
Abstract
Advances in sequencing technologies and bioinformatic analysis techniques have greatly improved our understanding of various classes of RNAs and their functions. Despite not coding for proteins, non-coding RNAs (ncRNAs) are emerging as essential biomolecules fundamental for cellular functions and cell survival. Interestingly, ncRNAs produced by viruses not only control the expression of viral genes, but also influence host cell regulation and circumvent host innate immune response. Correspondingly, ncRNAs produced by the host genome can play a key role in host–virus interactions. In this article, we will first discuss a number of types of viral and mammalian ncRNAs associated with viral infections. Subsequently, we also describe the new possibilities and opportunities that RNA genomics and next-generation sequencing technologies provide for studying ncRNAs in virology.
Acronyms:
Introduction
Understanding the role of RNA in viral replication, diversity and host interactions is critical for developing effective therapeutics for many deadly diseases. In the past, our understanding of the functional diversity and role of RNA within the cell was extremely limited. Due to its transitive nature and difficulty for its isolation compared to DNA, RNA was comparatively understudied with respect to DNA and the functional proteins of the cell. However, the rapid advances in sequencing technology have allowed us to better appreciate the great diversity of functional RNA molecules and the importance of RNA in fundamental cellular functions. One of the first breakthroughs appreciating the functional value of RNA occured in 1957 when Hoagland and colleagues discovered that a soluble RNA molecule was responsible for the transfer of activated amino acids to the growing peptide at the ribosome (Hoagland, Stephenson, Scott, Hecht, & Zamecnik, Citation1958). It was not until 1965 that the sequence of transfer RNA-alanine (tRNA-ala) was elucidated using enzymatic fragmentation by Holley and colleagues, representing the first sequencing of a nucleic acid biomolecule (Holley et al., Citation1965). From this point on, a great deal of research was produced identifying and sequencing various functional classes of RNAs, including messenger RNAs (mRNAs), that encode for proteins but also a number of classes of RNAs that are non-protein-coding and have other functions such as rRNAs (ribosomal RNAs, ribosomal formation) (Nissen, Hansen, Ban, Moore, & Steitz, Citation2000), tRNAs (transfer RNAs, protein translation) (Hoagland et al., Citation1958), snoRNAs (small nucleolar RNAs, chemical modifications) (Bachellerie, Cavaille, & Huttenhofer, Citation2002) and snRNAs (small nuclear RNA, mRNA splicing and processing) (Legrain, Seraphin, & Rosbash, Citation1988).
The emerging world of non-coding RNAs (ncRNAs)
Early efforts to understand the complexity of the genome were limited by the available sequencing technologies like capillary sequencing. These techniques were low throughput and enabled the reading of only a small number of short nucleic acid fragments at reasonable costs (Karger & Guttman, Citation2009). For example, it took more than 10 years and millions of dollars to sequence the genome of just one individual at the Human Genome Project (Hayden, Citation2014). During the last decade, the wide introduction of high throughput technologies such as massive parallel sequencing (next-generation sequencing) has reduced this time to hours (Hayden, Citation2014). As a result, it has been identified that only 2% of the human genome is protein-coding, while the remaining 98% of the DNA is not (Encode Project Consortium, Citation2007).
The application of new high throughput technologies to the analysis of RNA molecules within the cell (transcriptome) has led to large-scale projects like the Encyclopedia of DNA Elements (ENCODE) which has reported upwards of 80% of the genome can be assigned a biological function and that a large portion of the non-coding genome is actually transcribed (Encode Project Consortium, Citation2012). A number of these transcripts are further processed into lncRNAs (long non-coding RNAs) (Ma, Bajic, & Zhang, Citation2013) and miRNAs (microRNAs)(Starega-Roslan et al., Citation2011), both of which have important regulatory functions within the cell (Ambros, Citation2004). However, it is important to recognize the limitations of the functional annotation of the ENCODE project. The small absolute number of transcripts that arise from non-coding regions, evolutionary pressures (or lack thereof) on maintaining a large body of non-functional DNA, and lax criteria for functionality have been pointed out as weaknesses of claims to widely assign function across the genome. (Palazzo & Gregory, Citation2014; Palazzo & Lee, Citation2015) The prudent position is to assume that a ncRNA cannot be regarded as either functional or non-functional until specific studies on this ncRNA have been concluded.
Of the non-coding, non-repetitive part of the genome, many classes of functional non-coding RNAs have been thoroughly explored including: piwi-interacting RNA, promoter associated RNAs and others. Despite the increasing amount of study in the field, many functions of ncRNAs derived from the repetitive portion of the non-coding genome remain to be elucidated despite these repetitive genomic elements making up more than 60% of total human DNA (de Koning, Gu, Castoe, Batzer, & Pollock, Citation2011). Among the most populous of these repeats are Small Interspersed Nuclear Elements (SINEs). Interestingly, examining the origin of non-coding RNA sequences within the cell suggests that many non-coding RNAs, transposable elements, mammalian viruses and their hosts have complex evolutionary relationships (Piskurek & Jackson, Citation2012).
An ever-increasing number of novel viral ncRNAs
Many ncRNAs are produced by viruses, including the adenoviral non-coding RNA VAI, the non-structural ncRNAs of minute virus of mice (MVM) and ncRNAs within the human immunodeficiency virus-1 (HIV-1) genome. Some are important for self-regulation and replication. Others are likely to play roles in the interaction between the host and the virus controlling the cells response to infection (Lecellier et al., Citation2005; Yeung, Benkirane, & Jeang, Citation2007). Non-coding RNAs produced by viruses can be divided into two classes, microRNAs (miRNAs, miRs) and long non-coding RNAs (lncRNAs), although the existence of yet unidentified classes (as in case of mammalian genomes) cannot be excluded (Figure ).
Figure 1. A schematic overview of miRNA and lncRNA interplay between host and viral genome ncRNAs.
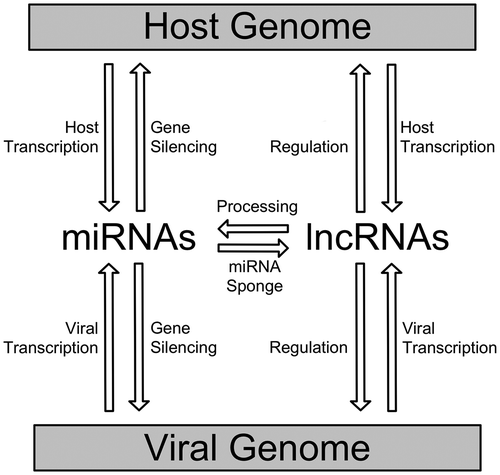
The miRNAs are produced from much longer transcripts and processed down into a ~22 nucleotide dsRNA effector that recruits the RISC (RNA-induced silencing complex) to initiate gene silencing. It is possible that through this process viral miRNAs can control protein translation of the host (Tycowski et al., Citation2015). Recently, viral miRNAs have been shown to play roles in evading the host immune response. For example, the human cytomegalovirus miRNAs repress the immune system by modulating host gene expression via silencing of important natural killer cell associated factors (Stern-Ginossar et al., Citation2007). Similarly, miRNAs produced by herpes simplex virus-1 reduce expression of tetherin and natural killer cell binding targets. Targeted silencing prevents the identification of infected cells, thereby facilitating the latent infection. The induction of apoptosis after infection may also be avoided by influencing the responsiveness of a cell to pro-apoptotic signals (Enk et al., Citation2016).
The Epstein-Barr virus (EBV) is linked to the development of various carcinoma and facilitates anti-apoptotic behavior by expression of miR-BART5. mirR-BART5 targets the mRNA of p53 upregulated modulator of apoptosis (PUMA) and significantly reduced the expression of PUMA in tissues with latent EBV infection (Choy et al., Citation2008). Long non-coding RNAs are much longer than miRNAs, ranging from 100 to 1000 nucleotides in length and can perform both pro-viral and anti-viral activities. For example, some viral lncRNAs are transcribed by polymerase III and are involved in inhibiting the host’s innate immune system (Kitajewski et al., Citation1986). Highly structured ncRNAs encoded by hepatitis C virus (HCV) achieve their pro-viral function function by acting as competitive inhibitors to protein kinase R (PKR) and prevent detection of the viral DNA (Gale & Katze, Citation1998). LncRNAs in HIV-1 exhibit unique regulatory dynamics that suggest a role in replication (Trypsteen et al., Citation2017) and denote the role of ncRNAs in helping viruses survive. Viruses may also express lncRNAs to mitigate the effects of host miRNAs to create conditions favourable to reproduction. Murine cytomegalovirus (MCMV) has difficulty replicating in the presence of high levels of miR-27a and miR-27b, and must accordingly produce a long ncRNA known as MCMV m169 to degrade the miRNAs and release repression. (Buck et al., Citation2010) Though viral ncRNAs are shown to be helpful in avoiding detection, they may also play a role in activating the host defenses. Epstein-Barr virus ncRNAs are transcribed by polymerase III in abundance and play a role in activating the immune response to EBV infection. (Samanta, Iwakiri, Kanda, Imaizumi, & Takada, Citation2006).
Virus–host interactions and mammalian ncRNAs
Out of more than 500 known human miRNAs, many miRNAs have been recently found to be connected with virus–host interactions and influence viral life cycle (Panwar, Omenn, & Guan, Citation2017). It is important to note that not all miRNA are helpful to the host despite being endogenously transcribed. In some cases, the upregulation or downregulation of microRNAs may directly benefit the invading pathogen. In HIV-1 positive individuals, downregulation of T-cell specific miRNAs occurs under high viral load (Houzet et al., Citation2008). Interestingly, Hepatitis C Virus (HCV) does not code for its own miRNA, but upon infection, HCV influences the expression of host miRNA, which in turn positively regulates the life cycle of HCV (Li, Jiang, & Peng, Citation2016). Additionally, mir-29 downregulates interferon (IFN) gamma, thus supressing the immune response to pathogens (Ma et al., Citation2011). On the other hand, production of miRNAs by a host cell can directly influence the ability of a virus to gain traction in the cell. IFNbeta upregulates miRs with targets in HCV in addition to downregulating miR-122 which is required for the completion of the viral life cycle (Pedersen et al., Citation2007) Compounding the complexity of viral-host interactions, the effects of miRNA-mediated silencing are not only unidirectional. Like other miRNAs that target retinoic acid-inducible gene I (RIG-I), miR-485 inhibits the ability of the cell to detect and therefore respond to infection. However, under increased viral loab miR-485 recognizes both the RIG-I mRNA as well as PB1 of influenza H5N1 (Ingle et al., Citation2015).
Longer ncRNAs also have diverse roles in the mammalian response to viral infection via numerous signaling pathways. The human lncRNA CMPK2 has been shown to be highly upregulated in response to rising levels of IFNalpha. Following the immune response, CMPK2 behaves as a negative regulator to modulate the interferon response and return the cell to homeostasis (Kambara et al., Citation2014). Similarly, the eosinophil granule ontogeny transcript (EGOT) also works against the immune response and is essential for HCV. However, instead of being activated by the IFN antiviral pathway, EGOT is activated by both RIG and protein kinase R ahead of viral replication (Carnero et al., Citation2016). Furthermore, flaviviral activation of the PERK-mediated unfolded protein response results in the transcriptional regulation of MALAT1 (Bhattacharyya & Vrati, Citation2015). While MALAT1 is known to be oncogenic, it remains to be seen if it has an antiviral function. Antiviral mechanisms of ncRNAs can also act outside of signal transduction pathways. GAS5 has been shown to impact the replication of HCV by tightly associating with the NS3 viral protein thereby inhibiting its function directly (Qian, Xu, Zhao, & Qi, Citation2016). Mammalian SINE-encoded ncRNAs also play a role in the response to viral stress. The 7SL-derived Alu element and tRNA-derived murine B2 are transcribed by polymerase III and are upregulated under the stress of viral infection (Panning & Smiley, Citation1993; Russanova, Driscoll, & Howard, Citation1995). Interestingly, these RNAs have been recently shown to regulate the stress response and transcription of immediate early genes by binding RNA Polymerase II and inhibiting its function (Ponicsan et al., Citation2013). Ordinarily, B2 transcripts repress polymerase II transcription of stress-responsive genes, called immediate early genes (IEGs), under non-stress conditions, until application of a stress stimulus leads to B2 RNA processing, at which point high polymerase II activity is observed on those genes (Zovoilis, Cifuentes-Rojas, Chu, Hernandez, & Lee, Citation2016). Upregulation of IEGs is of critical importance for the stress response and survival of the cell (Zovoilis et al., Citation2016). The upregulation of B2 also allows for broad binding of new targets, and specifically reduces the expression of housekeeping genes and prepares the cell for the rapid expression of immediate early genes (Yakovchuk, Goodrich, & Kugel, Citation2009) (Figure ). The Alu elements in humans also produce RNAs that bind and inhibit Polymerase II and thus may play a similar role as the B2 repeat does in mice (Walters, Kugel, & Goodrich, Citation2009).
Figure 2. An overview of gene expression regulation by the SINE B2 RNA. Recruitment of the protein EZH2 during cellular response to stress leads to B2 RNA cleavage and release of RNA Pol II transcription of IEGs. IEG: Immediate Early Genes.
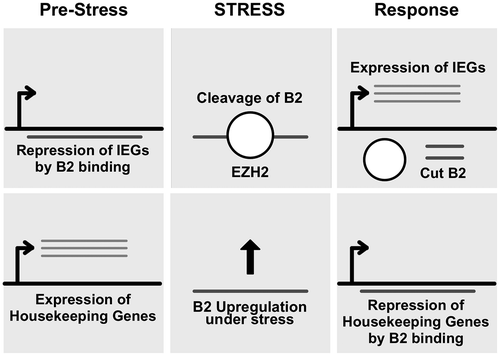
The B1 7SL-derived murine Alu orthologue (Kriegs, Churakov, Jurka, Brosius, & Schmitz, Citation2007) and B2 murine SINEs are upregulated in mouse fibroblast cells infected with minute virus of mice (MVM) (Williams, Tamburic, & Astell, Citation2004). The virus has two open reading frames that encode four proteins: two non-structural (NS1, NS2) and two structural (VP1, VP2). NS1 has multiple functions, including nickase, helicase, DNA binding and ATP hydrolysis activities, and cytotoxicity, in addition to the ability to induce cell cycle arrest. Differential display, primer extension and western blotting suggested that the presence of NS1 was necessary and sufficient for the upregulation of B2 SINE levels after 24 hours and was not caused by upregulation of other polymerase III transcription factors. The authors concluded that the nickase activity of NS1 could be responsible for exposing additional SINE elements by remodelling chromatin. Furthermore, other studies show the upregulation of B2 can also be induced by infection with SV-40, indicating a non-specific response to viral stress (Carey & Singh, Citation1988; Singh, Carey, Saragosti, & Botchan, Citation1985).
Numerous studies have determined that the Alu SINE is also upregulated under viral stress by transformation with HSV, Adenovirus and HIV-1 (Jang, Collins, & Latchman, Citation1992; Panning & Smiley, Citation1994; Russanova et al., Citation1995). Although the reported causes of SINE activation amongst these three viruses are not consistent, the change in expression of the Alu element was implied to be independent of the expression of 7SL RNA – the putative progenitor of the Alu (Kramerov & Vassetzky, Citation2011). While the specific mechanism of this induction is unknown, the variety of factors (viral and otherwise) that have been shown to influence SINE expression suggest that a general mechanism of induction, such as chromatin remodelling could be the main cause of the differential expression under stressed and unstressed conditions (Russanova et al., Citation1995). Thus, SINE ncRNAs (B2 and Alu RNAs) appear to have a central role in virus–host stress response and interactions.
Traditional and next-generation sequencing technologies
Studying transcriptomic changes in viruses and hosts is a difficult task based on the varying lengths, and processing events which have pushed forward the development of sequencing technologies during the last decades. A summary of these technologies is provided in Table . In 1977, Sanger et al. developed a sequencing technique involving the termination of an elongating strand of DNA which could generate reads under 1 Kb (kilobase) in length (Salas-Solano et al., Citation1998). This technology and subsequent improvements would mark the end of the first generation sequencing technologies. Another sequencing approach developed in 1997, pyrosequencing, is a ‘sequencing by synthesis’ technique that works by detecting the incorporation of nucleotides to a single stranded DNA template by DNA polymerase (Fakhrai-Rad, Pourmand, & Ronaghi, Citation2002). When the correct deoxynucleotide triphosphate (or ATP analogue) is incorporated into the strand, pyrophosphate is released and converted into ATP by ATP sulphurylase which is in turn is used by luciferase to convert luciferin to oxyluciferin that generates light upon conversion. The amount of light generated in the reaction can be used to identify whether a dNTP wash has resulted in successful incorporation within the strand, and successive wash cycles can be used to deduce the sequence of the template DNA. Pyrosequencing can produce reads between 300 and 500 nucleotides in length and has difficulty enumerating single-base polynucleotide repeats (Radford et al., Citation2012). Coupled with ultra-deep sequencing, pyrosequencing has been later utilized to characterize variation in complete HIV/SIV genomes, detecting genetic variations that make up less than 1% of the viral population quasispecies within a host (Bimber et al., Citation2010).
Table 1. Advantages and disadvantages of various sequencing technologies. Advantages and disadvantages are related to the sequencing chemistry behind each platform that influences approximate read lengths afforded by each technology as well as the error rate in base calling for the sequenced reads.
Illumina/Solexa sequencing is a fluorescence-based sequence-by-synthesis (SBS) technology developed in 1998, and currently the most commonly used of the second generation sequencing technologies in use. Next-generation sequencing, deep sequencing, massive parallel sequencing are some of the terms that have been used for this type of sequencing technology. It works by immobilizing DNA strands to a flow cell via complementary adaptor sequences and performing solid phrase PCR to produce large numbers of bound copies of the template sample (Radford et al., Citation2012). The actual sequencing is achieved by exciting and detecting fluorescence from reversibly terminating washes of fluorescently labelled dNTPs as they are incorporated into a new strand. Each read produced using Illumina sequencing is approximately 300–600 bp in length (Fadrosh et al., Citation2014). Based on its competitive pricing and coverage, Illumina is the most common technology of choice for a variety of sequencing projects (Caporaso et al., Citation2012).
Being a more recent sequencing technology released in 2010, ion semiconductor sequencing is a similar sequence-by-synthesis technology as pyrosequencing, that instead measures the release of hydrogen ions from the incorporation of dNTPs into a newly synthesized complimentary DNA strand. The main benefit of the system is the reduced operating cost and increased speed of data acquisition, but the system also retains the drawbacks of previously discussed sequence-by-synthesis technologies (Huse, Huber, Morrison, Sogin, & Welch, Citation2007), which may explain the small share of this technology in the current market of next-generation sequencing technologies.
The third generation of sequencing will be characterized by technologies capable of single-molecule sequencing in real time. As of September 2017, there are two players in this space: Pacific Biosciences Single Molecule Real Time (SMRT) sequencing, and Oxford Nanopore sequencing. The former utilizes a DNA polymerase bound to the bottom of a zero-mode waveguide. When a fluorescently labelled dNTP is incorporated by DNA polymerase, the dye is excited and triggers the detector. SMRT can produce reads several kilobases in length (Deurenberg et al., Citation2017).
Oxford Nanopore sequencing is also capable of producing very long reads (up to at least 21 Kb) and works by passing a biopolymer through a nanometre-width aperture by electrophoresis (Firnkes, Pedone, Knezevic, Doblinger, & Rant, Citation2010). The identity of a base is determined by the change in current across the pore which is characteristic to each of the nucleobases (Stoddart, Heron, Mikhailova, Maglia, & Bayley, Citation2009). Long read lengths also make detecting complete gene isoforms, splice variants, processing and cleavage, as well as easier de novo viral genome sequencing and assembly (Lu, Giordano, & Ning, Citation2016). Sequencing unfragmented nucleic acid polymers also allows for heterogeneous populations to be counted and binned with less bias and more accuracy as compared to short-read sequencing. When assembled into larger sequence parts, repetitive elements or sequences that share significant homology may underrepresent the diversity of a population if short reads are used for the assembly (Schatz, Delcher, & Salzberg, Citation2010). The ability to obtain long unfragmented sequences allows for reads to be more accurately assembled, allowing for the identification of sub-populations, or to allow transcripts of repetitive elements to be traced to the DNA that produced it. Extremely long-range sequencing results are invaluable for improving the resolution and quality of genome assemblies when paired with short read technologies. This hybrid approach has allowed for improvements in the assemblies of the genomes of HHV-1 and Homo sapiens (Chaisson et al., Citation2015; Karamitros et al., Citation2016).
Using next-generation sequencing applications for studying ncRNAs that influence viral life cycle
The specific chemistry of any of the NGS systems mentioned above, dictates the length of the read returned by the sequencing reaction. Accordingly, sequencing of RNA molecules, termed RNAseq, is a versatile tool that can obtain information on RNAs of different sizes. A small sample of applications and examples of RNAseq are discussed below and summarized in Table .
Table 2. Examples of size-specific RNAseq. Varying read lengths can be used for a variety of purposes. Long RNAseq can be used for differential expression, detection of viral integration and diagnosis of influenza. Short RNAseq can be used to observe processing events, and small RNAseq can be used to quantify differential expression of miRNAs. nt: nucleotides.
Small RNA-seq: MicroRNAs are arguably the most important class of small RNAs of ~22 nucleotides in length. Changes in miRNA expression are indicative of host-pathogen interactions, and result in phenotypic changes. Using deep sequencing, it has been shown that under infection with Kaposi’s sarcoma associated herpesvirus primary effusion lymphoma cells experience dysregulation of approximately 153 miRNAs, which may influence the progression of tumourigenesis (Viollet et al., Citation2015).
Short RNA-seq: Similarly, short RNAs (between 30 and 200 nucleotides) are also of importance to cellular regulation. Short-RNA seq reads can allow processing like cleavage to be detected. For example, the location of the cleavage of the viral infection-induced B2 RNA can be determined by aligning populations of fragmented reads against the complete sequences of the B2 element.
Long RNA-seq: Sequencing of still longer molecules can be informative in detecting genetic changes from viral integration, and diagnosis of influenza and additional viral pathogens within respiratory samples (Fischer et al., Citation2015; Khoury et al., Citation2013).
Other more specialized RNA genomics applications include cross-linking immunoprecipitation sequencing (CLIP-seq) and RNA immunoprecipitation sequencing (RIP-seq) for the identification of ncRNAs interacting with a protein, circRNA-seq for circular RNAs and capture hybridization analysis of RNA targets (CHART-seq) for identification of genomic regions interacting with a ncRNA (Figure ).
Figure 3. Common RNA genomics applications in virology. Genomic DNA is transcribed into RNA precursors for protein production (2–3%) and non-coding transcripts (74–90%). Abundance, identity and interaction data for RNA transcripts of varying lengths can be obtained with RNA genomic techniques.
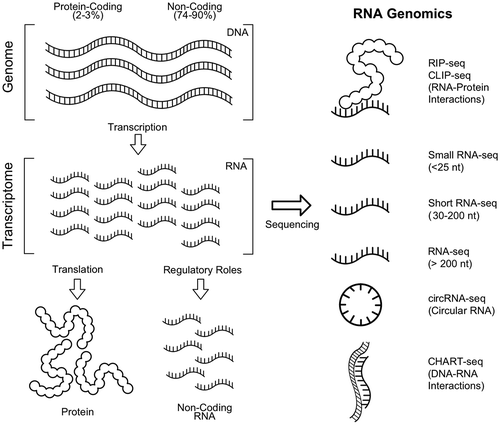
Thus, next-generation sequencing technologies have been deeply integrated into ncRNA virology research.
Bioinformatics considerations
Though viral genomes and transcriptomes are generally small, the amount of data produced with next-generation sequencing techniques should not be underestimated. Similarly, the sequence diversity within rapidly evolving viral populations also warrants careful consideration. Sequencing viral DNA or RNA genomes without fragmentation or cDNA intermediates can be highly informative of the position, orientation and origin of the read in context of the complete genome (Ozsolak & Milos, Citation2011). The long reads provided by single-molecule sequencing can showcase single nucleotide polymorphisms within a population and thereby reveal emerging quasispecies linked to changes in virulence or host evasion. This evolution can also be observed in high resolution by dynamic mapping against a reference genome when sequences are obtained over a time course. However, in some cases a reference genome may not be available, or may not be the correct choice for one’s research objectives.
A principal concern of those dealing with large amounts of RNA sequencing data is the choice to map reads against a pre-assembled reference genome or transcriptome, or to construct a genome/transcriptome de novo. De novo assembly is a computationally intensive process whereby reads are matched together, arranged into contigs, and finally organized into scaffolds to produce an assembled genome or transcriptome (R. Li et al., Citation2010). Constructing an assembly from the scratch is useful when the genome or transcriptome of interest is very plastic, and mapping to a reference would remove useful information (such as the position of enhancer elements) or otherwise introduce unwanted bias to the data. However, it is worth noting that studies have shown that de novo assemblies are often shorter than a comparable reference genome and often lack essential genes (Alkan, Sajjadian, & Eichler, Citation2011). This could be due in part to repetitive content leading to misaligned contigs. Advances in long-range sequencing should increase the quality and length of de novo assemblies, and algorithmic improvements should make it a more appealing choice. Despite the current and future benefits of de novo assembly, it is important to evaluate the results with respect to available standard reference genomes and understand the effects that experimental artefacts can have on the final assembly.
Future perspectives
RNA informatics and the resultant deluge of data promises to be a powerful tool to capture the complex interactions between host and pathogen and the continued co-evolution of host and virus. However, as sequencing depth and length constantly increases, next-generation sequencing approaches to virology will allow for even greater resolution of the viral and host transcriptomes and identification of transcript variants. The improved quality of sequencing data afforded by long reads will allow for a greater understanding of the changes in regulation of miRNAs, lncRNAs and variants of viral mRNAs as they relate to viral–host interactions. Consequently, improved bioinformatic techniques are necessary to process and interpret the new kinds of data made available by these advances in sequencing technologies, in addition to highly qualified bioinformatic experts to make the distinction between biologically relevant data, and experimental artefacts. The need for training of adequate numbers of such skilled bioinformaticians is imperative and will help to overcome the current data analysis bottleneck created due to the large amount of data that next-generation sequencing technologies have generated in virology research. Ultimately, application of bioinformatics tools in understanding the role of non-coding RNA will expand our knowledge of host–viral infections, and in doing so it will provide information on biomolecules that can be targeted to treat viral infections with greater specificity and efficacy than ever before.
Funding
This work was supported by the CI is a recipient of the University of Lethbridge School of Graduate Studies Tuition Award. TRP acknowledges NSERC Discovery [grant number RGPIN-2017-04003]. AZ acknowledges support by the Canada Research Chairs Program and the University of Lethbridge start-up grant programme.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Alkan, C., Sajjadian, S., & Eichler, E. E. (2011). Limitations of next-generation genome sequence assembly. Nature Methods, 8(1), 61–65. doi:10.1038/nmeth.1527
- Ambros, V. (2004). The functions of animal microRNAs. Nature, 431(7006), 350–355. doi:10.1038/nature02871
- Bachellerie, J. P., Cavaille, J., & Huttenhofer, A. (2002). The expanding snoRNA world. Biochimie, 84(8), 775–790.10.1016/S0300-9084(02)01402-5
- Bhattacharyya, S., & Vrati, S. (2015). The Malat1 long non-coding RNA is upregulated by signalling through the PERK axis of unfolded protein response during flavivirus infection. Scientific Reports, 5, 17794. doi:10.1038/srep17794
- Bimber, B. N., Dudley, D. M., Lauck, M., Becker, E. A., Chin, E. N., Lank, S. M., … O’Connor, D. H. (2010). Whole-genome characterization of human and simian immunodeficiency virus intrahost diversity by ultradeep pyrosequencing. Journal of Virology, 84(22), 12087–12092. doi:10.1128/JVI.01378-10
- Buck, A. H., Perot, J., Chisholm, M. A., Kumar, D. S., Tuddenham, L., Cognat, V., … Pfeffer, S. (2010). Post-transcriptional regulation of miR-27 in murine cytomegalovirus infection. RNA, 16(2), 307–315. doi:10.1261/rna.1819210
- Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Huntley, J., Fierer, N., … Knight, R. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. The ISME Journal, 6(8), 1621–1624. doi:10.1038/ismej.2012.8
- Carey, M. F., & Singh, K. (1988). Enhanced B2 transcription in simian virus 40-transformed cells is mediated through the formation of RNA polymerase III transcription complexes on previously inactive genes. Proceedings of the National Academy of Sciences U.S.A., 85(19), 7059–7063.10.1073/pnas.85.19.7059
- Carnero, E., Barriocanal, M., Prior, C., Pablo Unfried, J., Segura, V., Guruceaga, E., … Fortes, P. (2016). Long noncoding RNA EGOT negatively affects the antiviral response and favors HCV replication. EMBO Reports, 17(7), 1013–1028. doi:10.15252/embr.201541763
- Chaisson, M. J., Huddleston, J., Dennis, M. Y., Sudmant, P. H., Malig, M., Hormozdiari, F., … Eichler, E. E. (2015). Resolving the complexity of the human genome using single-molecule sequencing. Nature, 517(7536), 608–611. doi:10.1038/nature13907
- Choy, E. Y. W., Siu, K. L., Kok, K. H., Lung, R. W. M., Tsang, C. M., To, K. F., … Jin, D. Y. (2008). An Epstein-Barr virus-encoded microRNA targets PUMA to promote host cell survival. Journal of Experimental Medicine, 205(11), 2551–2560. doi:10.1084/jem.20072581
- de Koning, A. P., Gu, W., Castoe, T. A., Batzer, M. A., & Pollock, D. D. (2011). Repetitive elements may comprise over two-thirds of the human genome. PLoS Genetics, 7(12), e1002384. doi:10.1371/journal.pgen.1002384
- Deurenberg, R. H., Bathoorn, E., Chlebowicz, M. A., Couto, N., Ferdous, M., Garcia-Cobos, S., … Rossen, J. W. (2017). Application of next generation sequencing in clinical microbiology and infection prevention. Journal of Biotechnology, 243, 16–24. doi:10.1016/j.jbiotec.2016.12.022
- Encode Project Consortium. (2007). Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature, 447(7146), 799–816. doi:10.1038/nature05874EndFragment
- Encode Project Consortium. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature, 489(7414), 57–74. doi:10.1038/nature11247
- Enk, J., Levi, A., Weisblum, Y., Yamin, R., Charpak-Amikam, Y., Wolf, D. G., & Mandelboim, O. (2016). HSV1 MicroRNA modulation of GPI anchoring and downstream immune evasion. Cell Reports, 17(4), 949–956. doi:10.1016/j.celrep.2016.09.077
- Fadrosh, D. W., Ma, B., Gajer, P., Sengamalay, N., Ott, S., Brotman, R. M., & Ravel, J. (2014). An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome, 2(1), 6. doi:10.1186/2049-2618-2-6
- Fakhrai-Rad, H., Pourmand, N., & Ronaghi, M. (2002). Pyrosequencing? An accurate detection platform for single nucleotide polymorphisms. Human Mutation, 19(5), 479–485. doi:10.1002/humu.10078
- Firnkes, M., Pedone, D., Knezevic, J., Doblinger, M., & Rant, U. (2010). Electrically facilitated translocations of proteins through silicon nitride nanopores: Conjoint and competitive action of diffusion, electrophoresis, and electroosmosis. Nano Letters, 10(6), 2162–2167. doi:10.1021/nl100861c
- Fischer, N., Indenbirken, D., Meyer, T., Lutgehetmann, M., Lellek, H., Spohn, M., … Grundhoff, A. (2015). Evaluation of unbiased next-generation sequencing of RNA (RNA-seq) as a diagnostic method in influenza virus-positive respiratory samples. Journal of Clinical Microbiology, 53(7), 2238–2250. doi:10.1128/JCM.02495-14
- Gale, M., & Katze, M. G. (1998). Molecular mechanisms of interferon resistance mediated by viral-directed inhibition of PKR, the interferon-induced protein kinase. Pharmacology & Therapeutics, 78(1), 29–46. doi:10.1016/S0163-7258(97)00165-4
- Hayden, E.C. (2014). Technology: The $1000 genome. Nature, 507(7492), 294–295. doi:10.1038/507294a
- Hoagland, M. B., Stephenson, M. L., Scott, J. F., Hecht, L. I., & Zamecnik, P. C. (1958). A soluble ribonucleic acid intermediate in protein synthesis. The Journal of Biological Chemistry, 231(1), 241–257.
- Holley, R. W., Apgar, J., Everett, G. A., Madison, J. T., Marquisee, M., Merrill, S. H., … Zamir, A. (1965). Structure of a ribonucleic acid. Science, 147(3664), 1462–1465.10.1126/science.147.3664.1462
- Houzet, L., Yeung, M. L., de Lame, V., Desai, D., Smith, S. M., & Jeang, K. T. (2008). MicroRNA profile changes in human immunodeficiency virus type 1 (HIV-1) seropositive individuals. Retrovirology, 5, 118. doi:10.1186/1742-4690-5-118
- Huse, S. M., Huber, J. A., Morrison, H. G., Sogin, M. L., & Welch, D. M. (2007). Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biology, 8(7), R143. doi:10.1186/gb-2007-8-7-r143
- Ingle, H., Kumar, S., Raut, A. A., Mishra, A., Kulkarni, D. D., Kameyama, T., … Kumar, H. (2015). The microRNA miR-485 targets host and influenza virus transcripts to regulate antiviral immunity and restrict viral replication. Science Signaling, 8(406), 126–129. doi:10.1126/scisignal.aab3183
- Jang, K. L., Collins, M. K., & Latchman, D. S. (1992). The human immunodeficiency virus tat protein increases the transcription of human Alu repeated sequences by increasing the activity of the cellular transcription factor TFIIIC. Journal of Acquired Immune Deficiency Syndrome, 5(11), 1142–1147.
- Kambara, H., Niazi, F., Kostadinova, L., Moonka, D. K., Siegel, C. T., Post, A. B., … Valadkhan, S. (2014). Negative regulation of the interferon response by an interferon-induced long non-coding RNA. Nucleic Acids Research, 42(16), 10668–10680. doi:10.1093/nar/gku713
- Karamitros, T., Harrison, I., Piorkowska, R., Katzourakis, A., Magiorkinis, G., & Mbisa, J. L. (2016). De Novo assembly of human herpes virus type 1 (HHV-1) genome, mining of non-canonical structures and detection of novel drug-resistance mutations using short- and long-read next generation sequencing technologies. PLoS ONE, 11(6), e0157600. doi:10.1371/journal.pone.0157600
- Karger, B. L., & Guttman, A. (2009). DNA sequencing by CE. Electrophoresis, 30(Suppl 1), S196–S202. doi:10.1002/elps.200900218
- Khoury, J. D., Tannir, N. M., Williams, M. D., Chen, Y., Yao, H., Zhang, J., … Su, X. (2013). Landscape of DNA virus associations across human malignant cancers: Analysis of 3775 cases using RNA-Seq. Journal of Virology, 87(16), 8916–8926. doi:10.1128/JVI.00340-13
- Kitajewski, J., Schneider, R. J., Safer, B., Munemitsu, S. M., Samuel, C. E., Thimmappaya, B., & Shenk, T. (1986). Adenovirus VAI RNA antagonizes the antiviral action of interferon by preventing activation of the interferon-induced eIF-2 alpha kinase. Cell, 45(2), 195–200.10.1016/0092-8674(86)90383-1
- Kramerov, D. A., & Vassetzky, N. S. (2011). Origin and evolution of SINEs in eukaryotic genomes. Heredity, 107(6), 487–495. doi:10.1038/hdy.2011.43
- Kriegs, J. O., Churakov, G., Jurka, J., Brosius, J., & Schmitz, J. (2007). Evolutionary history of 7SL RNA-derived SINEs in supraprimates. Trends in Genetics, 23(4), 158–161. doi:10.1016/j.tig.2007.02.002
- Lecellier, C. H., Dunoyer, P., Arar, K., Lehmann-Che, J., Eyquem, S., Himber, C., … Voinnet, O. (2005). A cellular microRNA mediates antiviral defense in human cells. Science, 308(5721), 557–560. doi:10.1126/science.1108784
- Legrain, P., Seraphin, B., & Rosbash, M. (1988). Early commitment of yeast pre-mRNA to the spliceosome pathway. Molecular and Cellular Biology, 8(9), 3755–3760.10.1128/MCB.8.9.3755
- Li, R., Zhu, H., Ruan, J., Qian, W., Fang, X., Shi, Z., … Wang, J. (2010). De novo assembly of human genomes with massively parallel short read sequencing. Genome Research, 20(2), 265–272. doi:10.1101/gr.097261.109
- Li, H., Jiang, J. D., & Peng, Z. G. (2016). MicroRNA-mediated interactions between host and hepatitis C virus. World Journal of Gastroenterology, 22(4), 1487–1496. doi:10.3748/wjg.v22.i4.1487
- Lu, H., Giordano, F., & Ning, Z. (2016). Oxford nanopore MinION sequencing and genome assembly. Genomics Proteomics Bioinformatics, 14(5), 265–279. doi:10.1016/j.gpb.2016.05.004
- Ma, F., Xu, S., Liu, X., Zhang, Q., Xu, X., Liu, M., … Cao, X. (2011). The microRNA miR-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon-gamma. Nature Immunology, 12(9), 861–869. doi:10.1038/ni.2073
- Ma, L., Bajic, V. B., & Zhang, Z. (2013). On the classification of long non-coding RNAs. RNA Biology, 10(6), 925–933. doi:10.4161/rna.24604
- Nissen, P., Hansen, J., Ban, N., Moore, P. B., & Steitz, T. A. (2000). The structural basis of ribosome activity in peptide bond synthesis. Science, 289(5481), 920–930.10.1126/science.289.5481.920
- Ozsolak, F., & Milos, P. M. (2011). RNA sequencing: Advances, challenges and opportunities. Nature Reviews Genetics, 12(2), 87–98. doi:10.1038/nrg2934
- Palazzo, A. F., & Gregory, T. R. (2014). The case for junk DNA. PLoS Genetics, 10(5), e1004351. doi:10.1371/journal.pgen.1004351
- Palazzo, A. F., & Lee, E. S. (2015). Non-coding RNA: What is functional and what is junk? Frontiers in Genetics, 6(2), doi:10.3389/fgene.2015.00002
- Panning, B., & Smiley, J. R. (1993). Activation of RNA polymerase III transcription of human Alu repetitive elements by adenovirus type 5: Requirement for the E1b 58-kilodalton protein and the products of E4 open reading frames 3 and 6. Molecular and Cellular Biology, 13(6), 3231–3244.10.1128/MCB.13.6.3231
- Panning, B., & Smiley, J. R. (1994). Activation of RNA polymerase III transcription of human Alu elements by herpes simplex virus. Virology, 202(1), 408–417. doi:10.1006/viro.1994.1357
- Panwar, B., Omenn, G. S., & Guan, Y. (2017). miRmine: A database of human miRNA expression profiles. Bioinformatics, 33(10), 1554–1560. doi:10.1093/bioinformatics/btx019
- Pedersen, I. M., Cheng, G., Wieland, S., Volinia, S., Croce, C. M., Chisari, F. V., & David, M. (2007). Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature, 449(7164), 919–922. doi:10.1038/nature06205
- Piskurek, O., & Jackson, D. J. (2012). Transposable elements: From DNA parasites to architects of metazoan evolution. Genes, 3(3), 409–422. doi:10.3390/genes3030409
- Ponicsan, S. L., Houel, S., Old, W. M., Ahn, N. G., Goodrich, J. A., & Kugel, J. F. (2013). The non-coding B2 RNA binds to the DNA cleft and active-site region of RNA polymerase II. Journal of Molecular Biology, 425(19), 3625–3638. doi:10.1016/j.jmb.2013.01.035
- Qian, X., Xu, C., Zhao, P., & Qi, Z. (2016). Long non-coding RNA GAS5 inhibited hepatitis C virus replication by binding viral NS3 protein. Virology, 492, 155–165. doi:10.1016/j.virol.2016.02.020
- Radford, A. D., Chapman, D., Dixon, L., Chantrey, J., Darby, A. C., & Hall, N. (2012). Application of next-generation sequencing technologies in virology. Journal of General Virology, 93(Pt 9), 1853–1868. doi:10.1099/vir.0.043182-0
- Russanova, V. R., Driscoll, C. T., & Howard, B. H. (1995). Adenovirus type 2 preferentially stimulates polymerase III transcription of Alu elements by relieving repression: A potential role for chromatin. Molecular and Cellular Biology, 15(8), 4282–4290.10.1128/MCB.15.8.4282
- Salas-Solano, O., Carrilho, E., Kotler, L., Miller, A. W., Goetzinger, W., Sosic, Z., & Karger, B. L. (1998). Routine DNA sequencing of 1000 bases in less than one hour by capillary electrophoresis with replaceable linear polyacrylamide solutions. Analytical Chemistry, 70(19), 3996–4003.10.1021/ac980457f
- Samanta, M., Iwakiri, D., Kanda, T., Imaizumi, T., & Takada, K. (2006). EB virus-encoded RNAs are recognized by RIG-I and activate signaling to induce type I IFN. EMBO Journal, 25(18), 4207–4214. doi:10.1038/sj.emboj.7601314
- Schatz, M. C., Delcher, A. L., & Salzberg, S. L. (2010). Assembly of large genomes using second-generation sequencing. Genome Research, 20(9), 1165–1173. doi:10.1101/gr.101360.109
- Singh, K., Carey, M., Saragosti, S., & Botchan, M. (1985). Expression of enhanced levels of small RNA polymerase III transcripts encoded by the B2 repeats in simian virus 40-transformed mouse cells. Nature, 314(6011), 553–556.10.1038/314553a0
- Starega-Roslan, J., Krol, J., Koscianska, E., Kozlowski, P., Szlachcic, W. J., Sobczak, K., & Krzyzosiak, W. J. (2011). Structural basis of microRNA length variety. Nucleic Acids Research, 39(1), 257–268. doi:10.1093/nar/gkq727
- Stern-Ginossar, N., Elefant, N., Zimmermann, A., Wolf, D. G., Saleh, N., Biton, M., … Mandelboim, O. (2007). Host immune system gene targeting by a viral miRNA. Science, 317(5836), 376–381. doi:10.1126/science.1140956
- Stoddart, D., Heron, A. J., Mikhailova, E., Maglia, G., & Bayley, H. (2009). Single-nucleotide discrimination in immobilized DNA oligonucleotides with a biological nanopore. Proceedings of the National Academy of Sciences, 106(19), 7702–7707. doi:10.1073/pnas.0901054106
- Trypsteen, W., Mohammadi, P., Van Hecke, C., Mestdagh, P., Lefever, S., Saeys, Y., … De Spiegelaere, W. (2017). Corrigendum: Differential expression of lncRNAs during the HIV replication cycle: An underestimated layer in the HIV-host interplay. Scientific Reports, 7, 41112. doi:10.1038/srep41112
- Tycowski, K. T., Guo, Y. E., Lee, N., Moss, W. N., Vallery, T. K., Xie, M., & Steitz, J. A. (2015). Viral noncoding RNAs: More surprises. Genes & Development, 29(6), 567–584. doi:10.1101/gad.259077.115
- Viollet, C., Davis, D. A., Reczko, M., Ziegelbauer, J. M., Pezzella, F., Ragoussis, J., & Yarchoan, R. (2015). Next-generation sequencing analysis reveals differential expression profiles of MiRNA-mRNA target pairs in KSHV-infected cells. PLoS ONE, 10(5), e0126439. doi:10.1371/journal.pone.0126439
- Walters, R. D., Kugel, J. F., & Goodrich, J. A. (2009). InvAluable junk: The cellular impact and function of Alu and B2 RNAs. IUBMB Life, 61(8), 831–837. doi:10.1002/iub.227
- Williams, W. P., Tamburic, L., & Astell, C. R. (2004). Increased levels of B1 and B2 SINE transcripts in mouse fibroblast cells due to minute virus of mice infection. Virology, 327(2), 233–241. doi:10.1016/j.virol.2004.06.040
- Yakovchuk, P., Goodrich, J. A., & Kugel, J. F. (2009). B2 RNA and Alu RNA repress transcription by disrupting contacts between RNA polymerase II and promoter DNA within assembled complexes. Proceedings of the National Academy of Sciences, 106(14), 5569–5574. doi:10.1073/pnas.0810738106
- Yeung, M. L., Benkirane, M., & Jeang, K. T. (2007). Small non-coding RNAs, mammalian cells, and viruses: Regulatory interactions? Retrovirology, 4, 74. doi:10.1186/1742-4690-4-74
- Zovoilis, A., Cifuentes-Rojas, C., Chu, H. P., Hernandez, A. J., & Lee, J. T. (2016). Destabilization of B2 RNA by EZH2 activates the stress response. Cell, 167(7), 1788–1802.e1713. doi:10.1016/j.cell.2016.11.041