Abstract
This article covers the latest activities in mycotoxin analysis and the advances of its respective quality assurance. The majority of mycotoxin analyses carried out in the laboratories is still based on physicochemical methods, which are continually improved. For example, immunoaffinity columns and multifunctional clean–up columns have become of increasing importance and in some areas of mycotoxin analysis they have more or less displaced conventional liquid–liquid partitioning or column chromatography during clean–up. The need for rapid yes/no decisions on the other hand has led to a number of new screening methods. In particular, rapid and easy–to–use test kits based on immunoanalytical principles or the generation of artificial macromolecular receptors employed in molecularly imprinted polymers (MIPs) have made good progress. Further research in mycotoxin analysis is pursued in the field of biosensors and also the potential of infrared spectroscopic techniques as screening method has been demonstrated. In the area of multi mycotoxin analysis the most promising development was observed in mass spectrometry. At the same time, several interlaboratory studies in the field of mycotoxin analysis revealed problems proven by high between laboratory standard deviation and non–traceable results. This not only shows the necessity of reliable methods and well defined performance characteristics but also the need for appropriate calibrants of defined concentration and stated purity. A certified zearalenone (ZON) calibrant is already available and a certified calibrant containing various trichothecenes is currently under development. (Certified) reference materials are available for aflatoxins in a number of commodities, ochratoxin A (OTA) in wheat, deoxynivalenol (DON) in maize and wheat, and ZON in maize. With these measures important steps towards traceability of results in mycotoxin analysis have been achieved.
Introduction
General requirements for methods in mycotoxin analysis
All methods in mycotoxin analysis include sampling. Due to the high heterogeneity of mycotoxins in grains, sampling plans are usually followed to obtain representative samples. Heterogeneity of mycotoxins may vary strongly with the commodity and hence the kernel count, which is the amount of kernels per gram (JECFA Citation2001). Sampling plans for regulatory purposes have been designed for few mycotoxins only. A European Commission (EC) Directive, for example, specifies the sampling plan for the determination of the aflatoxins in foods (edible nuts and dried fruit) (European Commission Citation1998). However, there is still a need for harmonized and effective sampling plans especially in the area of surveillance and even more so in the area of Fusarium mycotoxins. It is more than likely that the analyst will receive a laboratory sample, which will be ground using a small laboratory mill. Only exceptional methods such as infrared spectroscopic techniques are able to draw data from the ground and homogenized sample (Kos et al. Citation2003). In all other cases, the toxins will be extracted from 20–100 g sample with mostly mixtures of water and relatively polar solvents (methanol, acetone, ethylacetate, etc.) according to the solubility of the target analyte (Cole and Cox Citation1981). The diluted extracts may be used for methods based on immunoanalytical principles (Barna-Vetro et al. Citation1997; Märtlbauer et al. Citation1991) but further clean-up is necessary for chromatographic separation. Liquid-liquid partitioning, solid-phase extraction (SPE), column chromatography, immunoaffinity columns (IAC), and multifunctional clean-up columns can be used for the purification of extracts in mycotoxin analysis. In some cases it is necessary to remove interfering lipids by extraction of the sample extract with n-hexane. The solvent is removed after clean-up and the sample is reconstituted in a solvent suitable for chromatographic analysis. A method of analysis for mycotoxins in grain should be simple, rapid, robust, accurate and selective to enable simultaneous determination. Above all, low tolerance levels in feed and food require sensitive methods. In the end, the analytical result has to be fit for purpose and the method has to be chosen accordingly. High performance liquid chromatography (HPLC) has become the workhorse of mycotoxin analysis. Coupled with a variety of detectors practically all mycotoxins have been separated and detected by HPLC. Fumonisins (Shephard Citation1998), aflatoxins (Coker Citation2000), zearalenone (ZON), and ochratoxin A (OTA) are routinely analysed by HPLC. Although there are robust and validated methods for the analysis of deoxynivalenol (DON) in cereals by HPLC (Trucksess et al. Citation1998), gas chromatographic (GC) methods with electron capture detection (ECD) (Scott et al. Citation1986) or sometimes even mass spectrometry (MS) are preferred (Krska et al. Citation2001). Screening methods are mostly based on thin layer chromatography, which is a very effective and simple technique. Nowadays, ELISAs (enzyme linked immunosorbent assays) have also found widespread use in mycotoxin determination. Test kits are available for practically all relevant mycotoxins. Mycotoxin test kit data bases are compiled on the homepages of AOAC International (www.aoac.org) and of the European Mycotoxin Awareness Network (www.mycotoxins.org).
Activities in mycotoxin research
Together with the sampling step, the variation associated with the sample clean-up makes up most of the uncertainty of a method. The analyst usually has no influence on the sampling, but to keep variation low during sample preparation, the clean-up is intended to be carried out in as few steps as possible. Formerly, liquid-liquid partitioning was employed to remove unwanted matrix components in the sample extract. This procedure, however, uses vast amounts of solvent, leads to losses and is time-consuming. Therefore, extracts containing mycotoxins were first preferably purified using column chromatography and are now purified by means of modern prepacked SPE columns. A variety of stationary phases (silica gel, aluminum oxide, Florisil, charcoal, and C8 or C18 revered-phases) or a mixture thereof have been used and shown their great potential. The aqueous sample extract is applied to the conditioned column and the analyte is trapped on the column. After a rinsing step to remove matrix compounds which are also trapped on the column, the analyte is eluted from the column with an organic solvent. For further concentration the solvent can be evaporated. Another variety of clean-up columns has established its place in mycotoxin analysis: rapid multifunctional MycoSep® columns have been especially developed for the clean-up of sample extracts. They contain a variety of adsorbents, e.g., charcoal, celite, polymers, ion-exchange resins among others. The material is packed in a plastic tube between filter discs with a rubber flange on the lower end containing a porous frit and a one-way valve. When the column is inserted into the tube containing the extract the flange seals tightly and pushes the extract through the packing material of the column. Within 20–30 seconds the purified trichothecene can be obtained on top of the plastic tube. Large molecules, proteins, fats, carbohydrates, and pigments are all adsorbed on the solid phase. Currently, MycoSep® columns are among the most frequently employed commercial columns for clean-up of DON and other A- and B-trichothecenes. Recoveries >80% can be achieved for most trichothecenes (Weingärtner et al. Citation1997) combining this sample clean-up with HPLC or GC methods. Only recently a new MycoSep® 229 Ochra column for the determination of OTA in wheat and other food stuffs has been added to the range of these clean-up columns (Buttinger et al. Citation2003). Immunoaffinity columns (IAC), too, have established their place in mycotoxin analysis and are increasingly being used by control laboratories. Analysis of ZON, for example, cannot be imagined without the use of IAC. There are also commercially available columns for the aflatoxins, fumonisins, type-A and -B trichothecenes such as DON, and OTA. Due to the enormous workload connected with the development of official methods, such as the organization of a collaborative study, official methods often lag behind the state-of-the-art. So while being state-of-the-art, IAC and multifunctional column clean-up are not included in most official methods.
Not only clean-up methods are continuously optimized, but also instrumental methods. An EC-funded project within the framework of the SMT-programme was organized by (Pettersson & Langseth Citation2002a, Citation2002b). In particular, gas chromatographic methods for nivalenol, DON, HT-2, and T-2 toxin were assessed. Several method problems were identified in that study:
| 1. | Higher trichothecene response for calibrants in presence of matrix than for pure calibrants; | ||||
| 2. | Non-linear calibration curves; | ||||
| 3. | Drifting response for trichothecenes; | ||||
| 4. | Carry-over or memory effects from previous samples; | ||||
| 5. | Matrix interference. | ||||
These problems were observed in different laboratories with different methods and type of equipment. Based on the results, strategies were sought to reduce or eliminate the identified method problems. At the end of the study, the following recommendations were made: Regular change to new deactivated liners in splitless injection, the use of matrix-matched calibration, and the use of internal GC standards, to name a few. Although good progress has been made, analysis of trichothecenes by GC is still liable to high variations in reproducibility and repeatability. On the other hand, in contrast to HPLC-UV-methods, GC-ECD enables the determination of several trichothecenes even in complex food matrices in the lower µg/kg range. The only official method for the determination of DON in wheat at levels ≥350 ng g−1 dates back to 1986 (first action) (AOAC Citation2002) where the equipment used is not state-of-the-art any more.
Yet there is a great need for development and standardization of an appropriate analytical method for DON and other trichothecenes which eliminates sources of variation. In its recent (14th) meeting, the task group “mycotoxins” of Working Group 5 of the European Committee (EC) for Standardization (CEN) has again postponed the publication of an official CEN method for DON as there is currently no method which is sufficiently robust for standardization. CEN, as the standardization body of the EC evaluates the performance criteria of a method, which are usually based on collaborative studies. Most of the CEN methods are also AOAC approved. Standardized methods for aflatoxins (EN 12955 Citation1999; EN 14123 Citation2001), OTA (EN 14132 Citation2003), fumonisins (EN 13585 Citation2001, EN 14352 Citation2004), and patulin (EN 14177 Citation2003) in various foods are available, and methods of analysis for trichothecenes in food and various other mycotoxins in feed are planned in the foreseeable future. A full compilation of official methods of analysis for mycotoxins can be found in (Gilbert and Anklam Citation2002).
New developments were observed in the analysis of moniliformin (MON). This highly toxic fungal metabolite is reported to occur worldwide in cereals. Yet, no regulatory limits and trade specifications have been established so far for this mycotoxin. To assess the occurrence of MON in cereals on a widespread scale, rapid, reliable, low-cost and sensitive methods of analysis are important. Most analytical methods for the determination of MON use C18 (Scott and Lawrence Citation1987), extrelut® (Lew et al. Citation1993), or a combination of reverse phase and SAX material (Sharman et al. Citation1991) to purify the sample extract followed or preceded by ion pair formation of the toxin. Recently, this method for MON in maize and wheat has been optimized using a clean-up with SAX column and subsequent PR-ion pair-HPLC separation and UV detection (Parich et al. Citation2003) and employed for surveys of MON in Norwegian grain (Uhlig et al. Citation2003). The method has an LOD of 39 µg kg-1 and recovery rates of 76 ± 9% for maize samples. A newly developed method for the determination of MON by ion chromatography shows an LOD of only 0.12 mg kg−1, but a recovery as high as 97 ± 3.5% (Kandler et al. Citation2002). In contrast to the two previously published ion chromatographic methods (Thiel et al. Citation1982; Thiel Citation1990), the MON peak is base line separated. An additional advantage of this method is the use of ultrapure water as extraction solvent only, and the simple clean-up using cation-exchanging resins and C18 cartridges. (Nadubinská et al. Citation2002) successfully employed this method for evaluating the contents of MON in maize ears from Slovakia.
Strong anion exchange clean-up of solvent extract was also commonly employed for the determination of fumonisins before analysis by HPLC-FLD. Several IUPAC-, AOAC- and SMT-collaborative studies have been carried out since 1993 in that field. Varying recoveries between 60 and 86% marked the performance of the proposed methods (Visconti et al. Citation1996). Immunoaffinity column clean-up with HPLC and FLD was finally studied collaboratively in an SMT-project and approved and standardised by CEN (Visconti et al. Citation2001).
Physicochemical analytical methods are usually intended to determine more than one single mycotoxin. However, mycotoxins constitute a structurally very inhomogeneous group. Hence, analytical procedures usually differ in extraction, clean up and end determination steps, depending on which group of mycotoxin is to be analysed. This problem is even enhanced, when the simultaneous analysis of several mycotoxins of different groups is attempted. First, methods using thin layer chromatographical techniques (Balzer et al. Citation1978; Johann and Dose Citation1983; Rottinghaus et al. Citation1982), and then GC-MS (Tanaka et al. Citation2000) and GC-ECD (Luo et al. Citation1992) methods have been developed for the simultaneous analysis of up to 15 mycotoxins in grains and other feed stuff. The clean-up procedure is the most challenging task of a multianalyte method. As for the chromatographic separation and detection steps, the method of Frisvad and Thrane which dates back to 1987 (Frisvad and Thrane Citation1987) is regarded as pioneer work in the area of multi-toxin analysis. The authors developed an HPLC-UV method for the analysis of 182 structurally different mycotoxins. The method uses a Nucleosil RP-18 HPLC column for a gradient elution with a mixture of acetonitrile and 0.05% trifluoroacetic acid in water as mobile phase.
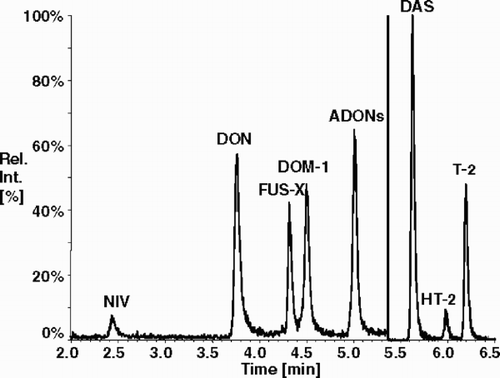
Today, the most promising development in the area of multi mycotoxin analysis can be observed in the area of mass spectrometry. HPLC-MS-(MS) has great potential for the simultaneous detection of various mycotoxins and their degradation products (Fuchs et al. Citation2002). HPLC-MS instruments, particularly using APCI (atmospheric pressure chemical ionisation) interfaces have been employed for the determination and identification of trichothecenes, including DON, at trace levels (Razzazi-Fazeli et al. Citation1999). Recently, Rundberget and Wilkins (Citation2002) developed and validated a method for the determination of Penicillium mycotoxins in food and feeds. Tandem MS with electro spray ionization (ESI) was successfully employed by (Driffield et al. Citation2003). The presented method was capable to simultaneously determine NIV, DON, ZON, OTA, and aflatoxin B1 in pig livers. HPLC-ion trap methods for the quantitative determination and structure elucidation of nine A- and B-trichothecenes was also proposed (Berger et al. Citation1999). shows the simultaneous determination of the A- and B-trichothecenes NIV, DON, Fusarenon-X, 3- acetyl-DON, 15-acetyl-DON, diacetoxyscirpenol (DAS), HT-2 and T-2 by HPLC–APCI–MS/MS in negative and positive ion modes at 450°C using deepoxy-deoxynivalenol (DOM-1) (Freudenschuss Citation2003) as internal standard. While a single HPLC peak was obtained for the sum of both 3- and 15-acetyl-DON, different fragmentation behavior also allows the separate determination and quantification of the individual mycotoxins, albeit at a lower sensitivity.
Figure 1. Total ion chromatogram of a spiked maize sample after clean-up with MycoSep® #227 column and subsequent gradient RP-HPLC with atmospheric pressure chemical ionization triple quadrupole mass spectrometry (HPLC–APCI–MS/MS) in multiple reaction monitoring (MRM) mode. Concentration of nivalenol (NIV), DON, Fusarenon-X (FUS-X), 3 Acetyl-DON + 15-acetyl-DON (= ADONs), Diacetoxyscirpenol (DAS), HT-2 and T-2 were determined at concentrations of 100 µg/kg each. DOM-1 was used as internal standard.
Developing rapid and innovative methods
Antibody-based approaches
The microtiterplate immunoassay (ELISA format) is one of the most frequently used rapid tests for mycotoxins. ELISAs are commercially available for many important mycotoxins (Aflatoxins, fumonisins, trichothecenes, ZON, OTA, citrinin). They are useful tools for screening and quantification and offer benefits with respect to speed and sensitivity. Drawbacks of these methods are cross reactivity and matrix dependence which often results in tremendous over-estimation (Schuhmacher et al. Citation1997; Josephs et al. Citation2001). Besides the common ELISA procedures there is an increasing demand for immunoassay techniques for field use offering protocols for quick and reliable results. One of these diagnostic tools is the dip-stick immunoassay, resembling ELISA constructs: Instead of microtiter plates, carrier membranes (usually polyvinylidene difluoride (PVDF), nylon and nitro-cellulose (NC)) are used to immobilize either the antibody or the antigen. Depending on the test format, one to three working steps have to be performed, requiring a total of about 30 min to 3 hours for obtaining the test results. Qualitative and semi-quantitative results can also be obtained by lesser experienced personnel. Multi-analyte dipstick immunoassays for various mycotoxins have been developed, however, with limited sensitivity (Schneider et al. Citation1995). Another interesting alternative rapid assay format for the detection of mycotoxins are lateral flow devices. These one-step tests were originally developed for the detection of large molecules in clinical analysis and take only a couple of minutes (2–3 min) to perform. The price of simplification and acceleration of antibody based test systems is a huge loss of sensitivity, therefore, extremely good antibodies are required. Although currently a lot of academic research can be observed towards the development of novel immunoanalytic tools for the determination of mycotoxins, none are suitable for widespread use. Additionally, they are not good enough for end product control of food at ∼100 µg/kg but may be useful for pre- and post-harvest control.
Other alternatives are immunoassays which are capable of antigen-antibody binding, but without any enzymatic labelling for the detection. These immunological biosensors comprise three components: An antibody of appropriate specificity for the analyte, a transducer to convert the recognition event into an acoustic, electrical, mechanical or optical signal and a detection, analysis, processing and display system. Amperometric sensors, potentiometric sensors, light-addressable potentiometric sensors, fluorescence evanescent-wave sensors or surface plasmon resonance (SPR) sensors are currently used for biosensing. The latter uses an electron charge density wave phenomenon that arises at the surface of a metallic film when light is reflected at the film under specific conditions. The resonance is a result of energy and momentum being transformed from incident photons into surface plasmons, and the resonance is sensitive to the refractive index of the medium on the opposite side of the film from the reflected light. Briefly, mycotoxin binds to immobilized antibodies on a gold film surface. The antigen-antibody interaction induces a change in the refractive index, which can be measured. The SPR technology for the monitoring of such biomolecular interactions has been used for several mycotoxins (Maragos Citation2002; Tudos et al. Citation2003; Schnerr et al. Citation2002). Yet cost is still high and reusability not optimized.
Molecular imprinted polymers
Mimicking antibodies is the basic idea of yet another technique, molecularly imprinted polymers (MIPs). MIPs provide an innovative synthetic receptor alternative to biological recognition elements applicable for sample clean-up, as selective separation matrix, and SPE extraction and enrichment material. During molecular imprinting, cross-linked polymers are formed by free-radical co-polymerization of functional monomers and cross-linker in the presence of an analyte serving as template. Hence, the formation of a mechanically and thermally stable polymer with synthetic receptor sites for template molecule recognition is enabled. After polymerization, the template is removed by liquid extraction (washing). Ideally, highly selective three-dimensional binding pockets complementary in size, shape, and functionality to the imprinted molecule remain in the polymer matrix. In the pre-polymerization mixture, the dissolved target analyte interacts by covalent, non-covalent, or metal coordination interactions with the functional monomer. This process is responsible for localizing the functional moieties of the target molecules during co-polymerization. It has been demonstrated that MIPs provide biomimetic recognition elements based on synthetic receptor sites capable of selective target binding/rebinding with comparable efficiency to substrate-enzyme or antibody-antigen interactions (Weiss et al. Citation2002). Imprinted polymers have successfully been used in a variety of analytical applications ranging from binding assays to chemical sensing (Molinelli et al. Citation2002).
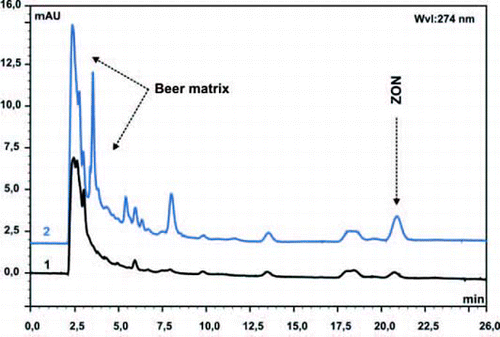
Solid phase extraction (SPE) based on molecularly imprinted polymers is an intriguing concept for molecule specific sample preparation and pre-concentration gaining increased interest in the fields of environmental, clinical and food/beverage analysis. Synthetic receptors for the mycotoxins OTA (Jodlbauer et al. Citation2002), DON and ZON (Weiss et al. Citation2003) have been reported for use in solid-phase extraction using the non-covalent self-assembly imprinting approach. In particular, approaches directly using DON and ZON as templates, and using the flavonoid quercetin as dummy template were investigated. MIPs were synthesized using 4-vinylpyridine, methacrylic acid or trifluoromethacrylic acid as functional monomers in conjunction with cross-linkers such as ethyleneglycol dimethacrylate or divinylbenzene in acetonitrile. The generated MIPs were characterized during chromatographic applications using the generated MIP as stationary phase confirming selectivity for the imprinted mycotoxins. Compared to a control column using non-imprinted materials, DON showed substantially higher retention times than the structural analogues 3-acetyl-DON, 15-acetyl-DON and fusarenon-X on the DON-imprinted column. Especially for rare and expensive templates such as DON and ZON, careful consideration of the imprinting procedure is required. ‘Dummy imprinting’ is a viable strategy enabling to avoid this problem using a structural analogue of the target analyte as template during the imprinting process. Due to related molecular sub-structures, a MIP SPE column based on a ‘dummy imprint’ with the flavonoid compound quercetin enabled selective enrichment of ZON from spiked beer samples as shown in with recovery rates >80%.
Figure 2. RP-HPLC-chromatograms of 1 ml aliquots after solid phase extraction of a beer sample (Coors Original) spiked with 1 µg/ml ZON. SPE with 100 mg cartridges of (1) control polymer and (2) quercetin imprinted polymer. 10 ml of beer was applied directly onto the SPE cartridges. Elution was performed with methanol containing 15% acetic acid. The Mobile phase: acetonitrile/water (1:1, v/v) with 0.1% acetic acid at a flow rate of 0.8 ml/min.
These preliminary results promise future applications of MIPs as highly selective matrix for separation and enrichment of mycotoxins from natural sources such as grain extracts and processed beverages such as beer, offering a viable alternative to classical sample clean-up procedures. However, the implementation of MIPs during large scale real-world applications still remains a big challenge.
Spectroscopic screening methods
A novel method based on mid (MIR)-infrared/attenuated total reflection (ATR), which enables the determination of fungal infection with Fusarium graminearum on corn within minutes has recently been developed (Kos et al. Citation2003). The sample was pressed onto the ATR crystal and reproducible pressure was applied. After the spectra were recorded, they were subjected to Principal Component Analysis (PCA) and classified using Cluster Analysis. Observed changes in the spectra reflected changes in protein, carbohydrate and lipid contents. DON served as reference parameter. The repeatability was highly improved by sieving prior to recording the spectra resulting in a better clustering in PCA score/score plots. The developed method enabled the separation of samples with a DON content as low as 310 µg/kg from non-contaminated (blank) samples. The investigated concentration range was 310–2.596 µg/kg for DON. The percentage of correctly classified samples was up to 100% for individual samples compared with a number of blank samples.
A Danish-Swedish project started in the year 2000 to investigate the potential of near-infrared transmittance (NIT) spectroscopy for the rapid estimation of deoxynivalenol (DON) in whole kernel barley and wheat (Pettersson and Aberg Citation2002, Aberg Citation2002). All spectra (570–1100 nm) were collected using the FOSS Infratec 1241 Grain Analyzer with the added color module, and reference analysis using HPLC or GC methods. Independent validation data showed best performance using artificial neural network (ANN) for calibration. Based on current findings, it appears that NIT could also be used as a screening tool to measure DON in cereals. For both, the MIR and the NIR spectrocopic approach further work on calibration development and the expansion of the calibration databases will reveal the true potential of IR-spectrocopy for this kind of application.
Quality assurance in analysis of Fusarium mycotoxins
Interlaboratory validations
Intercomparison studies play an important role for the quality of analytical data in regard to trueness and comparability. Whereas collaboratively studied methods of analysis for aflatoxins have been an issue already in the late 1970s (Schuller et al. Citation1976) and considerable efforts were made to improve the quality of analysis and quality assurance since then, collaborative efforts in the field of Fusarium mycotoxins are of more recent date. Particularly in Europe, several intercomparison studies were performed within projects commissioned by the EC in the last ten years. Extensive studies for the determination of the fumonisins for example resulted in long-lasting improvements of the quality of fumonisins analysis at European level (Visconti et al. Citation1995, Citation1996). In 1998, 14 European laboratories participated in an intercomparison study of trichothecene analysis (Pettersson and Langseth Citation2002a, Citation2002b). This study revealed alarming relative standard deviations (RSD) of up to 60%. In the course of the project, the main method problems were identified and recommendations were given for further trichothecene analysis. In 1996, (Schuhmacher et al. 1998) and 1998 (Krska 2000), two intercomparison studies with 28 participants were organized. In the first interlaboratory comparison in 1996, satisfactory RSDs of 15.0–27.7% for two spiked maize materials and 16.6% for naturally ZON contaminated maize were obtained. The good precision and accuracy were mainly attributed to the use of common ZON calibrants provided with the samples. The second intercomparison study of 1998 was carried out without the provision of a common ZON calibrant. In-house ZON calibrants were used by the participants and the interlaboratory RSDs rose to 27.7% for spiked maize and 40.5–41.2% for two naturally-ZON contaminated maize samples. These results indicate that a main problem in ZON analysis can be found in the use of in-house calibrants. In order to minimize this problem and therefore improve the quality and comparability of ZON analysis, a large-scale Standards, Measurement, and Testing (SMT) project of the European Commission (EC) dealing with the preparation and certification of a reference material for the determination of the mycotoxin ZON in maize was initiated.
The objective of the four-year project “ZONMAIZE” was the preparation and certification of both a blank maize material (<5 µg/kg ZON in maize) and a maize material naturally contaminated with ZON (c = 40–120 µg/kg ZON in maize) to be used as reference samples for the determination of ZON in maize. Within this project, also a ZON calibrant in acetonitrile for external calibration was produced and checked for purity, homogeneity, and stability (Krska et al. Citation2003). All materials are now available from the Institute for Reference Materials and Measurements of the Joint Research Center in Geel, Belgium (JRC-IRMM Geel). Two preliminary intercomparison studies were performed in the said project and in the final certification exercise an excellent RSD of 6% for the naturally ZON contaminated maize with a mean of 82.8 µg kg−1 could be achieved by the 25 participants who used different extraction-, clean-up- and detection methods. Similar improvements were observed for DON intercomparison studies (Josephs et al. Citation2001; Pettersson and Langseth Citation2002b; Schuhmacher et al. Citation1997), where a better agreement of the individual results (RSD = 19% instead of 32%) could be obtained after issuing a common calibrant.
Calibrants and reference materials
The good result achieved in the ZONMAIZE project also partly results from the thorough characterization of the calibrant (Josephs et al. Citation2003). Calibrants of defined concentrations are the basis of almost all analytical methods. Purity of pure mycotoxins is often claimed to be high (> 95%), but no information is given about bound solvent or water. Commercially available crystalline ZON was analysed (Krska et al. Citation2003) and a purity of 99.5% with a tolerance of ± 0.5% was finally attributed to the studied ZON. The concentration of calibrants should be checked by spectrophotometry. Different values for molar absorptivities in several solvents can be found in the literature. For spectrophotometric determination of A- and B-trichothtecenes, methanol is unsuitable for UV-determination due to the rather high absorption below λ = 220 nm. Therefore, the spectroscopic determination of type-A and -B trichothecenes in acetonitrile is recommended by Pettersson and Langseth (Citation2002a) as another result of the SMT project, which had enormous impact on the analysis of trichothecenes.
To check the purity of trichothecene calibrants and to develop a procedure for checking the purity was also within the scope of the project. Currently, the development of the ability to produce and certify calibrants for their concentrations of DON, 3-Ac-DON, 15-Ac-DON and NIV related to the monitoring of these mycotoxins in cereals, food and feed is the objective of “DONCALIBRANT”, a project within the 5th framework programme of the EU (G6RD-CT-2002-00853). This feasibility study determines the conditions and processes that are necessary to produce and certify solutions of DON and other relevant B-trichothecenes to be used as certified calibrants. The experience gained during an associated intercomparison study can be exploited directly by the participating laboratories to improve their overall performance of the determination of DON, 3-Ac-DON, 15-Ac-DON and NIV and should lead to improved comparability of measurement results for these toxins at a European level.
One of the latest results of this project stems from the elemental analysis of NIV. The required amount of carbons and hydrocarbons was not found, but exactly matched amounts for NIV-monohydrate. The difference in molecular weight between NIV and NIV-monohydrate is 5.4%. Consequently, 10 mg NIV-monohydrate contains only 9.46 mg NIV. Calibration with such insufficiently characterized calibrants results in systematic errors that could in future be avoided through a regular check by means of well characterized and in the course certified calibrants of these toxins. The availability of suitable CRMs for control purposes during analytical work is also a well-known need of many researchers, in particular when working in fields of health and consumer protection. They enable the traceability of results to appropriate measurement standards and comparability of data. To improve the quality of data, many laboratory managers rely on the use of certified reference materials as important elements in the quality assurance system of laboratories analysing mycotoxins. An overview of currently available (C)RMs and those in production can be found on the CORDIS-Homepage, the homepage of the IRMM (www.irmm.jrc.be), the IAEA (www.iaea.org), and the NIST (http://patapsco.nist.gov/srmcatalog/about/proram_info.htm) but also via the international data base for CRMs, COMAR (www.comar.bam.de), which compiles the most extensive catalogues of available RMs worldwide (www, accessed 2003). Many CRMs have become available in the past, but production costs are high.
Conclusions
In conclusion, there is a great need for sampling plans for mycotoxins other than aflatoxins for surveying purposes. There is a continuing trend towards quick and reliable methods giving rapid yes/no decisions or semi-quantitative results. Immunoassays which deliver quantitative or semi-quantitative results still represent the most frequently used rapid method. However, easy-to-use-methods are often either too expensive or show a lack of sensitivity. SPE and IAC-clean-up will become of increasing importance as sample preparation techniques before instrumental analysis. The great potential of LC-MS/MS for screening large amounts of samples for the presence of a number of mycotoxins has recently been demonstrated.
Related Research Data
References
References
- Åberg L Andren H Persson J-Å Pettersson H 2002 Rapid analysis of mycotoxins in grain with NIT. ICC Conference Budapest. May 2002
- AOAC 2002 In: Horwitz W, editor. Official methods of analysis of the association of official analytical chemists 17th ed. Current through revision Arlington, USA: Method Nr. 986;18
- Balzer , I , Bogdanic , C and Pepejnjak , S . 1978 . Rapid thin layer chromatographic method for determining aflatoxin B1, ochratoxin A, and zearalenone in corn . Journal of the Association of Official Analytical Chemists , 61 : 584 – 585 .
- Barna-Vetró , I , Gyöngyösi , À and Solti , L . 1997 . Immunodiagnostics for mycotoxin determination . Hungarian Agricultural Research , 2 : 16 – 19 .
- Berger , U , Oehme , M and Kuhn , F . 1999 . Quantitative determination and structural elucidation of type A- and B-trichothecenes by HPLC/ion trap multiple mass spectrometry . Journal of Agricultural and Food Chemistry , 47 : 4240 – 4245 .
- Buttinger G Knapp H Fuchs E Binder EM Krska R 2003 New rapid clean-up device for Ochratoxin A Proceedings of the International Symposium of Mycotoxins (Kagawa, Japan, 3–5 November 2003)
- Coker R 2000 Aflatoxins and mycotoxins: Chromatography In: Wilson ID, Adlard TR, Cooke M, CF, editors. Encyclopedia of separation science Poole Academic Press pp 1873–1888
- Cole R Cox R 1981 Handbook of Toxic Fungal Metabolites New York Academic Press
- Driffield , M , Hird , SJ and MacDonald , J . 2003 . The occurrence of a range of mycotoxins in animal offal food products by HPLC-MS/MS . Aspects of Applied Biology , 68 : 205 – 210 .
- EN 12955 1999 Foodstuffs. Determination of aflatoxin B1, and the sum of aflatoxins B1, B2, G1 and G2 in cereals, shell-fruits and derived products; High performance liquid chromatographic method with post column derivatization and immunoaffinity column clean-up
- EN 13585 2001 Foodstuffs. Determination of fumonisins B1 and B2 in maize; HPLC method with solid phase extraction clean-up
- EN 14123 2003 Foodstuffs. Determination of aflatoxin B1 and the sum of aflatoxin B1, B2, G1 and G2 in peanuts, pistachios, figs, and paprika powder; High performance liquid chromatographic method with postcolumn derivatization and immunoaffinity column clean-up
- EN 14132 2003 Foodstuffs. Determination of ochratoxin A in barley and roasted coffee; HPLC method with immunoaffinity column clean-up
- EN 14177 2003 Foodstuffs. Determination of patulin in clear and cloudy apple juice and puree; HPLC method with liquid/liquid partition clean-up
- EN 14352 2004 Foodstuffs. Determination of fumonisin B1 and B2 in maize based foods; HPLC method with immunoaffinity column clean-up
- European Commission . 1998 . Commission Directive 98/53/EC of 16 July 1998 laying down the sampling methods and the methods of analysis for the official control of the levels of certain contaminants in foodstuffs . Official Journal of the European Communities , L201 : 93 – 101 .
- Freudenschuss M 2003 www.biopure.at
- Frisvad , JC and Thrane , U . 1987 . Standardized high-performance liquid chromatography of 182 mycotoxins and other fungal metabolites based on alkylphenone retention indices and UV-VIS spectra (diodearray detection) . Journal of Chromatography , 404 : 195 – 214 .
- Fuchs , E , Binder , EM , Heidler , D and Krska , R . 2002 . Structural characterisation of metabolites after the microbial degradation of A-trichothecenes by the bacterial strain BBSH 797 . Food Additives and Contaminants , 19 : 379 – 386 .
- Gilbert , J and Anklam , E . 2002 . Validation of analytical methods for determining mycotoxins in foodstuffs . Trends in Analytical Chemistry , 21 : 468 – 486 .
- JECFA 2001 Joint FAO/WHO Expert Commitee on Food Additives, 56th Meeting, Geneva, 6–15 February 2001
- Jodlbauer , J , Maier , N and Lindner , W . 2002 . Towards ochratoxin. A selective molecularly imprinted polymers for solid-phase extraction . Journal of Chromatography A , 945 : 45 – 63 .
- Johann , H and Dose , K . 1983 . Multianalytical method for the routine determination of the aflatoxins B1, B2, G1 and G2, and of citrinin, ochratoxin A, patulin, penicillic acid and sterigmatocystin in molded foods . Fresenius Zeitschrift der Analytischen Chemie , 314 : 139 – 142 .
- Josephs , RD , Krska , R , MacDonald , S , Wilson , P and Pettersson , H . 2003 . Preparation of a calibrant to be used as a certified reference material for the determination of the Fusarium mycotoxin zearalenone . Journal of AOAC International , 86 : 50 – 60 .
- Josephs , RD , Schuhmacher , R and Krska , R . 2001 . International interlaboratory study for the determination of the Fusarium mycotoxins zearalenone and deoxynivalenol in agricultural commodities . Food Additives and Contaminants , 18 : 417 – 430 .
- Kandler , W , Nadubinská , M , Parich , A and Krska , R . 2002 . Determination of moniliformin in maize by ion chromatography . Analytical and Bioanalytical Chemistry , 374 : 1086 – 1090 .
- Kos , G , Lohninger , H and Krska , R . 2003 . Development of a method for the determination of Fusarium fungi on corn using mid-Infrared spectroscopy with attenuated total reflection and chemometrics . Analytical Chemistry , 75 : 1211 – 1217 .
- Krska , R , Baumgartner , S and Josephs , R . 2001 . The state-of-the-art in the analysis of type-A and -B trichothecende mycotoxins in cereals . Fresenius’ Journal of Analytical Chemistry , 371 : 285 – 299 .
- Krska , R , Welzig , E , Josephs , RD , Kandler , W , Pettersson , H , MacDonald , S , Charlton , A , Brereton , P , Hametner , C , Berner , D and Zöllner , P . 2003 . Purity assessment of crystalline zearalenone . Journal of AOAC International , 86 : 722 – 728 .
- Lew , H , Chelkowski , J , Wakulinski , W and Edinger , W . 1993 . Moniliformin in wheat and triticale grain . Mycotoxin Research , 9 : 66 – 71 .
- Luo , Y , Liu , F , Zheng , J and Yang , J . 1992 . Systematic extraction, separation, and detection of Fusarium mycotoxins in cereals . Junshi Yixue Kexueyuan Yuankan , 16 : 20 – 25 .
- Maragos , CM . 2002 . Novel assays and sensor platforms for the detection of aflatoxins . Advances in Experimental Medicine and Biology , 504 : 85 – 93 .
- Märtlbauer , E , Dietrich , R and Terplan , G . 1991 . Erfahrungen bei der Anwendung von Immunoassays zum Nachweis von Mykotoxinen in Lebensmitteln . Archiv für Lebensmittelhygiene , 42 : 3 – 6 .
- Molinelli , A , Weiss , R and Mizaikoff , B . 2002 . Advanced solid phase extraction using molecularly imprinted polymers for the determination of flavonoids in red wine . Journal of Agricultural and Food Chemistry , 50 : 1804 – 1808 .
- Nadubinská , M , Parich , A , Krska , R , Kandler , W , Sraboarová , A and Eged , S . 2002 . Contents of zearalenone, deoxynivalenol, moniliformin and fusaric acid in maize ears from Slovakia naturally contaminated with Fusarium spp . Journal of Applied Genetics , 43A : 133 – 140 .
- Parich A Schuch Boeira L Perez Castro S Krska R 2004 Determination of moniliformin using SAX column clean-up and HPLC/DAD-detection Mycotoxin Research (accepted)
- Pettersson , H and Aberg , L . 2002 . Rapid estimation of deoxynivalenol and Fusarium by near infared spectrocopy . Journal of Applied Genetics , 43A : 141 – 144 .
- Pettersson H Langseth W 2002a Intercomparison of trichothecene analysis and feasibility to produce certified calibrants and reference material. Final report I, Method Studies. BCR Information, Project Report EUR 20285/1 EN 1–82
- Pettersson H Langseth W 2002b Intercomparison of trichothecene analysis and feasibility to produce certified calibrants and reference material. Final report I, Homogeneity and Stability Studies, Final Intercomparison. BCRInformation, EU Project Report EUR 20285/2 EN 1–145
- Razzazi-Fazeli , E , Böhm , J and Luf , W . 1999 . Determination of nivalenol and deoxynivalenol in wheat using liquid chromatography-mass spectrometry with negative ion atmospheric pressure chemical ionisation . Journal of Chromatography A , 854 : 45 – 55 .
- Rottinghaus GE Olesen B Osweiler GD 1982 Rapid screening method for aflatoxin B1, zearalenone, ochratoxin A, T-2 toxin, diacetoxyscirpenol and vomitoxin Proceedings of the annual meeting – American Association of Veterinarian Laboratory Diagnostics. Coume Date 1982, 25th 477 482
- Rundberget , T and Wilkins , AL . 2002 . Determination of penicillium mycotoxins in foods and feeds using liquid chromatography-mass spectrometry . Journal of Chromatography A , 964 : 189 – 197 .
- Schneider , E , Usleber , E , Märtlbauer , E and Terplan , G . 1995 . Multimycotoxin dipstick enzyme immunoassay applied to wheat . Food Additives and Contaminants , 12 : 387 – 393 .
- Schnerr , H , Vogel , RF and Niessen , L . 2002 . A biosensor-based immunoassay for rapid screening of deoxynivalenol contamination in wheat . Food and Agricultural Immunology , 14 : 313 – 321 .
- Schuhmacher , R , Krska , R and Grasserbauer , M . 1997 . Interlaboratory comparison test for the determination of the Fusarium mycotoxins deoxynivalenol in wheat and zearalenone in maize . Fresenius’ Journal of Analytical Chemistry , 359 : 510 – 515 .
- Schuller , PL , Horwitz , W and Stoloff , L . 1976 . A review of sampling plans and collaboratively studied methods of analysis for aflatoxins . Journal of the Association of Official Analytical Chemists , 59 : 1315 – 1343 .
- Scott , PM , Kanhere , SR and Tarter , EJ . 1986 . Determination of nivalenol and deoxynivalenol in cereals by electron-capture gas chromatography . Journal of the Association of Official Analytical Chemists , 69 : 889 – 893 .
- Scott , PM and Lawrence , BA . 1987 . Liquid chromatographic determination and stability of the Fusarium mycotoxin monilifomin in cereal grains . Journal of AOAC , 70 : 850 – 853 .
- Sharman , M , Gilbert , J and Chelkowski , J . 1991 . A survey of the co-occurrence of the mycotoxin moniliformin in cereal samples from sources worldwide . Food Additives and Contaminants , 8 : 459 – 466 .
- Shephard , G . 1998 . Chromatographic determination of the fumonisin mycotoxins . Journal of Chromatography A , 815 : 31 – 39 .
- Tanaka , T , Yoneda , A , Inoue , S , Sugiura , Y and Ueno , Y . 2000 . Simultaneous determination of trichothecene mycotoxins and zearalenone in cereals by gas chromatography-mass spectrometry . Journal of Chromatography A , 882 : 23 – 28 .
- Thiel , PG , Meyer , CJ and Marasas , WFO . 1982 . Natural Occurrence of Moniliformin together with deoxynivalenol and zearalenone in transkeian Corn . Journal of Agricultural and Food Chemistry , 30 : 308 – 312 .
- Thiel , PG . 1990 . Determination of moniliformin by high-performance liquid chromatography . Journal of Environmental Pathology, Toxicology and Oncology , 10 : 162 – 165 .
- Trucksess , M , Page , S , Wood , G and Cho , T . 1998 . Determination of dexoynivalenol in white flour, whole wheat flour, and bran by solid phase extraction/liquid chromatography: Interlaboratory study . Journal of AOAC International , 81 : 880 – 886 .
- Tudos , AJ , Lucas-van den Bos , ER and Stigter , EC . 2003 . Rapid surface plasmon resonance-based inhibition assay of deoxynivalenol . Journal of Agricultural and Food Chemistry , 51 : 5843 – 5848 .
- Uhlig S Torp M Gutleb A Krska R 2003 The ability of Fusarium avenaceum to accumulate moniliformin in Norwegian grain. The World Mycotoxin Forum, Noordwijk aan Zee, The Netherlands, 17–18 February
- Visconti A Boenke A Doko MB Solfrizzo M Pascale M 1995 Occurrence of fumonisins in Europe and the BCR-measurement and testing projects Natural Toxins 3 269 274 discussion 280
- Visconti , A , Boenke , A , Solfrizzo , M , Pascale , M and Doko , MB . 1996 . European intercomparison study for the determination of the fumonisin content in two maize materials . Food Additives and Contaminants , 13 : 909 – 927 .
- Visconti A Solfrizzo M De Girolamo A Boenke A 2001 Determination of fumonisins at levels of interest for future EU legislation: Development of an analytical method and interlaboratory validation for maize flour and cornflakes. European Commission, BCR information, Report EUR 10451
- Weingärtner , J , Krska , R , Praznik , W and Grasserbauer , M . 1997 . Use of Mycosep multifunctional clean-up columns for the determination of trichothecenes in wheat by electrone capture gas-chromatography . Fresenius’ Journal of Analytical Chemistry , 357 : 1206 – 1210 .
- Weiss , R , Molinelli , A , Jakusch , M and Mizaikoff , B . 2002 . Imprinting of Flavonoid compounds and application for solid phase extraction (SPE) . Bioseparations , 10 : 379 – 387 .
- Weiss , R , Freudenschuss , M , Krska , R and Mizaikoff , B . 2003 . Improving the analysis of mycotoxins in beverages. Molecularly imprinted polymers for deoxynivalenol and zearalenone . Food Additives and Contaminants , 20 : 386 – 395 .