two tandemly arranged distinct scFvs that target two different antigens, and which are coupled to the same signaling endodomain (Grada et al., Citation2013). The TanCAR T-cell strategy is most suitable for mitigating antigen escape and has been used in glioblastoma therapy using two scFvs targeting HER2 and interleukin-13 receptor subunit alpha-2 (IL13Rα2) (Grada et al., Citation2013; Hegde et al., Citation2016).
6.2. Homing
The successful delivery of CAR T-cells to the tumor site after infusion is also a hurdle, especially for solid tumor treatment, because of the presence of highly restricted areas in solid tumors. The improved delivery of CAR T-cells into tumor sites has been achieved by modifying CAR T-cells with chemokine C-X-C motif receptor 2 (CXCR2), nerve growth factor receptor (NGFR) (NCT01740557), or CCR2b (Craddock et al., Citation2010; Di Stasi et al., Citation2009). Anti-VEGFR-2 CAR and IL-12-cotransduced T-cells have been shown to have enhanced migration capacity to the tumor area (Chinnasamy et al., Citation2012). Heparanase, an enzyme that degrades stroma, was proven to facilitate the delivery of CAR T-cells into a tumor (Caruana et al., Citation2015). Recently, some evidence has shown that CAR-engineered immune cells other than T-cells in mice are comparatively more effective (Han et al., Citation2015). More approaches to enhance the homing of CAR T-cells are currently under study, for example, blocking the inhibitory signal of migration (Buckanovich et al., Citation2008) or shaping the chaotic structure of tumor blood vessels via radiation (Ganss, Ryschich, Klar, Arnold, & Hammerling, Citation2002).
6.3. Persistence
It has been reported that the persistence and proliferation of CAR T-cells could be improved by the expression of naive markers, such as CD62L, in central memory T-cells (Berger et al., Citation2008; Hinrichs et al., Citation2009; Wang et al., Citation2012). Interestingly, a study showed that GD2-CAR T-cells not only induce complete tumor response in patients with active neuroblastoma but also extend the persistence of a low level of CAR T-cells in patients, and such persistence was associated with longer survival (Louis et al., Citation2011). Long-term follow-up was done on 19 patients who were infused with GD2-specific CAR T-cells, and the statistical analysis showed a significant association between prolonged persistence and the number of CD4 + cells and central memory cells (CD45RO + and CD62L + T-cell subsets) in the infused product (Louis et al., Citation2011). In addition, integration of the IL-12 and IL-15 genes into CAR T-cells can also enhance their proliferation and persistence (Hoyos et al., Citation2010; Pegram et al., Citation2015). Other alternative strategies include multiple cell infusion cycles and transcription activator-like effector nuclease (TALEN)-mediated editing approaches. A TALEN-mediated editing approach has been used in the large-scale production of CAR T-cells deficient in expression of both their TCRαβ and CD52 protein (Poirot et al., Citation2015), which is helpful in increasing the numbers of CAR T-cells in tumor sites. Moreover, the human origin scFv is the relatively better choice to prolong persistence compared with the non-human origin scFv (Jensen et al., Citation2010).
6.4. Immunosuppression
Compared to blood malignances, solid tumors have complicated microenvironments that potentially reduce the therapeutic efficacy of CAR T-cells. There are two main aspects of the microenvironment within a solid tumor that are related to decreased CAR T-cells efficacy. One is the intrinsic characteristics of the solid tumor microenvironment, such as, hypoxia, low pH, and the lack of arginine or tryptophan. The other aspect is the existence of many inhibitory pathways in the microenvironment of the solid tumor that inhibit the function of activated T-cells (Moon et al., Citation2014; Park et al., Citation2012; Zheng, Zha, & Gajewski, Citation2008). Many strategies have been applied to overcome these problems, including: (1) modifying CAR T-cells with the expression of a dominant-negative form of transforming growth factor β (TGFβ) receptor to overcome the inhibitory effects of TGFβ (Foster et al., Citation2008); (2) using NKG2D CAR T-cell therapy, which has effectively treated a heterogeneous group of tumors, although the role of NKG2D ligands in tumor immunity is contradictory (Spear, Barber, Rynda-Apple, & Sentman, Citation2013); (3) inhibiting the expression of the molecules that downregulate immune responses, such as CTLA-4 (Condomines et al., Citation2015); (4) reducing Fas-induced apoptosis (Dotti et al., Citation2005); and (5) facilitating the expression of survival genes such as Bcl-X(L) (Eaton, Gilham, O’Neill, & Hawkins, Citation2002). CAR T-cells can be protected from the hostile tumor microenvironment by the co-expression of CD25 (Chang, Silver, & Chen, Citation2015), CD28, and 4-1BB (Zhao et al., Citation2015) or the constitutive expression of the CD40 ligand on CAR T-cells (Curran et al., Citation2015). CAR T-cells engineered with inducible IL-12 expression in the tumor site (TRUCKs) not only resist the hostile environment of the tumor cells but also enhance antitumor activities (Cheadle et al., Citation2014; Chmielewski, Citation2015). A PD1-CD28 chimeric receptor could efficiently increase the proliferative capacity of CAR T-cells due to the enhanced cytokine secretion of CAR-T cells (Prosser, Brown, Shami, Forman, & Jensen, Citation2012). Moreover, tumor immunosuppression can be overcome by combining CAR T-cells with anti-PD1 antibodies (John et al., Citation2013; John, Kershaw, & Darcy, Citation2013) or with granulocyte-macrophage colony-stimulating factor neutralization (Burga et al., Citation2015). Chemotherapeutic agents including fludarabine and cyclophosphamide could be used before infusion of CAR T-cells to help CAR T-cells resist some immunosuppressive factors, such as indoleamine 2,3-dioxygenase (Ninomiya et al., Citation2015). The challenges for CAR T-cell therapy in solid tumors are summarized in Figure .
Figure 4. Challenges in CAR T-cell therapy.
7. Future prospects of CAR T-cells
CAR T-cells, which can proliferate and persist in the body of a patient after infusion, have been successfully used to treat hematological cancers including ALL, CLL, diffuse large B-cell lymphoma (DLBCL), FL, and multiple myelomas. However, their success rate for solid tumors is very low, with only a few encouraging trials reported, including those of tumors overexpressing mesothelin, neuroblastoma HER2, and EGFR (Feng et al., Citation2016; Jensen et al., Citation2010; Lamers et al., Citation2013). In the near future, strategies for the improvement of the therapeutic efficacy of CAR T-cell therapy for both hematologic malignancies and solid tumors will include: (1) modifying the intracellular domain of CARs to broaden the anti-tumor spectrum of CAR T-cells for minimizing the risk of antigen escape. The TanCAR T-cell strategy is most suitable to solve this problem and has been used in glioblastoma therapy (Grada et al., Citation2013; Hegde et al., Citation2016); (2) looking for new specific targets recognized by CAR T-cells: Some new targets are under investigation, including CD38 for B-cell malignancies (Mihara et al., Citation2009, 2010), CD70 for CD70-positive malignancies (Shaffer et al., Citation2011), and BCMA (Carpenter et al., Citation2013), CD74 (Sapra et al., Citation2005), CD138 (Guo et al., Citation2016) and CS1 (Chu et al., Citation2013) for multiple myeloma; (3) finding new effector cells expressing CARs. CARs can be expressed on other effector cells, e.g. NK cells. (Some groups have expressed second-generation CARs on NK cells (Chu et al., Citation2013). Details of this approach are out of the scope of this review); (4) enhancing the homing ability and persistence of CAR T-cells by expressing different receptors on CAR T-cells. Researchers have designed nanoparticle drug delivery systems for the delivery of CAR T-cells to the tumor site. Some studies have found that CAR T-cell death can be prevented by co-stimulation of OX40, CD137 and CD27 (Croft, Citation2003); (5) increasing the infiltration rate of CAR T-cells into solid tumors by the expression of cell migrating molecules such as cytokines or chemokines in the CAR T-cells; (6) controlling the hostile factors for CAR T-cells in the complex microenvironment by expressing negative dominant receptors in the CAR T-cells or by converting an immunosuppressive signal into a positive signal; (7) improving the safety of CAR T-cell therapy by optimizing viral and non-viral vectors used to generate CAR T-cells; (8) minimizing CAR T-cell toxicities by introducing different safety switches into CAR T-cells, such as an ICasp9, or HSV-tk switch (Di Stasi et al., Citation2011); (9) improving the efficacy of CAR T-cell therapy combining it with other therapies, such as chemotherapy, radiotherapy, and PD-1/PD-L1 antibodies, before or after infusion of CAR T-cells; (10) optimizing the techniques for T-cell selection and expansion; (11) improving the potential of CAR T-cells by optimizing culture conditions; (12) producing universal CAR T-cells by knocking out the endogenous αβ T-cell receptor and HLA genes in CAR T-cells from allogeneic donors (Torikai et al., Citation2012; Torikai et al., Citation2013); (13) lowering the cost of producing CAR T-cells to facilitate their large-scale production for future clinical application. Currently CAR-T-cells are still produced as a personal drug only for trial purposes; and (14) standardizing the production and dosage administration of CAR T-cells for future comparative clinical studies.
In conclusion, CAR T-cell therapy represents a potential paradigm shift in the treatment of cancer, yet many barriers and challenges still exist for its use in treating solid tumors. The optimization of conditions and technologies for generating CAR T-cells has the potential to make CAR T-cell treatment against cancer safer and more effective.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
This work was supported by the National Natural Science Foundation of China [grant number 81471772], [grant number 81773265]; Natural Science Foundation of Shaanxi Province [grant number GK201706002].
References
- Ahmed, N., Brawley, V. S., Hegde, M., Robertson, C., Ghazi, A., Gerken, C., ... Gottschalk, S. (2015). Human epidermal growth factor receptor 2 (HER2) -specific chimeric antigen receptor-modified T-cells for the immunotherapy of HER2-positive sarcoma. Journal of Clinical Oncology, 33(15), 1688–1696.
- Ahmed, N., Ratnayake, M., Savoldo, B., Perlaky, L., Dotti, G., Wels, W. S., ... Gottschalk, S. (2007). Regression of experimental medulloblastoma following transfer of HER2-specific T cells. Cancer Research, 67(12), 5957–5964.
- Altenschmidt, U., Kahl, R., Moritz, D., Schnierle, B. S., Gerstmayer, B., Wels, W., & Groner, B. (1996). Cytolysis of tumor cells expressing the Neu/erbB-2, erbB-3, and erbB-4 receptors by genetically targeted naive T lymphocytes. Clinical Cancer Research, 2(6), 1001–1008.
- Altvater, B., Landmeier, S., Pscherer, S., Temme, J., Juergens, H., Pule, M., & Rossig, C. (2009). 2B4 (CD244) signaling via chimeric receptors costimulates tumor-antigen specific proliferation and in vitro expansion of human T cells. Cancer Immunology, Immunotherapy, 58(12), 1991–2001.10.1007/s00262-009-0704-9
- Beatty, G. L., Haas, A. R., Maus, M. V., Torigian, D. A., Soulen, M. C., Plesa, G., ... June, C. H. (2014). Mesothelin-specific chimeric antigen receptor mRNA-engineered T-cells induce antitumor activity in solid malignancies. Cancer Immunology Research, 2(2), 112–120.
- Berger, C., Flowers, M. E., Warren, E. H., & Riddell, S. R. (2006). Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T-cells after allogeneic hematopoietic cell transplantation. Blood, 107(6), 2294–2302.
- Berger, C., Jensen, M. C., Lansdorp, P. M., Gough, M., Elliott, C., & Riddell, S. R. (2008). Adoptive transfer of effector CD8+T-cells derived from central memory cells establishes persistent T-cell memory in primates. Journal of Clinical Investigation, 118(1), 294–305.
- Bonifant, C. L., Jackson, H. J., Brentjens, R. J., & Curran, K. J. (2016). Toxicity and management in CAR T-cell therapy. Molecular Therapy – Oncolytics, 3, 16011.
- Brentjens, R. J., Davila, M. L., Riviere, I., Park, J., Wang, X., Cowell, L. G., ... Sadelain, M. (2013, March 20). CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Science Translational Medicine, 5(177), 177ra738.
- Brentjens, R. J., Riviere, I., Park, J. H., Davila, M. L., Wang, X., Stefanski, J., ... Sadelain, M. (2011). Safety and persistence of adoptively transferred autologous CD19-targeted T-cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood, 118(18), 4817–4828.
- Brentjens, R. J., Santos, E., Nikhamin, Y., Yeh, R., Matsushita, M., La Perle, K., ... Sadelain, M. (2007). Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clinical Cancer Research, 13(18), 5426–5435.10.1158/1078-0432.CCR-07-0674
- Brown, C. E., Badie, B., Barish, M. E., Weng, L., Ostberg, J. R., Chang, W. C., ... Jensen, M. C. (2015). Bioactivity and safety of IL13R alpha2-redirected chimeric antigen receptor CD8+ T-cells in patients with recurrent glioblastoma. Clinical Cancer Research, 21(18), 4062–4072.
- Buckanovich, R. J., Facciabene, A., Kim, S., Benencia, F., Sasaroli, D., Balint, K., ... Coukos, G. (2008). Endothelin B receptor mediates the endothelial barrier to T-cell homing to tumors and disables immune therapy. Nature Medicine, 14(1), 28–36.
- Buie, L. W., Pecoraro, J. J., Horvat, T. Z., & Daley, R. J. (2015). Blinatumomab: A first-in-class bispecific T-cell engager for precursor B-cell acute lymphoblastic leukemia. Annals of Pharmacotherapy, 49(9), 1057–1067.
- Burga, R. A., Thorn, M., Point, G. R., Guha, P., Nguyen, C. T., Licata, L. A., ... Katz, S. C. (2015). Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunology, Immunotherapy, 64(7), 817–829.
- Campana, D., Schwarz, H., & Imai, C. (2014). 4-1BB chimeric antigen receptors. The Cancer Journal, 20(2), 134–140.
- Carpenito, C., Milone, M. C., Hassan, R., Simonet, J. C., Lakhal, M., Suhoski, M. M., ... June, C. H. (2009). Control of large, established tumor xenografts with genetically retargeted human T-cells containing CD28 and CD137 domains. Proceedings of the National Academy of Sciences of the USA, 106(9), 3360–3365.
- Carpenter, R. O., Evbuomwan, M. O., Pittaluga, S., Rose, J. J., Raffeld, M., Yang, S., ... Kochenderfer, J. N. (2013). B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clinical Cancer Research, 19(8), 2048–2060.
- Caruana, I., Savoldo, B., Hoyos, V., Weber, G., Liu, H., Kim, E. S., ... Dotti, G. (2015). Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nature Medicine, 21(5), 524–529.
- Chang, Z. L., Silver, P. A., & Chen, Y. Y. (2015). Identification and selective expansion of functionally superior T-cells expressing chimeric antigen receptors. Journal of Translational Medicine, 13, 161.
- Cheadle, E. J., Gornall, H., Baldan, V., Hanson, V., Hawkins, R. E., & Gilham, D. E. (2014). CAR T-cells: Driving the road from the laboratory to the clinic. Immunological Reviews, 257(1), 91–106.
- Chinnasamy, D., Yu, Z., Kerkar, S. P., Zhang, L., Morgan, R. A., Restifo, N. P., & Rosenberg, S. A. (2012). Local delivery of interleukin-12 using T-cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clinical Cancer Research, 18(6), 1672–1683.
- Chmielewski, M. (2015). Abken H TRUCKs: The fourth generation of CARs. Expert Opinion on Biological Therapy, 15(8), 1145–1154.
- Chmielewski, M., Rappl, G., Hombach, A. A., & Abken, H. (2012). T cells redirected by a CD3zeta chimeric antigen receptor can establish self-antigen-specific tumor protection in the long term. Gene Therapy, 20, 177–186.
- Cho, H. S., Mason, K., Ramyar, K. X., Stanley, A. M., Gabelli, S. B., Denney, D. W., Jr., & Leahy, D. J. (2003). Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature, 421(6924), 756–760.
- Chow, K. K., Naik, S., Kakarla, S., Brawley, V. S., Shaffer, D. R., Yi, Z., ... Gottschalk, S. (2013). T cells redirected to EphA2 for the immunotherapy of glioblastoma. Molecular Therapy, 21, 629–637.
- Chu, J., Deng, Y., Benson, D. M. J., He, S., Hughes, T., Zhang, J., ... Yu, J. (2013). CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia, 28(4), 917–927.
- Ciceri, F., Bonini, C., Stanghellini, M. T., Bondanza, A., Traversari, C., Salomoni, M., ... Bordignon, C. (2009). Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): A non-randomised phase I-II study. The Lancet Oncology, 10(5), 489–500.
- Condomines, M., Arnason, J., Benjamin, R., Gunset, G., Plotkin, J., & Sadelain, M. (2015). Tumor-targeted human T-cells expressing CD28-based chimeric antigen receptors circumvent CTLA-4 inhibition. PLoS One, 10(6), e0130518.
- Craddock, J. A., Lu, A., Bear, A., Pule, M., Brenner, M. K., Rooney, C. M., & Foster, A. E. (2010). Enhanced tumor trafficking of GD2 chimeric antigen receptor T-cells by expression of the chemokine receptor CCR2b. Journal of Immunotherapy, 33(8), 780–788.
- Croft, M. (2003). Costimulation of T cells by OX40, 4-1BB, and CD27. Cytokine & Growth Factor Reviews, 14(3–5), 265–273.
- Cruz, C. R., Micklethwaite, K. P., Savoldo, B., Ramos, C. A., Lam, S., Ku, S., ... Dotti, G. (2013). Infusion of donor-derived CD19 redirected virus-specific T-cells for B-cell malignancies relapsed after allogeneic stem cell transplant: A phase 1 study. Blood, 122(17), 2965–2973.
- Curran, K. J., Seinstra, B. A., Nikhamin, Y., Yeh, R., Usachenko, Y., van Leeuwen, D. G., ... Brentjens, R. J. (2015). Enhancing antitumor efficacy of chimeric antigen receptor T-cells through constitutive CD40L expression. Molecular Therapy, 23(4), 769–778.
- Dai, H., Zhang, W., Li, X., Han, Q., Guo, Y., Zhang, Y., ... Han, W. (2015). Tolerance and efficacy of autologous or donor derived T-cells expressing CD19 chimeric antigen receptors in adult B-ALL with extramedullary leukemia. Oncoimmunology, 4(11), e1027469.
- Davies, D. M., Foster, J., Van der Stegen, S. J., Parente-Pereira, A. C., Chiapero-Stanke, L., Delinassios, G. J., ... Maher, J. (2012). Flexible targeting of ErbB dimers that drive tumorigenesis by using genetically engineered T-cells. Molecular Medicine, 18(4), 565–576.
- Davila, M. L., Riviere, I., Wang, X., Bartido, S., Park, J., Curran, K., ... Brentjens, R. (2014). Efficacy and toxicity management of 19-28z CAR T-cell therapy in B cell acute lymphoblastic leukemia. Science Translational Medicine, 6(224), 224ra25.
- Davis, S., Gale, N. W., Aldrich, T. H., Maisonpierre, P. C., Lhotak, V., Pawson, T., ... Yancopoulos, G. D. (1994). Ligands for EPH-related receptor tyrosine kinases that require membrane attachment or clustering for activity. Science, 266(5186), 8169.
- Depoil, D., Fleire, S., Treanor, B. L., Weber, M., Harwood, N. E., Marchbank, K. L., ... Batista, F. D. (2008). CD19 is essential for b cell activation by promoting B cell receptor-antigen micro cluster formation in response to membrane-bound ligand. Nature Immunology, 9, 63–72.
- Di Stasi, A., De Angelis, B., Rooney, C. M., Zhang, L., Mahendravada, A., Foster, A. E., ... Savoldo, B. (2009). T lymphocytes co-expressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood, 113(25), 6392–6402.
- Di Stasi, A., Tey, S. K., Dotti, G., Fujita, Y., Kennedy-Nasser, A., Martinez, C., ... Brenner, M. K. (2011). Inducible apoptosis as a safety switch for adoptive cell therapy. The New England Journal of Medicine, 365(18), 1673–1683.
- Doronin, I. I., Vishnyakova, P. A., Kholodenko, I. V., Ponomarev, E. D., Ryazantsev, D. Y., Molotkovskaya, I. M., & Kholodenko, R. V. (2014). Ganglioside GD2 in reception and transduction of cell death signal in tumor cells. BMC Cancer, 14, 295.
- Dotti, G., Savoldo, B., Pule, M., Straathof, K. C., Biagi, E., Yvon, E., ... Rooney, C. M. (2005). Human cytotoxic T lymphocytes with reduced sensitivity to Fas-induced apoptosis. Blood, 105(12), 4677–4684.
- Dubinski, D., Wölfer, J., Hasselblatt, M., Schneider-Hohendorf, T., Bogdahn, U., Stummer, W., ... Grauer, O. M. (2015). CD4+ T effector memory cell dysfunction is associated with the accumulation of granulocytic myeloid-derived suppressor cells in glioblastoma patients. Journal of Neuro-Oncology, 18(6), 807–818.
- Eaton, D., Gilham, D. E., O’Neill, A., & Hawkins, R. E. (2002). Retroviral transduction of human peripheral blood lymphocytes with Bcl-X(L) promotes in vitro lymphocyte survival in pro-apoptotic conditions. Gene therapy, 9(8), 527–535.
- Eshhar, Z., Waks, T., Bendavid, A., & Schindler, D. G. (2001). Functional expression of chimeric receptor genes in human T-cells. The Journal of Immunological Methods, 248(1–2), 67–76.
- Evans, A. G., Rothberg, P. G., Burack, W. R., Huntington, S. F., Porter, D. L., Friedberg, J. W., … Liesveld, J. L. (2015). Evolution to plasmablastic lymphoma evades cd19-directed chimeric antigen receptor T cells. British Journal of Haematology, 171, 205–209.
- Faitschuk, E., Nagy, V., Hombach, A. A., & Abken, H. (2016). A dual chain chimeric antigen receptor (CAR) in the native antibody format for targeting immune cells towards cancer cells without the need of an scFv. Gene Therapy, 23(10), 718–726.10.1038/gt.2016.48
- Feng, K., Guo, Y., Dai, H., Wang, Y., Li, X., Jia, H., & Han, W. (2016). Chimeric antigen receptor modified T-cells for the immunotherapy of patients with EGFR expressing advanced relapsed/refractory non-small cell lung cancer. Science China Life Sciences, 59(5), 468–479.
- Finney, H.M., Akbar, A. N., & Lawson, A. D. (2004). Activation of resting human primary T cells with chimeric receptors: Costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR chain. The Journal of Immunology, 172(1), 104–113.10.4049/jimmunol.172.1.104
- Foster, A. E., Dotti, G., Lu, A., Khalil, M., Brenner, M. K., Heslop, H. E., ... Bollard, C. M. (2008). Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-β receptor. Journal of Immunotherapy, 31, 500–505.
- Fritsch, E. F., Hacohen, N., & Wu, C. J. (2014). Personal neoantigen cancer vaccines: The momentum builds. Oncoimmunology, 6(3), e29311.
- Ganss, R., Ryschich, E., Klar, E., Arnold, B., & Hammerling, G. J. (2002). Combination of T-cell therapy and trigger of inflammation induces remodeling of the vasculature and tumor eradication. Cancer Research, 62(5), 1462–1470.
- Garfall, A. L., Maus, M. V., Hwang, W. T., Lacey, S. F., Mahnke, Y. D., Melenhorst, J. J., ... Stadtmauer, E. A. (2015). Chimeric antigen receptor T-cells against CD19 for multiple myeloma. The New England Journal of Medicine, 373(11), 1040–1047.
- Geiger, T. L., Nguyen, P., Leitenberg, D., & Flavell, R. A. (2001). Integrated src kinase and costimulatory activity enhances signal transduction through single-chain chimeric receptors in T lymphocytes. Blood, 98(8), 2364–2371.
- Gorlick, R., Huvos, A. G., Heller, G., Aledo, A., Beardsley, G. P., Healey, J. H., & Meyers, P. A. (1999). Expression of HER2/erbB-2 correlates with survival in osteosarcoma. Journal of Clinical Oncology, 17(9), 2781–2788.
- Grada, Z., Hegde, M., Byrd, T., Haffer, D. R., Ghazi, A., Brawley, V. S., ... Ahmed, N. (2013). TanCAR: A novel bispecific chimeric antigen receptor for cancer immunotherapy. Molecular Therapy – Nucleic Acids, 2, e105.
- Gross, G., Waks, T., & Eshhar, Z. (1989). Expression of Immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proceedings of the National Academy of Sciences of the USA, 86(24), 10024–10028.
- Grupp, S. A., Kalos, M., Barrett, D., Aplenc, R., Porter, D. L., Rheingold, S. R., ... June, C. H. (2013). Chimeric antigen receptor-modified T-cells for acute lymphoid leukemia. The New England Journal of Medicine, 368(16), 1509–1518.
- Guo, B., Chen, M., Han, Q., Hui, F., Dai, H., Zhang, W., ... Han, W. (2016). CD138-directed adoptive immunotherapy of chimeric antigen receptor (CAR)-modified T cells for multiple myeloma. Journal of Cellular Immunotherapy, 2, 28–35.
- Hacein-Bey-Abina, S., Von Kalle, C., Schmidt, M., McCormack, M. P., Wulffraat, N., Leboulch, P., ... Cavazzana-Calvo, M. (2003). LMO2-associated clonal T-cell proliferation in two patients after gene therapy for SCID-X1. Science, 302, 415–419.
- Hacein-Bey-Abina, S., Garrigue, A., Wang, G. P., Soulier, J., Lim, A., Morillon, E., ... Cavazzana-Calvo, M. (2008). Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. Journal of Clinical Investigation, 118(9), 3132–3142.
- Han, J., Chu, J., Keung Chan, W., Zhang, J., Wang, Y., Cohen, J. B., ... Yu, J. (2015). CAR-engineered NK Cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Scientific Reports, 5, 11483.
- Harris, D. T., & Kranz, D.M. (2015). Adoptive T cell therapies: A comparison of T cell receptors and chimeric antigen receptors. Trends Pharmacol Sc., 37(3), 220–230.
- Haso, W., Lee, D. W., Shah, N. N., Stetler-Stevenson, M., Yuan, C. M., Pastan, I. H., ... Orentas, R. J. (2013). Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood, 121(7), 1165–1174.
- Hassan, R., Bera, T., & Pastan, I. (2004). Mesothelin: A new target for immunotherapy. Clin Cancer Res., 10(12 Pt 1), 3937–3942.
- Hegde, M., Corder, A., Chow, K. K., Mukherjee, M., Ashoori, A., Kew, Y., ... Ahmed, N. (2013). Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Molecular Therapy, 21(11), 2087–2101.10.1038/mt.2013.185
- Hegde, M., Mukherjee, M., Grada, Z., Pignata, A., Landi, D., Navai, S. A., ... Ahmed, N. (2016). Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. Journal of Clinical Investigation, 126(8), 3036–3052.
- Heimberger, A. B., Suki, D., Yang, D., Shi, W., & Aldape, K. (2005). The natural history of EGFR and EGFRvIII in glioblastoma patients. Journal of Translational Medicine, 3, 38.
- Hinrichs, C. S., Borman, Z. A., Cassard, L., Gattinoni, L., Spolski, R., Yu, Z., ... Restifo, N. P. (2009). Adoptively transferred effector cells derived from naive rather than central memory CD8+ T-cells mediate superior antitumor immunity. Proceedings of the National Academy of Sciences of the USA, 106(41), 17469–17474.
- Hombach, A., Hombach, A. A., & Abken, H. (2010). Adoptive immunotherapy with genetically engineered T-cells: Modification of the IgG1 Fc ‘spacer’ domain in the extracellular moiety of chimeric antigen receptors avoids ‘off-target’ activation and unintended initiation of an innate immune response. Gene Therapy, 17(10), 1206–1213.10.1038/gt.2010.91
- Hombach, A. A., Heiders, J., Foppe, M., Chmielewski, M., & Abken, H. (2012). OX40 co-stimulation by a chimeric antigen receptor abrogates CD28 and IL-2 induced IL-10 secretion by redirected CD4 (+) T-cells. Oncoimmunology, 1(4), 458–466.
- Hoyos, V., Savoldo, B., Quintarelli, C., Mahendravada, A., Zhang, M., Vera, J., ... Dotti, G. (2010). Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia, 24(6), 1160–1170.
- Hudecek, M., Schmitt, T. M., Baskar, S., Lupo-Stanghellini, M. T., Nishida, T., Yamamoto, T. N., ... Riddell, S. R. (2010). The B-cell tumor-associated antigen ROR1 can be targeted with T cells modified to express a ROR1-specific chimeric antigen receptor. Blood, 116(22), 4532–4541.
- Hudziak, R. M., Schlessinger, J., & Ullrich, A. (1987). Increased expression of the putativegrowth factor receptor p185HER2 causes transformation and tumorigenesis of NIH 3T3 cells. Proceedings of the National Academy of Sciences of the USA, 84(20), 7159–7163.
- Hynes, N. E., & MacDonald, G. (2009). ErbB receptors and signaling pathways in cancer. Current Opinion in Cell Biology, 21(2), 177–184.
- Imai, C., Mihara, K., Andreansky, M., Nicholson, I. C., Pui, C. H., Geiger, T. L., & Campana, D. (2004). Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia, 18(4), 676–684.10.1038/sj.leu.2403302
- Irving, B. A., & Weiss, A. (1991). The cytoplasmic domain of the T-cell receptor ζ chain is sufficient to couple to receptor associated signal transduction pathways. Cell, 64(5), 891–901.10.1016/0092-8674(91)90314-O
- Jeha, S. (2001). Tumor lysis syndrome. Seminars in Hematology, 38(4 Suppl 10), 4–8.
- Jensen, M. C., Popplewell, L., Cooper, L. J., DiGiusto, D., Kalos, M., Ostberg, J. R., & Forman, S. J. (2010). Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T-cells in humans. Biology of Blood and Marrow Transplantation, 16(9), 1245–1256.
- John, L. B., Kershaw, M. H., & Darcy, P. K. (2013). Blockade of PD-1 immunosuppression boosts CAR T-cell therapy. Oncoimmunology, 2(10), e26286.
- John, L. B., Devaud, C., Duong, C. P., Yong, C. S., Beavis, P. A., Haynes, N. M., ... Darcy, P. K. (2013). Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T-cells. Clinical Cancer Research, 19(20), 5636–5646.
- Kalos, M., Levine, B. L., Porter, D. L., Katz, S., Grupp, S. A., Bagg, A., & June, C. H. (2011). T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Science Translational Medicine, 3(95), 95ra73.
- Kalos, M., Frey, N. V., Grupp, S. A., Loren, A. W., Jemison, C., Gilmore, J., ... June, C. H. (2013). Chimeric antigen receptor modified T cells directed against CD19 (CTL019 cells) have long-term persistence and induce durable responses in relapsed, refractory CLL. Blood, 122(21), 4162.
- Katz, S. C., Burga, R. A., McCormack, E., Wang, L. J., Mooring, W., Point, G. R., ... Junghans, R. P. (2015). Phase I Hepatic Immunotherapy for metastases study of intra-arterial chimeric antigen receptor-modified T-cell therapy for CEA+ liver metastases. Clinical Cancer Research, 21(14), 3149–3159.
- Kershaw, M. H., Westwood, J. A., Parker, L. L., Wang, G., Eshhar, Z., Mavroukakis, S. A., ... Hwu, P. (2006). A phase I study on adoptive immunotherapy using gene-modified T-cells for ovarian cancer. Clinical Cancer Research, 12(20 Pt 1), 6106–6115.
- Kinch, M. S., & Carles-Kinch, K. (2003). Overexpression and functional alterations of the EphA2 tyrosine kinase in cancer. Clinical & Experimental Metastasis, 20(1), 59–68.
- Kochenderfer, J. N., & Rosenberg, S. A. (2011). Chimeric antigen receptor- modified T cells in CLL. The New England Journal of Medicine, 365, 1937–1938.
- Kochenderfer, J. N., Wilson, W. H., Janik, J. E., Dudley, M. E., Stetler-Stevenson, M., Feldman, S. A., ... Rosenberg, S. A. (2010). Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T-cells genetically engineered to recognize CD19. Blood, 116(20), 4099–4102.
- Kochenderfer, J. N., Dudley, M. E., Feldman, S. A., Wilson, W. H., Spaner, D. E., Maric, I., ... Rosenberg, S. A. (2012). B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T-cells. Blood, 119(12), 2709–2720.
- Kochenderfer, J. N., Dudley, M. E., Carpenter, R. O., Kassim, S. H., Rose, J. J., Telford, W. G., ... Rosenberg, S. A. (2013). Donor-derived CD19- targeted T-cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood, 122(25), 4129–4139.
- Kochenderfer, J. N., Dudley, M. E., Kassim, S. H., Somerville, R. P., Carpenter, R. O., Stetler-Stevenson, M., ... Rosenberg, S. A. (2015). Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T-cells expressing an anti-CD19 chimeric antigen receptor. Journal of Clinical Oncology, 33(6), 540–549.
- Kofler, D. M., Chmielewski, M., Rappl, G., Hombach, A., Riet, T., Schmidt, A., ... Abken, H. (2011). CD28 costimulation impairs the efficacy of a redirected T-cell antitumor attack in the presence of regulatory T cells which can be overcome by preventing Lck activation. Molecular Therapy, 19(4), 760–767.10.1038/mt.2011.9
- Kowolik, C., Topp, M., Gonzalez, S., Pfeiffer, T., Olivares, S., Gonzalez, N., ... Cooper, L. J. (2006). CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Research, 66(22), 10995–11004.10.1158/0008-5472.CAN-06-0160
- Krug, C., Birkholz, K., Paulus, A., Schwenkert, M., Schmidt, P., Hoffmann, N., ... Schaft, N. (2015). Stability and activity of MCSP-specific chimeric antigen receptors (CARs) depend on the scFv antigen-binding domain and the protein backbone. Cancer Immunology, Immunotherapy, 64, 1623–1635.
- Lamers, C. H., Sleijfer, S., Vulto, A. G., Kruit, W. H., Kliffen, M., Debets, R., ... Oosterwijk, E. (2006). Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: First clinical experience. Journal of Clinical Oncology, 24(13), e20–e22.
- Lamers, C. H. J., Willemsen, R., van Elzakker, P., van Steenbergen-Langeveld, S., Broertjes, M., Oosterwijk-Wakka, J., ... Gratama, J. W. (2011). Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood, 117(1), 72–82.10.1182/blood-2010-07-294520
- Lamers, C. H., Sleijfer, S., van Steenbergen, S., van Elzakker, P., van Krimpen, B., Groot, C., ... Gratama, J. W. (2013). Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T-cells: Clinical evaluation and management of on-target toxicity. Molecular Therapy, 21(4), 904–912.
- Lee, D. W., Kochenderfer, J. N., Stetler-Stevenson, M., Cui, Y. K., Delbrook, C., Feldman, S. A., ... Mackall, C. L. (2015). T-cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet, 385(9967), 517–528.
- Li, Y. S., Hayakawa, K., & Hardy, R. R. (1993). The regulated expression of B lineage associated genes during B cell differentiation in bone marrow and fetal liver. Journal of Experimental Medicine, 178(3), 951–960.
- Li, Y. S., Wasserman, R., Hayakawa, K., & Hardy, R. R. (1996). Identification of the earliest B lineage stage in mouse bone marrow. Immunity, 5(6), 527–535.
- Liu, X., Jiang, S., Fang, C., Yang, S., Olalere, D., Pequignot, E. C., ... Zhao, Y. (2015). Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T-cells exhibit an increased therapeutic index against tumors in mice. Cancer Research, 75(17), 3596–3607.
- Loskog, A., Giandomenico, V., Rossig, C., Pule, M., Dotti, G., & Brenner, M. K. (2006). Addition of the CD28 signaling domain to chimeric T-cell receptors enhances chimeric T-cell resistance to T regulatory cells. Leukemia, 20(10), 1819–1828.10.1038/sj.leu.2404366
- Louis, C. U., Savoldo, B., Dotti, G., Pule, M., Yvon, E., Myers, G. D., ... Brenner, M. K. (2011). Antitumor activity and long-term fate of chimeric antigen receptor-positive T-cells in patients with neuroblastoma. Blood, 118(23), 6050–6056.
- Luo, X., Xie, H., Long, X., Zhou, M., Xu, Z., Shi, B., Jiang, H., ... Li, Z. (2013). EGFRvIII mediates hepatocellular carcinoma cell invasion by promoting S100 calcium binding protein A11 expression. PLoS One, 8(12), e83332.
- Maher, J. (2014). Clinical immunotherapy of B-cell malignancy using CD19-targeted CAR T-cells. Current Gene Therapy, 14(1), 35–43.
- Mardiros, A., Forman, S. J., & Budde, L. E. (2015). T cells expressing CD123 chimeric antigen receptors for treatment of acute myeloid leukemia. Current Opinion in Hematology, 22(6), 484–488.
- Marktel, S., Magnani, Z., Ciceri, F., Cazzaniga, S., Riddell, S. R., Traversari, C., ... Bonini, C. (2003). Immunologic potential of donor lymphocytes expressing a suicide gene for early immune reconstitution after hematopoietic T-cell-depleted stem cell transplantation. Blood, 101(4), 1290–1298.
- Maude, S. L., Frey, N., Shaw, P. A., Aplenc, R., Barrett, D. M., Bunin, N. J., ... Grupp, S. A. (2014). Chimeric antigen receptor T-cells for sustained remissions in leukemia. The New England Journal of Medicine, 371(16), 1507–1517.
- Maude, S. L., Teachey, D. T., Porter, D. L., & Grupp, S. A. (2015). CD19- targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood, 125(26), 4017–4023.
- Maus, M. V. (2015). Designing CAR T cells for glioblastoma. Oncoimmunology, 4(12), e1048956.
- Maus, M. V., & Levine, B. L. (2016). Chimeric antigen receptor T-cell therapy for the community oncologist. Oncologist, 21(5), 608–617.
- Maus, M. V., Haa, A. R., Beatty, G. L., Albelda, S. M., Levine, B. L., Liu, X., ... June, C. H. (2013). T-cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunology Research, 1, 26–31.
- Mihara, K., Yanagihara, K., Takigahira, M., Imai, C., Kitanaka, A., Takihara, Y., ... Kimura, A. (2009). Activated T-cell-mediated immunotherapy with a chimeric receptor against CD38 in b-cell non-hodgkin lymphoma. Journal of Immunotherapy, 32(7), 737–743.
- Mihara, K., Yanagihara, K., Takigahira, M., Kitanaka, A., Imai, C., Bhattacharyya, J., ... Kimura, A. (2010). Synergistic and persistent effect of T-cell immunotherapy with anti-CD19 or anti-CD38 chimeric receptor in conjunction with rituximab on B-cell non-Hodgkin lymphoma. British Journal of Haematology, 151(1), 37–46.
- Milone, M. C., Fish, J. D., Carpenito, C., Carroll, R. G., Binder, G. K., Teachey, D., ... June, C. H. (2009). Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T-cells and increased anti-leukemic efficacy in vivo. Molecular Therapy, 17(8), 1453–1464.
- Moon, E. K., Wang, L. C., Dolfi, D. V., Wilson, C. B., Ranganathan, R., Sun, J., ... Albelda, S. M. (2014). Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor–transduced human T cells in solid tumors. Clinical Cancer Research, 20, 4262–4273.
- Morgan, R. A., Dudley, M. E., Wunderlich, J. R., Hughes, M. S., Yang, J. C., Sherry, R. M., ... Rosenberg, S. A. (2006). Cancer regression in patients after transfer of genetically engineered lymphocytes. Science, 314(5796), 126–129.10.1126/science.1129003
- Morgan, R. A., Yang, J. C., Kitano, M., Dudley, M. E., Laurencot, C. M., & Rosenberg, S. A. (2010). Case report of a serious adverse event following the administration of T-cells transduced with a chimeric antigen receptor recognizing ERBB2. Molecular Therapy, 18(4), 843–851.
- Moritz, D., & Groner, B. (1995). A spacer region between the single chain antibody and the CD3 zeta-chain domain of chimeric T-cell receptor components is required for efficient ligand binding and signaling activity. Gene Therapy, 2(8), 539–546.
- Muniappan, A., Banapour, B., Lebkowski, J., & Talib, S. (2000). Ligand-mediated cytolysis of tumor cells: Use of heregulin-ζ chimeras to redirect cytotoxic T lymphocytes. Cancer Gene Therapy, 7(1), 128–134.10.1038/sj.cgt.7700100
- Ninomiya, S., Narala, N., Huye, L., Yagyu, S., Savoldo, B., Dotti, G., ... Ramos, C. A. (2015). Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T-cells and is downregulated by lymphodepleting drugs. Blood, 125(25), 3905–3916.
- Pameijer, C. R. J., Navanjo, A., Meechoovet, B., Wagner, J. R., Aguilar, B., Wright, C. L., ... Jensen, M. C. (2007). Conversion of a tumor-binding peptide identified by phage display to a functional chimeric T-cell antigen receptor. Cancer Gene Therapy, 14(1), 91–97.10.1038/sj.cgt.7700993
- Park, J. R., Digiusto, D. L., Slovak, M., Wright, C., Naranjo, A., Wagner, J., ... Jensen, M. C. (2007). Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Molecular Therapy, 15(4), 825–833.
- Park, H. J., Kusnadi, A., Lee, E. J., Kim, W. W., Cho, B. C., Lee, I. J., ... Ha, S. J. (2012). Tumor-infiltrating regulatory T cells delineated by upregulation of PD-1 and inhibitory receptors. Cellular Immunology, 278, 76–83.
- Park, J. H., Riviere, I., Wang, X., Bernal, Y., Purdon, T., Halton, E., ... Brentjens, R. J. (2015). Efficacy and safety of CD19-targeted 19-28z CAR modified T cells in adult patients with relapsed or refractory B-ALL. Journal of Clinical Oncology, 33(15), 7010.
- Pegram, H. J., Lee, J. C., Hayman, E. G., Imperato, G. H., Tedder, T. F., Sadelain, M., & Brentjens, R. J. (2012). Tumor-targeted T-cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood, 119(18), 4133–4141.
- Pegram, H. J., Purdon, T. J., van Leeuwen, D. G., Curran, K. J., Giralt, S. A., Barker, J. N., & Brentjens, R. J. (2015). IL-12-secreting CD19-targeted cord blood-derived T-cells for the immunotherapy of B-cell acute lymphoblastic leukemia. Leukemia, 29(2), 415–422.
- Philip, B., Kokalaki, E., Mekkaoui, L., Thomas, S., Straathof, K., Flutter, B., ... Pule, M. (2014). A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood, 124(8), 1277–1287.
- Poirot, L., Philip, B., Schiffer-Mannioui, C., Le Clerre, D., Chion-Sotinel, I., Derniame, S., ... Smith, J. (2015). Multiplex genome-edited T-cell manufacturing platform for ‘off-the-shelf’ adoptive T-cell immunotherapies. Cancer Research, 75(18), 3853–3864.
- Porter, D. L., Hwang, W. T., Frey, N. V., Lacey, S. F., Shaw, P. A., Loren, A. W., ... June, C. H. (2015). Chimeric antigen receptor T-cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Science Translational Medicine, 7(303), 303ra139.
- Porter, D. L., Levine, B. L., Kalos, M., Bagg, A., & June, C. H. (2011). Chimeric antigen receptor-modified T-cells in chronic lymphoid leukemia. The New England Journal of Medicine, 365(8), 725–733.
- Prosser, M. E., Brown, C. E., Shami, A. F., Forman, S. J., & Jensen, M. C. (2012). Tumor PD-L1 co-stimulates primary human CD8(+) cytotoxic T-cells modified to express a PD1:CD28 chimeric receptor. Molecular Immunology, 51(3–4), 263–272.
- Pule, M. A., Straathof, K. C., Dotti, G., Heslop, H. E., Rooney, C. M., & Brenner, M. K. (2005). A chimeric T-cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T-cells. Molecular Therapy, 12(5), 933-941.
- Pule, M. A., Savoldo, B., Myers, G. D., Rossig, C., Russell, H. V., Dotti, G., ... Brenner, M. K. (2008). Virus-specific T-cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nature Medicine, 14(11), 1264–1270.
- Ritchie, D. S., Neeson, P. J., Khot, A., Peinert, S., Tai, T., Tainton, K., ... Prince, H. M. (2013). Persistence and efficacy of second generation CAR T-cell against the LeY antigen in acute myeloid leukemia. Molecular Therapy, 21(11), 2122–2129.
- Roybal, K. T., Rupp, L. J., Morsut, L., Walker, W. J., McNally, K. A., Park, J. S., & Lim, W. A. (2016). Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell, 164, 770–779.
- Sadelain, M., Brentjens, R., & Rivière, I. (2013). The basic principles of chimeric antigen receptor design. Cancer Discovery, 3(4), 388–398.10.1158/2159-8290.CD-12-0548
- Sampson, J. H., Heimberger, A. B., Archer, G. E., Aldape, K. D., Friedman, A. H., Friedman, H. S., ... Bigner, D. D. (2010). Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. Journal of Clinical Oncology, 31(28), 4722–4729.
- Sapra, P., Stein, R., Pickett, J., Qu, Z., Govindan, S. V., Cardillo, T. M., ... Goldenberg, D. M. (2005). Anti-CD74 antibody-doxorubicin conjugate, IMMU-110, in a human multiple myeloma xenograft and in monkeys. Clinical Cancer Research, 11(14), 5257–5264.
- Sato, T., Neschadim, A., Konrad, M., Fowler, D. H., Lavie, A., & Medin, J. A. (2007). Engineered human tmpk/AZT As a novel enzyme/prodrug axis for suicide gene therapy. Molecular Therapy, 15(5), 962–970.10.1038/mt.sj.6300122
- Savoldo, B., Rooney, C. M., Di Stasi, A., Abken, H., Hombach, A., Foster, A. E., ... Dotti, G. (2007). Epstein Barr virus-specific cytotoxic T lymphocytes expressing the anti-CD30ζ artificial chimeric T-cell receptor for immunotherapy of Hodgkin disease. Blood, 110(7), 2620–2630.
- Savoldo, B., Ramos, C. A., Liu, E., Mims, M. P., Keating, M. J., Carrum, G., ... Dotti, G. (2011). CD28 co-stimulation improves expansion and persistence of chimeric antigen receptor-modified T-cells in lymphoma patients. Journal of Clinical Investigation, 121(5), 1822–1826.
- Scheuermann, R. H., & Racila, E. (1995). CD19 antigen in leukemia and lymphoma diagnosis and immunotherapy. Leukemia & Lymphoma, 18, 385–397.
- Scholler, J., Brady, T. L., Binder-Scholl, G., Mitsuyasu, R. T., Bernstein, W. B., Aronson, N. E., ... June, C. H. (2012). Decade-long safety and function of retroviral-modified chimeric antigen receptor T-cells. Science Translational Medicine, 4(132), 132ra53.
- Schuster, S. J., Svoboda, J., Nasta, S., Porter, D. L., Mato, A., Shah, G. D., ... June, C. H. (2015). Phase IIa trial of chimeric antigen receptor modified T cells directed against CD19 (CTL019) in patients with relapsed or refractory CD19+ lymphomas. Journal of Clinical Oncology, 33(15), 8516.
- Serrano-Olvera, A., Duenas-Gonzalez, A., Gallardo-Rincon, D., Candelaria, M., & De la Garza-Salazar, J. (2006). Predictive and therapeutic implications of HER2 in invasive epithelial ovarian cancer. Cancer Treatment Reviews, 32(3), 180–190.
- Shaffer, D. R., Savoldo, B., Yi, Z., Chow, K. K., Kakarla, S., Spencer, D. M., ... Gottschalk, S. (2011). T-cells redirected against CD70 for the immunotherapy of CD70-positive malignancies. Blood, 117(16), 4304–4314.
- Shaffer, D. R., Zhou, P., & Gottschalk, S. (2014). Foreign or domestic CARs: Receptor ligands as antigen-binding domains. Medical Sciences, 2(1), 23–36.
- Sharifzadeh, Z., Rahbarizadeh, F., Shokrgozar, M. A., Ahmadvand, D., Mahboudi, F., Jamnani, F. R., & Moghimi, S. M. (2013). Genetically engineered T cells bearing chimeric nanoconstructed receptors harboring TAG-72-specific camelid single domain antibodies as targeting agents. Cancer Letters, 334(2), 237–244.10.1016/j.canlet.2012.08.010
- Shengnan, Y., Li, A., Liu, Q., Li, T., Yuan, X., Han, X., & Wu, K. (2017). Chimeric antigen receptor T cells: A novel therapy for solid tumors. Journal of Hematology and Oncology, 10, 78.
- Simons, F. E. (2009). Anaphylaxis: Recent advances in assessment and treatment. The Journal of Allergy and Clinical Immunology, 124(4), 625–636.
- Singh, R., & Paterson, Y. (2007). Immunoediting sculpts tumor epitopes during immunotherapy. Cancer Research, 67(5), 1887–1892.
- Slamon, D. J., Godolphin, W., Jones, L. A., Holt, J. A., Wong, S. G., Keith, D. E., ... Ullrich, A. (1989). Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science, 244(4905), 707–712.
- Song, D. G., Ye, Q., Poussin, M., Harms, G. M., Figini, M., & Powell, D. J. (2012). CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood, 119(3), 696–706.10.1182/blood-2011-03-344275
- Spear, P., Barber, A., Rynda-Apple, A., & Sentman, C. L. (2013). NKG2D CAR T-cell therapy inhibits the growth of NKG2D ligand heterogeneous tumors. Immunology and Cell Biology, 91(6), 435–440.
- Tey, S. K., Dotti, G., Rooney, C. M., Heslop, H. E., & Brenner, M. K. (2007). Inducible caspase 9 suicide gene to improve the safety of allodepleted T-cells after haploidentical stem cell transplantation. Biology of Blood and Marrow Transplantation, 13(8), 913–924.
- Thomis, D. C., Marktel, S., Bonin, C., Traversari, C., Gilman, M., Bordignon, C., & Clackson, T. (2001). A Fas-based suicide switch in human T-cells for the treatment of graft-versus-host disease. Blood, 97(5), 1249–1257.
- Thompson, S. K., Sullivan, T. R., Davies, R., & Ruszkiewicz, A. R. (2011). Her-2/neu gene amplification in esophageal adenocarcinoma and its influence on survival. Annals of Surgical Oncology, 18(7), 2010–2017.
- Till, B. G., Jensen, M. C., Wang, J., Chen, E. Y., Wood, B. L., Greisman, H. A., ... Press, O. W. (2008). Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T-cells. Blood, 112(6), 2261–2271.
- Till, B. G., Jensen, M. C., Wang, J., Qian, X., Gopal, A. K., Maloney, D. G., ... Press, O. W. (2012). CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: Pilot clinical trial results. Blood, 119(17), 3940–3950.
- Tintinalli, J. E. (2010). Emergency medicine: A comprehensive study guide (emergency medicine (Tintinalli)). New York, NY: McGraw-Hill Companies, pp. 177-182. ISBN 0-07-148480-9.
- Topp, M. S., Gökbuget, N., Stein, A. S., Zugmaier, G., O’Brien, S., Bargou, R. C., ... Kantarjian, H. M. (2015). Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: A multicentre, single-arm, phase 2 study. Lancet Oncology, 16(1), 57–66.
- Torikai, H., Reik, A., Liu, P. Q., Zhou, Y., Zhang, L., Maiti, S., ... Cooper, L. J. (2012). A foundation for universal T-cell based immunotherapy: T-cells engineered to express a CD19-specific chimeric antigen- receptor and eliminate expression of endogenous TCR. Blood, 119(24), 5697–5705.
- Torikai, H., Reik, A., Soldner, F., Warren, E. H., Yuen, C., Zhou, Y., … Cooper, L. J. (2013). Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood, 122(8), 1341–1349.
- Traversari, C., Marktel, S., Magnani, Z., Mangia, P., Russo, V., Ciceri, F., ... Bordignon, C. (2007). The potential immunogenicity of the TK suicide gene does not prevent full clinical benefit associated with the use of TK-transduced donor lymphocytes in HSCT for hematologic malignancies. Blood, 109(11), 4708–4715.
- Van der Stegen, S. J., Hamieh, M., & Sadelain, M. (2015). The pharmacology of second-generation chimeric antigen receptors. Nature Reviews Drug Discovery, 14(7), 499–509.
- Vera, J., Savoldo, B., Vigouroux, S., Biagi, E., Pule, M., Rossig, C., ... Dotti, G. (2006). T lymphocytes redirected against the κ light chain of human immunoglobulin efficiently kill mature B lymphocyte-derived malignant cells. Blood, 108(12), 3890–3897.
- Wang, X., Chang, W. C., Wong, C. W., Colcher, D., Sherman, M., Ostberg, J. R., ... Jensen, M. C. (2011). A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablationof engineered cells. Blood, 118(5), 1255–1263.
- Wang, J., Jensen, M., Lin, Y., Sui, X., Chen, E., Lindgren, C. G., ... Press, O. W. (2007). Optimizing adoptive polyclonal T-cell immunotherapy of lymphomas, using a chimeric T-cell receptor possessing CD28 and CD137 costimulatory domains. Human Gene Therapy, 18(8), 712–725.
- Wang, X., Naranjo, A., Brown, C. E., Bautista, C., Wong, C. W., Chang, W. C., ... Jensen, M. C. (2012). Phenotypic and functional attributes of lentivirus-modified CD19-specific human CD8+ central memory T-cells manufactured at clinical scale. Journal of Immunotherapy, 35(9), 689–701.
- Wang, J., Press, O. W., Lindgren, C. G., Greenberg, P., Riddell, S., Qian, X., ... Jensen, M. C. (2004). Cellular immunotherapy for follicular lymphoma using genetically modified CD20-specific CD8+ cytotoxic T lymphocytes. Molecular Therapy, 9(4), 577–586.
- Wang, Q. S., Wang, Y., Lv, H. Y., Han, Q. W., Fan, H., Guo, B., ... Han, W. D. (2015). Treatment of CD33-directed chimeric antigen receptor-modified T-cells in one patient with relapsed and refractory acute myeloid leukemia. Molecular Therapy, 23(1), 184–191.
- Wang, Y., Zhang, W. Y., Han, Q. W., Liu, Y., Dai, H. R., Guo, Y. L., ... Wang, Q. S. (2014). Effective response and delayed toxicities of refractory advanced diffuse large B-cell lymphoma treated by CD20-directed chimeric antigen receptor-modified T-cells. Journal of Clinical Immunology, 155(2), 160–175.
- Weiss, A., & Littman, D. R. (1994). Signal transduction by lymphocyte antigen receptors. Cell, 76(2), 263–274.
- Wilkie, S., Picco, G., Foster, J., Davies, D. M., Julien, S., Cooper, L., ... Maher, J. (2008). Retargeting of human T-cells to tumor-associated MUC1: The evolution of a chimeric antigen receptor. Journal of Immunology, 180(7), 4901–4909.
- Wu, C. Y., Roybal, K. T., Puchner, E. M., Onuffer, J., & Lim, W. A. (2015). Remote control of therapeutic T-cells through a small molecule–gated chimeric receptor. Science, 350(6258), aab4077.
- Yarden, Y., & Sliwkowski, M. X. (2001). Untangling the ErbB signalling network. Nature Reviews Molecular Cell Biology, 2(2), 127–137.
- Zhang, J. G., Kruse, C. A., Driggers, L., Hoa, N., Wisoff, J., Allen, J. C., ... Jadus, M. R. (2008). Tumor antigen precursor protein profiles of adult and pediatric brain tumors identify potential targets for immunotherapy. Journal of Neuro-Oncology, 88(1), 65–76.
- Zhang, T., Wu, M. R., & Sentman, C. L. (2012). An NKp30-based chimeric antigen receptor promotes T cell effector functions and antitumor efficacy in vivo. The Journal of Immunology, 189(5), 2290–2299.10.4049/jimmunol.1103495
- Zhao, Z., Condomines, M., van der Stegen, S. J., Perna, F., Kloss, C. C., Gunset, G., ... Sadelain, M. (2015). Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T-cells. Cancer Cell, 28(4), 415–428.
- Zheng, Y., Zha, Y., & Gajewski, T. F. (2008). Molecular regulation of T-cell anergy. EMBO Reports, 9, 50–55.
- Zhong, X. S., Matsushita, M., Plotkin, J., Riviere, I., & Sadelain, M. (2010). Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3 kinase/AKT/Bcl-XL activation and CD8+ T-cell-mediated tumor eradication. Molecular Therapy, 18(2), 413–420.
- Zhou, X., Di Stasi, A., Tey, S. K., Krance, R. A., Martinez, C., Leung, K. S., ... Dotti, G. (2014). Long-term outcome after haploidentical stem cell transplant and infusion of T-cells expressing the inducible caspase 9 safety transgene. Blood, 123(25), 3895–3905.
- Zhou, G., & Levitsky, H. (2012). Towards curative cancer immunotherapy: Overcoming post therapy tumor escape. Clinical and Developmental Immunology, 2012, 124187.