Abstract
Adenoviral replacement of the p53 gene has already been proved effective for the treatment of various tumours, including malignant gliomas. However, it is difficult to treat malignant glioma with p53 gene therapy alone because of problems with resistance or a less-than-satisfactory response to the treatment. This study investigated whether heat shock at 43°C (mild hyperthermia) augments the cytotoxic effect of p53 gene transfer on malignant glioma cells expressing wild-type p53 (D54) or mutant p53 (U373-MG and U251-MG). The combination of mild hyperthermia and adenoviral p53 over-expression had an additive inhibitory effect on cellular proliferation in all three cell lines studied. Further, both cell cycle analysis and a DNA fragmentation assay showed that apoptosis was induced by p53 over-expression alone but not by heat shock at 43°C alone. However, p53 over-expression followed by mild hyperthermia additively increased the proportion of cells in which apoptosis was induced, regardless of the endogenous p53 status of the tumour cells. Interestingly, a caspase-independent mechanism was observed to be involved in the p53-induced apoptosis in U251-MG and D54 cells. Taken together, the findings showed that combining adenoviral p53 transfer with mild hyperthermia inhibits the proliferation of malignant glioma cells in an additive manner, irrespective of their endogenous p53 status, suggesting a novel treatment strategy for this malignancy.
Introduction
Malignant gliomas are the most common neoplasms of the central nervous system. However, even in the face of aggressive treatments, including radical surgery, γ-irradiation and chemotherapy, the median survival of patients with the most malignant type, glioblastoma multiforme, is less than 1 year from diagnosis Citation[1]. This calls for the exploration of novel or alternative treatment strategies to improve the now dismal prognosis in patients with these tumours.
Like many other cancers, the malignant progression of gliomas is closely associated with the multi-step accumulation of genetic abnormalities. The most frequent gene mutation seen in malignant glioma cell lines and primary brain tumours, but not in normal brain tissues, is a mutation of the tumour-suppressor p53 gene Citation[2]. Therefore, replacement of the mutated p53-gene with the wild-type gene or over-expression of the wild-type gene is considered an effective alternative treatment to conventional anti-cancer treatments. Indeed, there is accumulating evidence showing the anti-tumour effect of p53 transfer on malignant glioma cells Citation[3], Citation[4]. However, one difficulty with this approach is that tumour cells with the wild-type p53 are highly resistant to p53 gene transfer Citation[5]. Moreover, a recent clinical study suggested that it is difficult to eradicate malignant glioma by means of p53 over-expression alone Citation[6]. Therefore, p53 gene therapy may be most effective in combination with other therapy.
At the same time, some patients with malignant glioma treated with local hyperthermia have been found to have heat-resistant tumours Citation[7]. This has pointed to the need to develop a novel adjuvant therapy to make these tumours more sensitive to hyperthermia.
The study described here investigated the combined effect of mild hyperthermia (heat shock at 43°C) and adenoviral p53 gene transfer on malignant glioma cells expressing wild-type p53 (D54) and mutant p53 (U373-MG and U251-MG).
Materials and methods
Cells
Human malignant glioma U251-MG and U373-MG cells were purchased from the American Type Culture Collection (Rockville, MD). Human malignant glioma D54 cells were kindly provided by Dr Chuan-Yuan Li (Department of Radiation Oncology, Duke University Medical Center, NC, USA). Cells were cultured in Dulbecco's modified Eagle medium (Life Technologies, Inc., Carlsbad, CA) supplemented with 10% foetal bovine serum (Life Technologies, Inc.), 4 mM glutamine, 100 units ml−1 penicillin and 100 µg ml−1 streptomycin. The three cell lines were routinely tested for Mycoplasma contamination by using the MycoFluor™ Mycoplasma detection kit (M-7006: Molecular Probes, Inc., Eugene, OR) and consistently tested negative for the organism.
Heating method
The cell culture plates were incubated at 43°C in a waterbath (Thermo Minder, Mini-80: TAITEC, Koshigaya, Saitama, Japan), as previously described Citation[8]. This temperature was selected on the basis of several considerations. As was recently reported, heating at 43°C is not sufficient to induce cell death, but heating at 45°C causes the permanent death of malignant glioma cells Citation[8], However, while it is important to make the tumour cells at the tumour–normal brain border zone susceptible to cell death because these are the cells most actively dividing, which heat shock at 43°C will achieve, it is also important to avoid damage to the surrounding normal brain cells, which temperatures of 45°C or higher would cause.
Adenoviral vector
The recombinant Ad5CMV-p53 or Ad5CMV-green fluorescent protein (GFP)-containing cytomegalovirus (CMV) promoter, p53 or GFP complement cDNAs and SV40 polyadenylation signal were constructed by inserting the expression cassette into the E1-deleted region of a modified Ad5 genome. Viral stocks were propagated in 293 cells and purified through CsCl2 banding, as described previously Citation[9]. Both vectors were prepared in the Vector Core Laboratory of the W. M. Keck Center for Cancer Gene Therapy at The University of Texas M.D. Anderson Cancer Center (Houston, TX).
Treatment of cells with p53 gene transfer and heat-shock treatment
At 24 h after the seeding of cells, they were infected with Ad5CMV-p53 or Ad5CMV-GFP at a multiplicity of infection (MOI) of 1–20. Then, 48 h after the infection, the cells were heated at 43 or 37°C for 2 h. Finally, 48 h after the heat-shock treatment, the cells were assayed. To determine the best sequence of combination of p53 over-expression and heat-shock treatment, the sequence in which p53 gene transfer was performed after heat-shock treatment or in which both treatments were given concurrently was also investigated.
Cell viability assay
Cell viability was determined by a standard 3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyltetrazolium bromide (MTT) assay Citation[10]. Briefly, 1.5 or 3 × 103 cells were plated in 96-well plates and treated with p53 gene transfer followed by heat-shock treatment as mentioned above. Forty-eight hours after the heat-shock treatment, MTT (0.5 mg ml−1) was added and the cells were incubated for 2 h at 37°C. Formazan crystals were dissolved with dimethyl sulphoxide (DMSO) and the colour intensity was measured using an enzyme-linked immunosorbent assay reader at 570 nm (reference filter, 690 nm). Survival fractions were calculated by dividing the mean cell viability of treated cells by that of untreated cells. Graphs show the mean ± standard deviation (SD).
Cell cycle analysis
Non-adherent and trypsinized adherent cells were collected, washed twice with phosphate-buffered saline (PBS) and fixed overnight with 70% ethanol. The cells were resuspended in PBS containing 40 µg ml−1 of DNase-free RNaseA and 50 µg ml−1 propidium iodide. The cell cycle distribution was measured using a FACScan flow cytometer (Becton Dickinson, San Jose, CA). The DNA content was quantified by CellQuest software (Becton Dickinson) Citation[9].
DNA fragmentation assay
Non-adherent and trypsinized adherent cells were collected, washed, fixed with 4% paraformaldehyde and permeabilized with 70% ethanol and then DNA-nick-end labelled with TdT and FITC-dUTP. The fluorescence intensity was determined using a FACScan flow cytometer (Becton Dickinson) and analysed by CellQuest software (Becton Dickinson) Citation[9]. As positive controls for DNA fragmentation, the cells that were treated with 5 µg ml−1 cisplatin for the first 24 h and the ones that were treated with DNase I were used.
Caspase inhibition assay
At 24 h after the seeding of cells at a density of 3 × 103 per well in 96-well plates, the cells were pre-treated with 10 μM of the pan-caspase inhibitor, benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (zVAD-fmk) (Promega, Madison, WI) or an equivalent content of DMSO (control treatment) for 3 h. Then, the medium was replaced with the one with or without Ad/CMV5-p53 or Ad/CMV5-GFP (control vector) as well as with either 10 μM zVAD-fmk or an equivalent content of DMSO (control treatment). After a 24-h exposure to the adenoviral vector, the medium was replaced with the one with 10 μM zVAD-fmk or an equivalent content of DMSO (control treatment). After an additional 24 h, the cells were heated at 43°C for 2 h or kept at 37°C for 2 h. Then, 48 h after the heat treatment, cell viability was determined in the MTT assay as described above.
Immunoblotting assay
Cells were trypsinized and washed twice in PBS without calcium and magnesium. The cells were lysed in buffer A Citation[10] mM Tris (pH 7.4), 1 mM EGTA, 1 mM EDTA, 50 mM NaCl, 5 mM β-glycerophosphate, 1% Triton X-100, 0.5% NP40, 1 mM sodium vanadate, 1 mM dithiothreitol, 1 mM phenylmethylsulphonyl fluoride and 10 µg ml−1 each of leupeptin and aprotinin]. Insoluble material was removed by centrifugation at 14 000 rpm for 5 min at 4°C. Proteins were separated by electrophoresis in sodium dodecyl sulphate-polyacrylamide gels and then transferred to a nitrocellulose paper. β-actin was also studied simultaneously to confirm equivalent protein loading. The residual binding sites were blocked by incubating the filter in 5% dry milk in PBS/0.05% Tween 20 for 1 h. Then, the blots were incubated with anti-p21 monoclonal antibody (MAb) (BD Biosciences Phramingen, San Diego, CA), or anti-β-actin MAb (Sigma-Aldrich Inc., St Louis, MO) at 4°C overnight. After the blots were washed twice with PBS/0.05% Tween 20, they were incubated with anti-mouse IgG peroxidase conjugate (Amersham Biosciences UK Limited, Buckinghamshire, UK). The antigen–antibody complexes were then visualized by chemiluminescence (ECL detection system; Amersham Biosciences UK Limited).
Statistical analysis
Data are expressed as the mean ± SD. Statistical analysis was performed using the StatView software (StatView-J 5.0: SAS Institute Inc., Cary, NC). All data were compared between two or more groups using an unpaired t-test. A p-value of <0.05 was considered statistically significant.
Results
Effect of heat-shock treatment and p53 over-expression on cell viability
First, the optimal MOI of Ad5CMV-p53 for each cell line was determined in an MTT assay. The following MOIs of Ad5CMV-p53 significantly inhibited cell proliferation without non-specific toxicity: 1–5 MOI for U373-MG cells, 5–10 MOI for U251-MG cells and 10–15 MOI for D54 cells ().
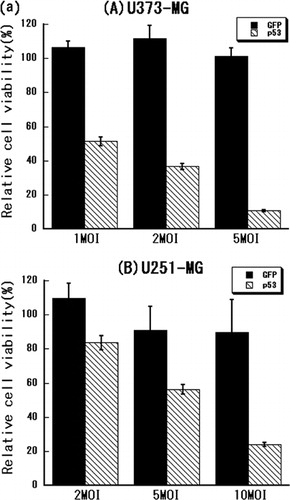
Figure 1. (a) Treatment of malignant glioma cells with adenoviral p53 gene transfer. (A) U373-MG, (B) U251-MG and (C) D54 glioma cells were treated with Ad5CMV-GFP or Ad5CMV-p53 at the MOI indicated on the X-axis for 72 h and an MTT assay was performed. (b) Treatment with adenoviral p53 over-expression, heat shock or their combination. (A) U373-MG, (B) U251-MG and (C) D54 glioma cells were treated with Ad5CMV-GFP or Ad5CMV-p53 for 48 h, heat-treated at 37 or 43°C for 2 and 48 h later an MTT assay was performed.
The MOI that inhibited cell viability by ∼50% was 1 for U373-MG cells, 5 for U251-MG cells and 10 for D54 cells. These findings indicated that the D54 cells with the wild-type p53 were more resistant to p53 over-expression than were the mutant p53-expressing U373-MG and U251-MG cells, consistent with previous observations Citation[5]. Therefore, 1, 5, and 10 MOI were used in the subsequent experiments in U373-MG, U251-MG and D54 cells, respectively.
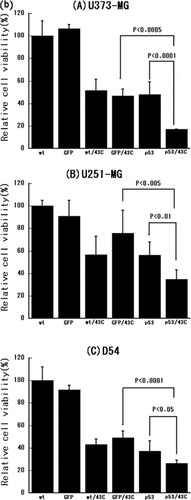
The combined effect of heat shock at 43°C and p53 over-expression in an MTT assay was investigated next. As shown in , the viability of all three cell lines was significantly inhibited by heat shock at 43°C alone. Moreover, the combination of heat-shock treatment and p53 over-expression had a statistically significant additive effect in all three tumour cell lines (p < 0.0001 in U373-MG, p < 0.01 in U251-MG and p < 0.05 in D54).
Effect of heat-shock treatment and p53 over-expression on cell cycle
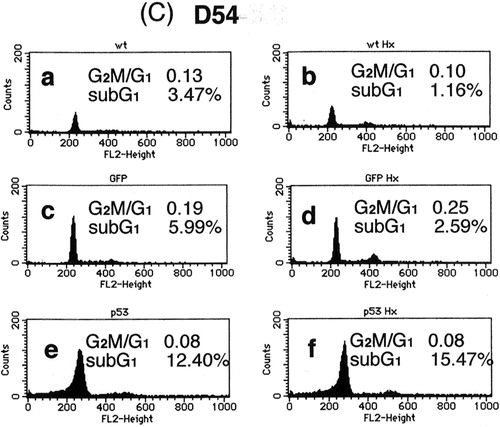
Next, the effect of heat-shock treatment alone, p53 over-expression alone and their combination on the cell cycle were assessed. shows that, characteristically of apoptosis, the sub-G1 population was increased by p53 over-expression in all three cell lines. In contrast, no such increase was induced by 43°C heat-shock treatment alone. However, the combination of heat-shock treatment and p53 over-expression additively increased the sub-G1 population in all three cell lines over that induced by p53 over-expression alone. These findings, together with those from the MTT assay, indicate that mild hyperthermia (heat-shock at 43°C) and p53 over-expression additively inhibit cell survival at least in part by inducing apoptosis.
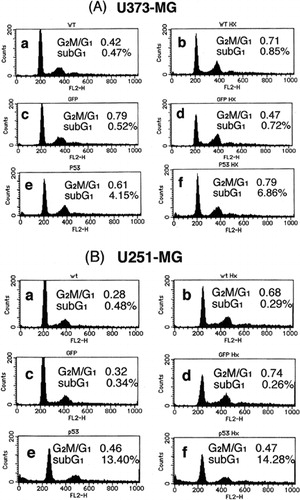
Figure 2. Cell cycle analysis of (a) U373-MG, (b) U251-MG and (c) D54 glioma cells after p53 over-expression, heat-shock treatment or their combination. (a) No treatment, (b) heat-shock treatment at 43°C for 2 h only, (c) Ad5CMV-GFP only, (d) Ad5CMV-GFP + heat-shock treatment at 43°C for 2 h, (e) Ad5CMV-p53 only and (f) Ad5CMV-p53 + heat-shock treatment at 43°C for 2 h.
Effect of heat-shock treatment and p53 over-expression on DNA fragmentation
To confirm the induction of apoptosis by p53 over-expression alone and in combination with heat-shock treatment, the extent of DNA fragmentation in treated cells was investigated. As shown in , DNA fragmentation was observed in 21.3, 17.7 and 11.8% of U373-MG, U251-MG and D54 cells infected with Ad5CMV-p53, respectively, but no significant DNA fragmentation was induced by Ad5CMV-GFP infection (1.1, 4.8 and 1.9%, respectively), or by heat-shock treatment alone (0.4, 3.4 and 7.2%, respectively). These results were consistent with the data from the cell cycle analysis. However, as predicted, the combination of heat-shock treatment and p53 over-expression additively increased the extent of DNA fragmentation in all tumour cells (64.2, 21.3 and 15.2% for U373-MG, U251-MG and D54 cells, respectively) over that produced by p53 over-expression alone. These results were consistent with both the cell viability and cell cycle findings, thus indicating that the combination of the two treatments additively inhibited cell survival at least partially by inducing apoptosis.
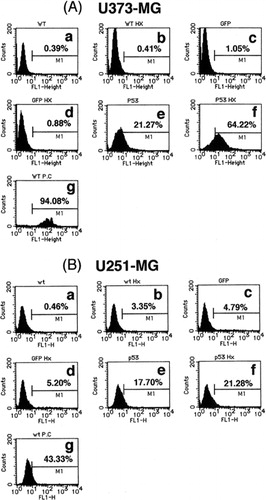
Figure 3. Flow cytometric analysis of (a) U373-MG, (b) U251-MG and (c) D54 glioma cells with TUNEL staining for detection of DNA fragmentation. (a) No treatment, (b) heat-shock treatment at 43°C for 2 h only, (c) Ad5CMV-GFP only, (d) Ad5CMV-GFP + heat-shock treatment at 43°C for 2 h, (e) Ad5CMV-p53 only, (f) Ad5CMV-p53 + heat-shock treatment at 43°C for 2 h and (g) treatment with DNase I as a positive control.
Caspase dependency of cellular damage induced by heat-shock treatment and p53 over-expression
To determine whether a caspase-dependent pathway mediates the combined effect of heat-shock treatment and p53 over-expression, the effect of caspase-inhibition on treated cells was examined. As shown in , the cell growth inhibited by p53 over-expression alone or in combination with the heat-shock treatment was significantly restored by the pan-caspase inhibitor in U373-MG cells (p < 0.05 or p < 0.005, respectively) but not in the U251-MG and D54 cells. These results suggest that a caspase-independent pathway is involved in the apoptosis induced by p53 over-expression alone or in combination with heat-shock treatment in U251-MG and D54 cells.
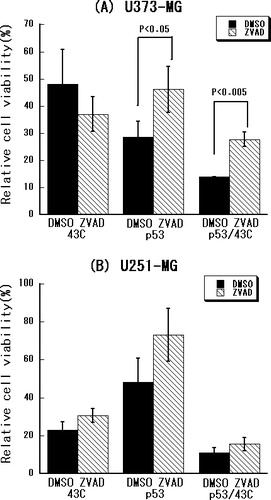
Figure 4. Caspase inhibition as shown by an MTT assay performed in (a) U373-MG, (b) U251-MG and (c) D54 glioma cells using the pan-caspase inhibitor zVAD-fmk along with adenoviral p53 gene transfer and/or heat shock at 43°C.
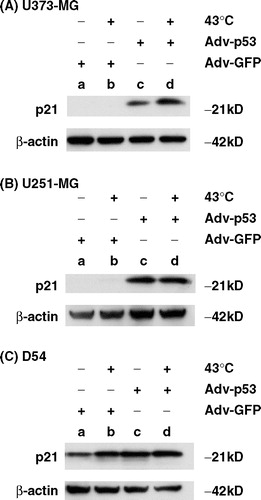
p21 protein expression after heat-shock treatment and p53 over-expression
Finally, an immunoblotting assay was performed to determine whether expression of the p21 protein is up-regulated by p53 over-expression or heat-shock treatment at 43°C or both. As shown in , p21 protein expression was up-regulated by p53 over-expression alone in U373-MG and U251-MG cells and additively increased by heat-shock treatment plus p53 over-expression in U373-MG cells. On the other hand, untreated D54 cells expressed the p21 protein to some extent and expression was slightly enhanced by p53 over-expression or heat-shock treatment alone or by their combination. These results indicate that the combination of p53 over-expression and heat-shock treatment is associated with up-regulation of the p21 protein.
Figure 5. Immunoblotting assay for the expression of p21 protein after p53 over-expression, heat-shock treatment or their combination. Cell lysates were prepared from (a) U373-MG, (b) U251-MG and (c) D54 cells, which were treated with (a) Ad5CMV-GFP only, (b) Ad5CMV-GFP + heat-shock treatment at 43°C for 2 h, (c) Ad5CMV-p53 only and (d) Ad5CMV-p53 + heat-shock treatment at 43°C for 2 h. Proteins were subjected to immunoblot analysis using anti-p21 antibody. Anti-β-actin antibody was used to confirm equivalent protein loading.
Discussion
It can be concluded from the findings in the present study that the combination of heat shock at 43°C and adenoviral p53 over-expression additively inhibited cellular proliferation, at least in part by inducing apoptosis, in three human malignant glioma cell lines studied—U373-MG, U251-MG and D54. This indicates that each modality works by an independent mechanism. Moreover, involvement of the caspase-dependent pathway seems to depend on the cell type rather than on the endogenous p53 status of the tumour cells.
Although hyperthermia alone has been found to induce apoptosis in human tongue squamous cell carcinoma cells Citation[11], A172 human malignant glioma cells Citation[12], and rat yolk sac tumour cell lines Citation[13], as has been reported previously, it does not have this effect in glioma cell lines except in the A172 line Citation[14]. Instead, heat shock at 43°C induces autophagy with mild G2/M-arrest in U251-MG and U87-MG cells Citation[8]. The findings in the present study confirm that heat-shock therapy, especially mild 43°C heating, does not induce apoptosis in glioma cells.
One of the most important issues in the combination of heat-shock treatment and p53 over-expression is the sequence in which they are given. This is because one of the problems with heat-shock treatment is that it can induce thermo-tolerance by inducing heat-shock protein (HSP), such as HSP 27, HSP 70 or HSP 90 Citation[15], Citation[16]. Heat-shock treatment may also damage cellular metabolism, which then inhibits gene transcription and the synthesis of p53 and other proteins that play major roles in the apoptosis pathway Citation[17]. Therefore, it is reasonable to treat tumour cells first with p53 over-expression and then with heat shock. In fact, in this experiment, cell viability was successfully reduced when p53 gene transfer was done before heat-shock treatment. However, the combined effect was not significant when p53 gene transfer was performed after heat-shock treatment or when both treatments were given concurrently (data not shown). Recently, several investigations have shown that HSP inhibitors, such as geldanamycin (an HSP90 inhibitor), make cancer cells more susceptible to the cellular damage induced by chemotherapy Citation[18] or irradiation Citation[19]. Therefore, consideration might be given to administering an HSP inhibitor to augment the anti-tumour effect of this combination therapy.
Although it was found that hyperthermia does not antagonize the effect of the exogenous p53 protein introduced through p53 over-expression, Guan et al. Citation[20], reported that, while heat shock induces its own p53 response, it also prevents the induction of the irradiation-mediated p53 response. Moreover, these researchers noted that the activation of p53 induced by heat is rapid and short-lived compared with p53 activation after exposure to ionizing radiation Citation[20]. If this is the case in malignant glioma cells, it may be that the heat-induced p53 response interferes with the function of the exogenous p53 over-expression. However, this was not observed in this study.
A particular concern is that the endogenous wild-type p53 protein can be functionally inactivated in cancer cells through its interaction with various viral or cellular proteins, including the MDM2 cellular oncoprotein Citation[21], Citation[22]. However, when the level of the exogenous p53 protein exceeds that of the endogenous protein, the p53 protein inactivation mechanism may be overcome, with the result that the p53 protein is able to function and cells undergo apoptosis. Indeed, this was borne out in this study, in which cell cycle and DNA fragmentation assays showed that p53 gene transfer at a relatively high MOI could cause apoptosis, even in wild-type p53-expressing D54 cells.
It is also interesting that the apoptosis induced by p53 over-expression in the D54 cells with the wild-type p53 was caspase-independent. Autophagic cell death is the most studied type of cell death in cells that show caspase-independent cell deaths. In autophagic cell death, lysosomes play a major role in the pathway Citation[23]. It has previously been reported that mild heat-shock treatment, such as that used in the study described here, induces autophagic growth arrest Citation[8]. Nonetheless, the authors do not know a lot about the caspase-independent mechanism responsible for the cell death resulting from p53 over-expression. It is important to elucidate this mechanism because of the possibility that caspase-independent apoptosis results from p53 gene transfer in wild-type p53 cells.
It is believed that, even in mutant p53 cell lines in which the p53 gene is mutated, the p21 pathway, which is downstream from p53, may be preserved and, therefore, that the transactivation of p21 may still be induced. Consistent with this, as the immunoblotting assay revealed, the p21 protein was up-regulated after exogenous p53 over-expression, even in mutant p53 tumour cells. This was also borne out in a study of Zhou et al. Citation[24], who reported that some of the COOH-terminal mutants of p53 can transcriptionally transactivate p21 and inhibit cell growth.
As shown in and , although there is a clear increase in DNA fragmentation in U373-MG cells produced by p53 over-expression alone and in combination with the heat-shock treatment, the results of the cell cycle analysis did not strongly support this. This discrepancy may be due to the fact that the sub-G1 content does not necessarily reflect the actual percentage of apoptotic cells: it only indicates the existence of cells that are now undergoing apoptosis. That is, even though most of the cells are actually dying as a result of apoptosis, only a partial increase in the sub-G1 population is seen in the cell cycle analysis.
In conclusion, p53 over-expression induced apoptosis in the three glioma cell lines used in the study, although mutant p53-expressing tumour cells were more sensitive to p53 over-expression than tumour cells expressing wild-type p53. On the other hand, while the heat-shock treatment at 43°C did not induce apoptosis in tumour cells, combination treatment consisting of p53 gene transfer followed by heat-shock treatment additively inhibited cellular proliferation, suggesting that a separate mechanism is at work in each modality. Particularly noteworthy was the fact that this additive effect was seen regardless of the endogenous p53 status of the tumour cells. Taken together, these findings suggest that adenoviral p53 gene transfer followed by hyperthermia may be a promising approach for the management of human malignant gliomas.
Acknowledgements
We are grateful to Ms Fumie Higuchi, Ms Nobuko Kiyama and Ms Sachiko Moriyama for technical assistance and to Ms Hiroko Toju for the literature search. We thank Ms Beth Notzon for editing the manuscript. This work was supported by the grant of Ministry of Education, Culture, Sports, Science and Technology in Japan (No. 15390430) and the Core grant at the Vector Core Laboratory at M.D. Anderson (CA-16672).
Related Research Data
References
- Laws ER, Parney IF, Huang W, Anderson F, Morris AM, Asher A, Lillehei KO, Bernstein M, Brem H, Sloan A, Berger MS, Chang S. the Glioma Outcomes Investigators. Survival following surgery and prognostic factors for recently diagnosed malignant glioma: Data from the Glioma Outcome Project. Journal of Neurosurgery 2003; 99: 467–473
- Nozaki M, Tada M, Kobayashi H, Zhang CL, Sawamura Y, Abe H, Ishii N, Van Meir EG. Roles of the functional loss of p53 and other genes in astrocytoma tumorigenesis and progression. Neurooncology 1999; 1: 124–137
- Gomez-Manzano C, Fueyo J, Kyritsis AP, Steck PA, Roth JA, McDonnel TJ, Steck KD, Levin VA, Yung WK. Adenovirus-mediated transfer of the p53 gene produces rapid and generalized death of human glioma cells via apoptosis. Cancer Research 1996; 56: 694–699
- Kock H, Harris MP, Anderson SC, Machemer T, Hancock W, Sutjipto S, Wills KN, Gregory RJ, Shepard HM, Westphal M, Maneval DC. Adenovirus-mediated p53 gene transfer suppresses growth of human glioblastoma cells in vitro and in vivo. International Journal of Cancer 1996; 67: 808–815
- Lang FF, Yung WK, Sawaya R, Tofilon PJ. Adenovirus-mediated p53 gene therapy for human gliomas. Neurosurgery 1999; 45: 1093–1104
- Lang FF, Bruner JM, Fuller GN, Aldape K, Prados MD, Chang S, Berger MS, McDermott MW, Kunwar SM, Junck LR, Chandler W, Zwiebel JA, Kaplan RS, Yung WK. Phase I trial of adenovirus-mediated p53 gene therapy for recurrent glioma: Biological and clinical results. Journal of Clinical Oncology 2003; 21: 2508–2518
- Uzuka T, Tanaka R, Takahashi H, Kakinuma K, Matsuda J, Kato K. Planning of hyperthermic treatment for malignant glioma using computer simulation. International Journal of Hyperthermia 2001; 17: 114–122
- Komata T, Kanzawa T, Nashimoto T, Aoki H, Endo S, Nameta M, Takahashi H, Yamamoto T, Kondo S, Tanaka R. Mild heat shock induces autophagic growth arrest, but not apoptosis in U251-MG and U87-MG human malignant glioma cells. Journal of Neurooncology 2004; 68: 101–111
- Komata T, Kondo Y, Koga S, Ko SC, Chung LWK, Kondo S. Combination therapy of malignant glioma cells with 2-5A-antisense telomerase RNA and recombinant adenovirus p53. Gene Therapy 2000; 7: 2071–2079
- Kondo Y, Komata T, Kondo S. Combination therapy of 2-5A antisense against telomerase RNA and cisplatin for malignant gliomas. International Journal of Oncology 2001; 18: 1287–1292
- Yasumoto J, Kirita T, Takahashi A, Ohnishi K, Imai Y, Yuki K, Ohnishi T. Apoptosis-related gene expression after hyperthermia in human tongue squamous cell carcinoma cells harboring wild-type or mutated-type p53. Cancer Letters 2004; 204: 41–51
- Ohnishi T, Ohnishi K, Takahashi A. Glycerol restores heat-induced p53-dependent apoptosis of human glioblastoma cells bearing mutant p53. BMC Biotechnology 2002; 2: 6
- Islam MS, Mitsuhashi N, Akimoto T, Sakurai H, Hasegawa M, Ishikawa H, Niibe H. Hyperthermia-induced apoptosis in two rat yolk sac tumor cell lines with different radiothermosensitivity in vitro. Oncology Reports 2001; 8: 501–507
- Fuse T, Yoon KW, Kato T, Yamada K. Heat-induced apoptosis in human glioblastoma cell line A172. Neurosurgery 1998; 42: 843–849
- Kato K, Hasegawa K, Goto S, Inagguma Y. Dissociation as a result of phosphorylation of an aggregated form of the small stress protein, hsp27. Journal of Biological Chemistry 1994; 269: 11274–11278
- Ohnishi T, Matsumoto H, Takahashi A, Shimura M, Majima HJ. Accumulation of mutant p53 and hsp72 by heat treatment, and their association in a human glioblastoma cell line. International Journal of Hyperthermia 1995; 11: 663–671
- Ahern TJ, Casal JI, Petsko GA, Klibanov AM. Control of oligomeric enzyme thermostability by protein engineering. Proceedings of the National Academy of Sciences (USA) 1987; 84: 675–679
- Arlander SJ, Eapen AK, Vroman BT, McDonald RJ, Toft DO, Karnitz LM. Hsp90 inhibition depletes Chk1 and sensitizes tumor cells to replication stress. Journal of Biological Chemistry 2003; 278: 52572–52577
- Machida H, Matsumoto Y, Shirai M, Kubota N. Geldanamycin, an inhibitor of Hsp90, sensitizes human tumour cells to radiation. International Journal of Radiation Biology 2003; 79: 973–980
- Guan J, Stavridi E, Leeper DB, Iliakis G. Effects of hyperthermia on p53 protein expression and activity. Journal of Cell Physiology 2002; 190: 365–374
- Capoulade C, Bressac-de Paillerets B, Lefrere I, Ronsin M, Feunteun J, Tursz T, Wiels J. Overexpression of MDM2, due to enhanced translation, results in inactivation of wild-type p53 in Burkitt's lymphoma cells. Oncogene 1998; 16: 1603–1610
- Momand J, Jund D, Wilczyinski S, Niland J. The MDM2 gene amplification database. Nucleic Acids Research 1998; 26: 3453–3459
- Lockshin RA, Zakeri Z. Caspase-independent cell deaths. Current Biology 2002; 14: 727–733
- Zhou X, Wang XW, Xu L, Hagiwara K, Nagashima M, Wolkowicz R, Zurer I, Rotter V, Harris CC. COOH-terminal domain of p53 modulates p53-mediated transcriptional transactivation, cell growth, and apoptosis. Cancer Research 1999; 59: 843–848