Abstract
Interleukin-12 (IL-12) is a pro-inflammatory cytokine that has shown great promise as a therapeutic agent in experimental models of infectious disease and cancer. However, it is also a highly toxic molecule and for that reason has not been accepted readily into the clinic. A replication-deficient adenoviral vector was designed to deliver the feline interleukin-12 gene into tumour cells. The interleukin-12 gene has been placed under control of a heat inducible promoter, human heat shock promoter 70b, with the intent of spatially and temporally controlling the expression of IL-12, thus limiting its toxicity. In vitro, the transfection efficiency of the adenoviral vector, the effect of multiplicity of infection and the production of biologically active feline IL-12 were studied in the infected cells in response to a range of temperatures. This adenoviral vector will be a useful tool to examine the effects of intra-tumoural IL-12 delivery in a spontaneously occurring feline soft tissue sarcoma model.
Introduction
The aim of cancer gene immunotherapy is the resolution of primary tumours and the development of a systemic immune response directed against metastases and micro-metastases. Interleukin-12 (IL-12) is one of the most widely studied immunoregulatory cytokines and has been found to eradicate tumours, metastases, elicit long-term anti-tumour immunity Citation[1–6] and possess anti-angiogenic properties Citation[7–10]. The IL-12 protein is a 70 kDa heterodimer (p70) consisting of two covalently linked sub-units, p35 and p40. It is produced by antigen-presenting cells including dendritic cells and macrophages in response to infection. In turn, IL-12 induces the synthesis of interferon-γ (IFN-γ) by T-cells and NK cells. It also enhances cell mediated immunity (CMI) by generating and potentiating the activity of cytotoxic T-lymphocytes (CTL) and lymphokine activated killer (LAK) cells Citation[11], Citation[12]. There is also evidence for a role of IL-12 in humoral immunity Citation[13]. Interleukin-12 is postulated to exert its anti-tumour effect through NK cells Citation[7], Citation[14–17], NKT cells Citation[18], Citation[19] and T-cells Citation[20–23]. Interferon-γ, a downstream product of IL-12 action, is also thought to play a major role in the anti-tumour actions of IL-12 Citation[1], Citation[24–29].
Various methods of IL-12 administration have been studied and described in the literature including systemic administration Citation[25], Citation[30–32], intra-tumoural injection of IL-12 plasmid DNA Citation[5], genetic engineering approaches Citation[33], Citation[34] and gene therapy techniques Citation[6], Citation[17], Citation[35–39]. The role of interleukin-12 in the anti-cancer arsenal has been limited due to its toxicities. In mice daily administration of 0.1–10 μg of recombinant murine IL-12 for up to 2 weeks resulted in liver function abnormalities, muscle degeneration, gastrointestinal toxicity and hematopoietic changes including anaemia, neutropenia, lymphopenia and thrombocytopenia Citation[40]. In humans, the toxic effects are mostly IFN-γ dependent and include fever, chills, fatigue, headache, nausea, vomiting, cough, myalgia, dizziness, insomnia, anaemia, neutropenia, lymphocytopenia, thrombocytopenia, hyperglycemia, liver function test abnormalities, rhinitis, stomatitis and colitis Citation[41]. In a phase II clinical trial in renal carcinoma using recombinant human IL-12, two deaths were reported in 17 patients treated Citation[42], Citation[43]. One approach that can be used to avoid the systemic toxicity of IL-12 is to use local intra-tumoural IL-12 gene therapy. This approach should yield high tumour levels of IL-12 with little or no systemic IL-12 exposure, thereby avoiding consequent toxicities.
A promising gene delivery method harnesses the heat shock response, an evolutionarily conserved response to cellular stress involving the rapid and transient increase in the cellular production of heat shock proteins, most importantly hsp70 and hsp90. These proteins have protective functions in keeping other key proteins from being denatured by heat or other stresses and, hence, are also called molecular chaperones Citation[44–46]. The heat shock promoter, hsp70, has been found to be one of the most efficient inducible regulatory sequences Citation[47–49]. A further level of spatial specificity of IL-12 expression can be achieved if the hsp promoter containing gene construct is injected intra-tumourally and the expression of the construct is restricted to the tumour by delivering hyperthermia only to that area. This approach has been shown to improve selectivity of expression to the heated tumour, whereas intra-tumoural administration of expression constructs under control of a constitutive promoter can lead to ectopic expression of transgene in distant organs, such as the liver, spleen and lung Citation[36].
Adenoviruses have been developed for use as gene delivery vehicles Citation[50–52] and they offer certain advantages which make them attractive candidates for this role. Under experimental conditions, early clinical vaccination with wild-type (wt) live adenovirus showed no significant side effects, demonstrating the relative safety of adenoviruses as vectors for in vivo gene therapy. They efficiently infect and transfer genes in a wide variety of cell types including dividing and non-dividing cells. Strong immunogenicity of adenoviral vectors may enhance tumour-killing in cancer gene therapy. Furthermore, adenoviruses are easy to manipulate by classical recombinant DNA techniques and they have the added advantage that they can be produced in large quantities in cell culture. Unlike retroviruses, adenoviruses do not integrate into the host genome. This gives adenoviral vectors an important safety feature for in vivo gene therapy.
This is important because there is no need for stable integration into the host genome in case of anti-cancer therapy, unlike diseases involving inborn errors of metabolism where a stable integration is desired.
With this background, the Adenovirus heat shock promoter feline interleukin-12 construct was designed and its properties studied in vitro with respect to its ability to produce IL-12 in Crandell feline kidney cells and the downstream effects of IL-12 in inducing IFN-γ production in peripheral blood mononuclear cells. This work was done in support of an immunogene therapy trial conducted in pet cats with vaccine-associated sarcomas. This trial was conducted as a prelude to initiation of human trials using a similar gene therapy approach.
Materials and methods
Cells used
Crandell Feline Kidney (CrFK) cells were cultured in DMEM supplemented with 10% foetal bovine serum (FBS) and 1% penicillin-streptomycin (CrFK media). These cells were grown in six-well tissue culture plates. It was determined that a minimum of 3 × 105 cells per well was needed to allow subsequent growth. These were the primary cells infected with the adenoviral construct and exponential growth in cell number was seen over the next 2–3 days of the experiment.
Peripheral blood mononuclear cells (PBMC) were obtained from specific pathogen free cats maintained at Colorado State University and isolated by density gradient centrifugation (Histopaque 1077, Sigma). These cells were plated in six-well tissue culture plates at a density of 1 × 106 cells per well in RPMI 1640 medium supplemented with 20% FBS, 1% penicillin-streptomycin, 2% glutamine and 1 × 10−5 M 2-mercaptoethanol (LBT media). Supernatants from CrFK cells, containing the IL-12 protein, were used to stimulate production of IFN-γ in these PBMC.
Design of vector
The AdEasy system (Stratagene, La Jolla, CA) was used to construct the Ad hsp fIL12. The two sub-units (p35 and p40) of the feline IL-12 were amplified from cDNA of CrFK cell line and sequence-verified. They were then connected into one gene expression unit by use of a flexible linker sequence (Gly4Ser). A 400-bp hsp70B promoter was then used to control the expression of the modified feline IL-12 gene. The whole hsp-fIL12 gene expression cassette was then transferred into an adenovirus shuttle plasmid. The shuttle plasmid was co-transfected into 293 cells to derive AdhspfIL12. Amplification and isolation of the virus was achieved following standard protocols Citation[53], Citation[54].
AdCA35 expressing LacZ was kindly provided by Dr Frank Graham (McMaster University).
Flow cytometry
CrFK cells were infected with an adenoviral construct with the feline IL-12 gene and enhanced green fluorescent protein (eGFP) placed under control of the constitutively expressing CMV promoter (Ad CMV fIL-12 + eGFP). This vector was used to determine the transfection efficiency of the adenoviral vector by detecting green fluorescence using the Coulter EPICS XL-MCL (Fullerton, CA) flow cytometer.
Real time PCR
Real-time reverse transcriptase PCR was used to detect and quantitatively express the production of feline interleukin-12, interferon-γ and GAPDH mRNA.
Sequences for the primers and probes were obtained from the literature Citation[55] and purchased from MWG Biotech (High Point, NC).
Feline GAPDH;
Forward GAPDH.57f, GCC GTG GAA TTT GCC GT;
Reverse GAPDH.138r, GCC ATC AAT GAC CCC TTC AT;
Probe GAPDH.77p, CTC AAC TAC ATG GTC TAC ATG TTC CAG TAT GAT TCC A;
IL 12 p40;
Forward IL12.253f, TGG CTT CAG TTG CAG GTT CTT;
Reverse IL12.333r, TGG ACG CTA TTC ACA AGC TCA;
Probe IL12.283p, CGG TTT GAT GAT GTC CCT GAT GAA GAA GCT;
IFN-γ;
Forward IFN.141f, TGG TGG GTC GCT TTT CGT AG;
Reverse IFN.225r, GAA GGA GAC AAT TTG GCT TTG AA; and
Probe IFN.152p, CAT TTT GAA GAA CTG GAA AGA GGA GAG TGA TAA AAC AAT.
The reporter dye attached covalently at the 5′ end was FAM (6-carboxyfluorescin) and the quencher bound to the 3′ end was TAMRA (6-carboxytetramethylrhodamine).
Real time PCR was performed using the Applied Biosystems ABI Prism 7000 (Foster City, CA). The amplification protocol was: 2 min at 50°C, 10 min at 95°C, 40 cycles of 15 s at 95°C and 60 s at 60°C. Cycle threshold values were obtained from the ABI software and the 2−ΔΔCt method Citation[56] was used to determine the relative expression of the genes of interest. Briefly, a standard housekeeping gene (e.g. GAPDH, β-actin, β2 microglobulin) is chosen as an internal control gene. This serves to normalize the amount of cDNA loaded for each reaction. An untreated control is selected as the ‘calibrator’. The relative expression data is then obtained as the fold change in gene expression normalized to the chosen endogenous reference gene and relative to the untreated control.
Prior to using the 2−ΔΔCt method for relative quantification, its use was validated for the primer and probe sequences and PCR conditions being employed in the experimental conditions. The methods and acceptable results are described in the literature Citation[56], Citation[57]. Briefly, the target cDNA was serially diluted and real time PCR performed. Cycle threshold values for the serially diluted samples are plotted on a graph with the log dilution on the x-axis and cycle threshold values on the y-axis. The slope of the line is used to obtain the efficiency of PCR amplification using the formula: Efficiency = 10(−1/slope) − 1.
Adenoviral infection
Crandell cells were plated in six-well plates in 2 ml of CrFK media at a density of 3 × 105 cells/well. Twenty-four hours later cells in one of the representative wells were trypsinized and counted using a haemocytometer. Total number of cells was thus obtained and the volume of adenoviral construct stock solution needed for the desired MOI was determined. This volume was dissolved in 300 μl of sterile PBS and added to each well after drawing off the media for a period of 30 min at room temperature after which fresh media was added back to all wells. The plates were then placed back in the incubator at 37°C and 5% CO2. Twenty-four hours were allowed for adenoviral infection and then the cells were subjected to hyperthermia. Cells were heated in a VWR Water Bath Model 1235PC. Heating was carried out in six-well plates that were placed in the water bath such that only the wells remained immersed in the heated water and water did not enter the plates. After placing the six-well tissue culture plates in the water bath, 8–10 min were allowed for temperature equilibration and then the plates were subsequently heated for 60 min.
PBMC stimulation
Naïve feline peripheral blood mononuclear cells (PBMC) were obtained from specific pathogen free cats and stimulated using 5 μg ml−1 Concavalin A for 48 h to upregulate the IL-12 receptor. These are the target cells for IL-12 protein in vivo, which leads to the production of IFN-γ. Supernatants were collected from the Ad hsp fIL12 infected CrFK cells at specified time-points after being heated at 41°C for 60 min. One hundred microlitres of these supernatants, containing IL-12 protein, were then added to Concavalin A stimulated PBMCs. Twelve hours after addition of the supernatants, PBMCs were lysed and real time PCR was carried out.
To assess presence of a dose-response relationship, CrFK cells infected with AdhspfIL12 were heated at 41°C for 60 min, supernatants were collected at 6 and 12 h post-HT and 100 or 200 μl was added to the PMBC media. Twelve hours later PBMCs were lysed, RNA extracted and cDNA synthesized. Real-time PCR were performed for IFN-γ mRNA levels.
Cell lysis and RNA Isolation
At standardized time points after heat treatment the cells were lysed and RNA isolated using TRIzol Reagent (GibcoBRL) as per manufacturer's protocol. RNA purity was assessed spectrophotometrically.
cDNA synthesis
Total isolated RNA was treated with DNase I (Invitrogen) to remove any genomic DNA. cDNA synthesis was then carried out using Superscript II RNase H− Reverse Transcriptase (Invitrogen, Carlsbad, CA). Each reaction mixture contained 10 μl of RNA solution to which was added 4 μl First strand buffer (5×), 1 μl dNTP (10 mM), 1 μl DTT (0.1 M), 0.25 μl Rnase out (40 U μl−1), 0.25 μl SuperScript II (200 U μl−1), 2 μl of random hexamers (300 ng μl−1) and 1.5 μl DNase, RNase free water giving a total of 20 μl per reaction mixture. This was incubated at 42°C for 50 min following which 30 μl of DNase, RNase free water was added to the mix and the enzymes inactivated by placing the reaction tubes on a 95°C heat-block for 5 min. The cDNA was then stored at −20°C until the time of RT-PCR.
Results
Transfection efficiency
CrFK cells infected with the Ad CMV fIL12 + eGFP construct at MOIs of 0, 5, 10, 20, 30 and 40 were split into two groups. In the first group the media containing any free-floating virus was left in the wells over all 3 days of the experiment. In the second group, after 24 h of infection, the CrFK media containing any free virus was removed, the cells washed with PBS and fresh media added.
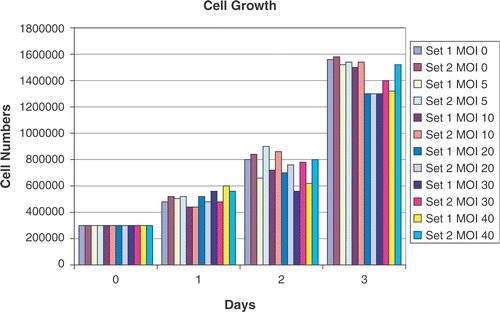
Cell counts were carried out to assess if adenoviral infection, per se, has any effect on cell growth. As shown in , there appears to be no major effect of adenoviral infection on exponential cell growth.
Figure 1. Cell growth in response to adenoviral infection. Cells numbers were counted in two sets of cells over a period of 3 days after plating. Each count is the average of three counts performed using a Coulter cell counter. In set 1, the viral construct added to the cells was left in the media over the duration of the experiment. In set 2, after 24 h of infection, the attached CrFK cells were washed with PBS to remove any free-floating virus and fresh media replaced. No major effect of adenoviral infection was noted on exponential cell growth at various MOIs (0, 5, 10, 20, 30 and 40).
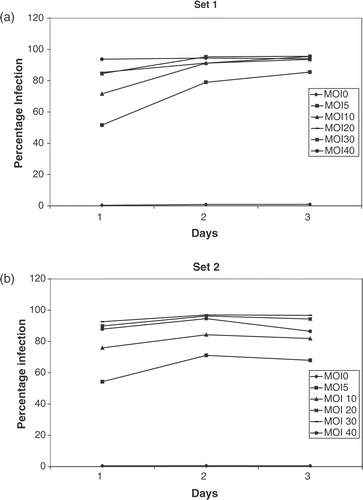
In the cells in sets 1 and 2 ( and ), the percentage of infected cells is a reflection of the MOI on day 1. By the second day, 90–95% of cells in both groups are infected. By day 3, the percentage of cells infected remains constant in set 1 but decreased slightly in set 2 where the extra virus had been washed off. These results show that the adenoviral construct is a highly efficient method of gene transfer in vitro.
Figure 2. Transfection efficiency of adenoviruses. CrFK cells were infected with the Ad CMV fIL-12 + eGFP construct and green fluorescence detected flow cytometrically over 3 days, starting 24 h after addition of virus (day 1). A high transfection efficiency (90–95%) was noted at MOIs > 20. The high percentage of infected cells was maintained over the 3 days in set 1 (a). There was a slight decrease in percentage infection on day 3 in cells where the free-floating virus had been washed off (b).
Validation of the 2−ΔΔCt method for relative quantification of feline cytokine mRNA
The 2−ΔΔCt method allows the amount of target to be normalized to an endogenous reference and expressed relative to a calibrator. Criteria for the use of this method have been described in the literature Citation[56], Citation[57]. For the three genes of interest the following slopes were obtained IL-12 = −3.1557, IFN-γ = −3.5056 and GAPDH = −3.3798 suggesting a high level of PCR amplification (for 100% efficiency, ideal slope = −3.3213) Citation[57]. Using the second method Citation[56], pairwise comparisons were made between IL-12–GAPDH and IFN-γ–GAPDH. For an ideal situation of slope = 0, slopes were obtained of 0.097 for IL12–GAPDH and 0.095 for IFN-γ–GAPDH. Thus, it was established that using the primer-probe sequences and PCR conditions, there was a high PCR amplification and the 2−ΔΔCt method could be used for relative quantification.
Temperature dependent increase in IL-12 mRNA expression
CrFK cells were infected at an MOI = 20. Twenty-four hours later, cells were heated in a water bath at the desired temperature. Each temperature experiment was carried out two-to-three times, each time in triplicate. The results are shown in and . Each graph shows the trends for increase and decrease in IL-12 mRNA expression in the CrFK cells infected with Ad hsp feline IL-12 construct relative to IL-12 mRNA expression in cells infected with Ad LacZ or no adenoviral infection. The maximum relative expression of IL-12 mRNA was noted at 6 h post-heating with a rapid decline over the next few hours. There was also a temperature-dependent increase in mRNA levels noted. Experiments were also done to determine the trend for the time points between 6–12 hours post-HT. The relative expression at 6, 8, 10 and 12 h showed a decreasing trend (data not shown). The heated cells show a biphasic response, with the initial peak due to hyperthermia followed by a rapid decline and again a rising trend at later time-points. However, in the non-heated cells only a steady increase is seen in the IL-12 mRNA levels without the initial peak. The phenomenon of increasing relative expression in non-heated cells and the secondary increase in heated cells at later time points can be ascribed to heat shock promoter induction by cell crowding and nutritional stress (Siddiqui et al. manuscript submitted). Relatively more IL-12 was produced in the unheated cells than any of the heated cells at the 48 h time point. The density of the unheated cells was slightly increased (1 × 106 cells/well) as compared to the heat treated cells (7 × 105 cells/well) which may account for some of the increase in IL-12 production. Additionally, it has been shown that HSP70A mRNA production secondary to heat shock promoter signalling results in a transient period of decreased responsiveness to subsequent triggering, a term referred to as thermotolerance Citation[58], Citation[59]. It is possible that the initial induction of the hsp promoter with heat had rendered these cells less responsive to the subsequent triggering induced by nutritional stress.
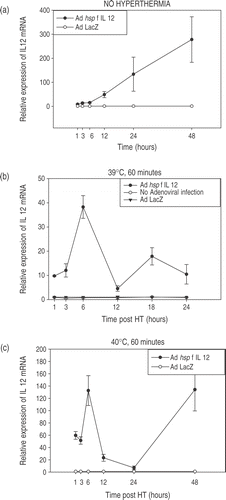
Figure 3. Temperature-dependent IL-12 expression. Profiles of temperature-dependent changes in IL-12 mRNA expression at specified time-points after hyperthermia. Cells were infected using the Ad hsp fIL-12 construct at MOI = 20. (a) Shows the minimal levels of expression at the early time points in unheated cells (kept in an incubator at 37°C, 5% CO2). (b−e) Show the profiles for heating at 39°C, 40°C, 41°C and 42°C for a duration of 60 min. Each temperature experiment was performed 2–3 times each in triplicate. The maximal expression of mRNA is seen at 6 h post-HT in all cases with a rapid decrease and then a secondary increase at 48 h. There is an ∼3.5-fold increase in maximal expression between 39°C, 40°C and 41°C. However, a supra-linear increase is seen at 42°C (note: log scale on y-axis). (f) Shows the maximal relative expression of IL-12 at the 6-h post-HT time point relative to temperature.
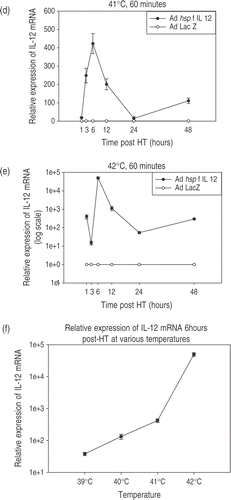
Table I. CrFK were infected at a MOI = 20 with the Ad hsp fIL-12 gene construct and heated at various non-cytotoxic temperatures for 60 min. The IL-12 mRNA relative expression relative to non-infected CrFK cells is presented as average ± SD. Each temperature experiment was performed 2–3 times each in triplicate.
There was an ∼3.5-fold increase in expression at the 6-h post-HT time point between 39°C, 40°C and 41°C. However, there was a supralinear increase seen at 42°C.
At 43°C and 44°C there is direct hyperthermia induced cytotoxicity as determined by trypan blue dye exclusion (). This is not desirable for these purposes as cell viability is essential. Only intact cellular machinery will be able to produce IL-12.
Figure 4. Assessment of cell viability at higher temperatures. The trypan blue dye-exclusion test was used to assess cellular viability after being heated at 43°C and 44°C for 60 min. Direct hyperthermia induced cytotoxicity reduced the percentage of viable cells to ∼88% for 43°C and ∼73% for 44°C as early as 1 h post-HT and this percentage remained constant up to 24 h post-HT.
Effect of multiplicity of infection
CrFK cells infected at MOIs of 5, 10, 20, 30 and 40 were heated at 40°C. A dose-dependent increase in maximal relative expression was seen ( and ). The curve, however, tends to flatten out at high MOIs.
Figure 5. Effect of multiplicity if infection (MOI). CrFK cells were infected at various MOIs and heated at 40°C for 60 min. A dose-response relationship is seen (a) with the curve for maximum relative expression tending to flatten out at higher MOIs (b).
Production of IFN-γ from IL-12 target cells
This study wanted to ascertain that the IL-12 mRNA being detected using the real time PCR was actually being translated into functional IL-12 protein. It can be seen in that maximal production of IFN-γ in feline PBMC occurred when they were stimulated with CrFK supernatant collected at 18 h post-HT. This suggests that the maximum IL-12 protein would have been present at that time-point. It has also been seen that the maximal mRNA levels are seen 6 h post-HT. Hence, it takes ∼12 h for IL-12 protein synthesis after mRNA transcription.
Figure 6. Induction of IFN-γ. Functionality of IL-12 was assessed by adding 100 μl of supernatants obtained from Ad hsp fIL-12 infected CrFK cells to Con A stimulated naïve feline PBMCs. These CrFK cells had been heated at 41°C for 60 min and supernatants were collected at 6, 12, 18 and 24 h post-HT. The PBMCs were lysed 12 h later (to allow action of IL-12 upon its target cells and the production of IFN-γ mRNA). The maximum induction was seen in the PBMCs in which supernatant from the 18 h post-HT CrFK cells had been added, suggesting that this sample contained the maximum amount of IL-12 protein.
Another experiment was done to see if there is a dose-response effect in IFN-γ production from the PBMCs in response to different doses of IL-12. Results shown in suggest a dose-response relationship in the induction of IFN-γ. The supernatant collected at the 6 h post-HT time point would have little IL-12 protein; therefore, no difference is seen for the relative expression but there would be more IL-12 protein present in the supernatants at the 12 h time point and a difference in relative expression of IFN-γ mRNA is noted. Hence, the IL-12 being produced in cells infected with the construct is functional and is able to induce production of its downstream product in a dose-dependent manner.
Table II. Supernatants from Ad hsp fIL-12 infected cells heated at 41°C for 60 min containing the IL-12 protein were collected at 6 and 12 h post-HT. These supernatants were added to naïve feline PBMCs in volumes of 100 or 200 μl to assess any dose response effect for IL-12 protein in inducing production of IFN-γ. At the earlier time point (6 h), when there would have been little IL-12 protein production, no difference was seen for the two doses, but by the 12 h time point an ∼2-fold increase in relative expression of IFN-γ mRNA was seen for the higher dose of added IL-12 protein.
Discussion
Interleukin 12, a pro-inflammatory cytokine, has been found to have significant anti-cancer activity in vitro and in vivo Citation[1–10], Citation[37], Citation[49], Citation[60]. However, there have been limited clinical successes largely owing to the toxicity profile of IL-12 and mortalities in Phase II trials when administered systemically Citation[42], Citation[43]. As an alternative to using systemic administration of this potent cytokine, one elected to test a replication defective adenoviral gene therapy approach to achieve relatively high intra-tumoural transgene expression, while minimizing systemic exposure to the cytokine. The feline interleukin-12 gene was placed under the control of a highly inducible heat shock promoter (400-bp hsp70B promoter) as an additional means of controlling cytokine expression. To answer important methodological questions that relate to optimizing the use of this approach clinically, a series of in vitro experiments were conducted. Important considerations were: (1) the MOI required to obtain significant levels of transgene expression, (2) timing of hyperthermia treatment after introduction of the adenovirus, (3) the effects of temperature on level of gene expression, (4) whether factors other than hyperthermia can alter levels of gene expression and (5) whether the gene product produced is functional.
Real time reverse transcriptase PCR was chosen as the method used to evaluate cytokine mRNA levels in part because the antibodies needed to determine protein levels of these feline cytokines are not readily available. Real time PCR offers several advantages, the most important being the ability to express results in quantitative terms rather than qualitative estimates and it is very sensitive to differences in cDNA copy numbers between samples. The 2−ΔΔCt method Citation[56] used to calculate relative expression of cytokine mRNA levels allows mathematical normalization of the amount of cDNA used per reaction and, hence, the reported fold expression is independent of the number of cells from which the mRNA is isolated.
GAPDH was used as the housekeeping gene. It is an important glycolytic pathway enzyme. It is used as an internal standard as its synthesis occurs in all nucleated cell types since it is necessary for survival. Despite recent criticisms about the use of GAPDH as the internal control Citation[61–63], other systematic studies have shown that it still remains one of the most stable control genes in various cell types Citation[55], Citation[64]. Importantly, its expression is not known to be affected by temperature Citation[65].
Adenoviral vectors are highly efficient in transferring genes of interest into target cells and the administration of adenoviruses in humans is well tolerated up to doses of 1011 particle units, demonstrating the relative safety of adenoviruses as vectors for in vivo gene therapy Citation[66]. In the laboratory, adenoviruses are easy to manipulate by classical recombinant DNA techniques and can be produced in large quantities in cell culture. They efficiently infect and transfer genes in a wide variety of cell types including dividing and non-dividing cells. Despite all these advantages, the coxsackievirus group B and adenovirus receptor (CAR) necessary for adenoviral entry is expressed at low levels by some tumours and the transfection efficiency of adenoviral vectors has been found highly dependent on the CAR receptor status in tumours Citation[67–69]. It has also been shown that CAR expression levels are inversely proportional to tumour grade in some systems Citation[70], Citation[71]. Because of this, adenoviruses have been modified to make them more permissive for cell entry by binding to non-CAR surface molecules such as integrins Citation[72] and the epidermal growth factor receptor Citation[73]. One will need to address this issue of tumour cell infectivity in a feline sarcoma model system.
Twenty-four hours were allowed for optimal adenoviral infection of the cells based on reports in literature Citation[48], Citation[60] and then the cells were heated. The maximal IL-12 mRNA expression was seen at 6 ho post-HT followed by a rapid decline which is consistent with that reported in the literature Citation[74]. Unheated cells produced IL-12 during the 48 h of culture which it was subsequently shown is due to progressive nutritional stress during in vitro growth (Siddiqui et al., manuscript submitted). It is unclear whether this same phenomenon would take place within the tumour micro-environment, but it seems unlikely to contribute to extra-tumoural IL-12 expression in well-perfused tissues in vivo.
A gradual 3.5-fold increase in the maximal expression of IL-12 mRNA was seen with temperature for 39°C, 40°C and 41°C. These fold increases seem to reflect the Arrhenius relationship for effectiveness of cell killing using hyperthermia Citation[75]. A 4-fold decrease in effectiveness is seen with each degree centigrade fall in temperature below the breakpoint. However, the level of gene expression seen at 42°C was much greater than that at 41°C. The reason for this supra-linear increase is unclear. At 43°C and 44°C there was significant cell killing from direct cytotoxic effects of hyperthermia on the CrFK cells. Also, in a clinical situation, it is difficult achieving temperatures greater than 42°C. Hence, a complete profile of IL-12 expression at these temperatures was not studied. It is essential for the cellular machinery to remain viable after heating to allow production of immunomodulatory cytokines.
Initially the cells were divided into three groups, those infected with the Ad hsp fel IL12 construct, those infected with Ad LacZ (control) and cells not infected with any virus. It was established that the IL-12 mRNA expression levels were similar in the non-infected and Ad LacZ infected cells. In later experiments, only two groups were studied, Ad hsp fel IL12 infected and Ad LacZ infected to control for any difference in expression due to adenoviral infection per se.
A logarithmic increase was seen with increasing MOI for the maximal expression at 6 h and the curve tended to plateau at higher MOI. The decrease with time was more gradual at higher MOIs (20, 30 and 40) than at lower MOIs (5 and 10). These findings serve to emphasize the importance of titrating the dose to be injected into tumours in in vivo studies with the tumour volume to try and achieve an optimum MOI. A similar dose response was not seen in the study by Borrelli et al. Citation[48]. They had studied enhanced GFP expression using a CMV promoter and they attributed the non-proportionality to the non-linear CMV promoter activity. This study did not specifically address multiple heat treatments to increase the duration of IL-12 production, but others have shown sustained high levels of luciferase expression with repeated hsp promoter induction Citation[76]. These investigators did note a degree of thermotolerance where the subsequent heat treatment needed to be increased or delayed for maximal re-stimulation. Given the potential toxicity of IL-12, the risks and benefits of prolonged IL-12 expression would need to be studied in vivo for the particular expression system.
The PBMC stimulation experiments showed that there is an ∼12 h time-interval between maximal mRNA expression and maximal protein expression. This is also consistent with earlier reports using the hsp promoter Citation[48], Citation[60].
This study demonstrates that the heat shock promoter is a highly inducible promoter which can be triggered by hyperthermia. This promoter then causes the production of feline IL-12 in a temperature and multiplicity of infection (MOI) dependent manner. The IL-12 protein is effective in inducing the production of IFN-γ from feline peripheral blood mononuclear cells. This expression system, thus, offers as attractive approach to local cancer immunotherapy while minimizing the potential systemic toxicity of interleukin 12.
References
- Brunda MJ, Luistro L, Hendrzak JA, Fountoulakis M, Garotta G, Gately MK. Role of interferon-gamma in mediating the antitumor efficacy of interleukin-12. Journal of Immunotherapy Emphasis Tumor Immunology 1995; 17: 71–77
- Puisieux I, Odin L, Poujol D, Moingeon P, Tartaglia J, Cox W, Favrot M. Canarypox virus-mediated interleukin 12 gene transfer into murine mammary adenocarcinoma induces tumor suppression and long-term antitumoral immunity. Human Gene Therapy 1998; 9: 2481–2492
- Seetharam S, Staba MJ, Schumm LP, Schreiber K, Schreiber H, Kufe DW, Weichselbaum RR. Enhanced eradication of local and distant tumors by genetically produced interleukin-12 and radiation. International Journal of Oncology 1999; 15: 769–773
- Golab J, Zagozdzon R. Antitumor effects of interleukin-12 in pre-clinical and early clinical studies (Review). International Journal of Molecular Medicine 1999; 3: 537–544
- Shi F, Rakhmilevich AL, Heise CP, Oshikawa K, Sondel PM, Yang NS, Mahvi DM. Intratumoral injection of interleukin-12 plasmid DNA, either naked or in complex with cationic lipid, results in similar tumor regression in a murine model. Molecular Cancer Therapy 2002; 1: 949–957
- Liu Y, Ehtesham M, Samoto K, Wheeler CJ, Thompson RC, Villarreal LP, Black KL, Yu JS. In situ adenoviral interleukin 12 gene transfer confers potent and long-lasting cytotoxic immunity in glioma. Cancer Gene Therapy 2002; 9: 9–15
- Yao L, Sgadari C, Furuke K, Bloom ET, Teruya-Feldstein J, Tosato G. Contribution of natural killer cells to inhibition of angiogenesis by interleukin-12. Blood 1999; 93: 1612–1621
- Voest EE, Kenyon BM, O’Reilly MS, Truitt G, D'Amato RJ, Folkman J. Inhibition of angiogenesis in vivo by interleukin 12. Journal of the National Cancer Institute 1995; 87: 581–586
- Duda DG, Sunamura M, Lozonschi L, Kodama T, Egawa S, Matsumoto G, Shimamura H, Shibuya K, Takeda K, Matsuno S. Direct in vitro evidence and in vivo analysis of the antiangiogenesis effects of interleukin 12. Cancer Research 2000; 60: 1111–1116
- Coughlin CM, Salhany KE, Wysocka M, Aruga E, Kurzawa H, Chang AE, Hunter CA, Fox JC, Trinchieri G, Lee WM. Interleukin-12 and interleukin-18 synergistically induce murine tumor regression which involves inhibition of angiogenesis. Journal of Clinical Investigations 1998; 101: 1441–1452
- Kobayashi M, Fitz L, Ryan M, Hewick RM, Clark SC, Chan S, Loudon R, Sherman F, Perussia B, Trinchieri G. Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. Journal of Experimental Medicine 1989; 170: 827–845
- Gately MK, Wolitzky AG, Quinn PM, Chizzonite R. Regulation of human cytolytic lymphocyte responses by interleukin-12. Cell Immunology 1992; 143: 127–142
- Metzger DW, McNutt RM, Collins JT, Buchanan JM, Van Cleave VH, Dunnick WA. Interleukin-12 acts as an adjuvant for humoral immunity through interferon-gamma-dependent and -independent mechanisms. European Journal of Immunology 1997; 27: 1958–1965
- Pham-Nguyen KB, Yang W, Saxena R, Thung SN, Woo SL, Chen SH. Role of NK and T cells in IL-12-induced anti-tumor response against hepatic colon carcinoma. International Journal of Cancer 1999; 81: 813–819
- Kodama T, Takeda K, Shimozato O, Hayakawa Y, Atsuta M, Kobayashi K, Ito M, Yagita H, Okumura K. Perforin-dependent NK cell cytotoxicity is sufficient for anti-metastatic effect of IL-12. European Journal of Immunology 1999; 29: 1390–1396
- Watanabe M, Fenton RG, Wigginton JM, McCormick KL, Volker KM, Fogler WE, Roessler PG, Wiltrout RH. Intradermal delivery of IL-12 naked DNA induces systemic NK cell activation and Th1 response in vivo that is independent of endogenous IL-12 production. Journal of Immunology 1999; 163: 1943–1950
- Rakhmilevich AL, Janssen K, Hao Z, Sondel PM, Yang NS. Interleukin-12 gene therapy of a weakly immunogenic mouse mammary carcinoma results in reduction of spontaneous lung metastases via a T-cell-independent mechanism. Cancer Gene Therapy 2000; 7: 826–838
- Cui J, Shin T, Kawano T, Sato H, Kondo E, Toura I, Kaneko Y, Koseki H, Kanno M, Taniguchi M. Requirement for Valpha14 NKT cells in IL-12-mediated rejection of tumors. Science 1997; 278: 1623–1626
- Zilocchi C, Stoppacciaro A, Chiodoni C, Parenza M, Terrazzini N, Colombo MP. Interferon gamma-independent rejection of interleukin 12-transduced carcinoma cells requires CD4+ T cells and Granulocyte/Macrophage colony-stimulating factor. Journal of Experimental Medicine 1998; 188: 133–143
- North RJ, Kirstein DP. T-cell-mediated concomitant immunity to syngeneic tumors. I. Activated macrophages as the expressors of nonspecific immunity to unrelated tumors and bacterial parasites. Journal of Experimental Medicine 1977; 145: 275–292
- Berendt MJ, North RJ, Kirstein DP. The immunological basis of endotoxin-induced tumor regression. Requirement for a pre-existing state of concomitant anti-tumor immunity. Journal of Experimental Medicine 1978; 148: 1560–1569
- Le HN, Lee NC, Tsung K, Norton JA. Pre-existing tumor-sensitized T cells are essential for eradication of established tumors by IL-12 and cyclophosphamide plus IL-12. Journal of Immunology 2001; 167: 6765–6772
- Saffran DC, Horton HM, Yankauckas MA, Anderson D, Barnhart KM, Abai AM, Hobart P, Manthorpe M, Norman JA, Parker SE. Immunotherapy of established tumors in mice by intratumoral injection of interleukin-2 plasmid DNA: Induction of CD8+ T-cell immunity. Cancer Gene Therapy 1998; 5: 321–330
- Segal JG, Lee NC, Tsung YL, Norton JA, Tsung K. The role of IFN-gamma in rejection of established tumors by IL-12: source of production and target. Cancer Research 2002; 62: 4696–4703
- Nastala CL, Edington HD, McKinney TG, Tahara H, Nalesnik MA, Brunda MJ, Gately MK, Wolf SF, Schreiber RD, Storkus WJ. Recombinant IL-12 administration induces tumor regression in association with IFN-gamma production. Journal of Immunology 1994; 153: 1697–1706
- Zou JP, Yamamoto N, Fujii T, Takenaka H, Kobayashi M, Herrmann SH, Wolf SF, Fujiwara H, Hamaoka T. Systemic administration of rIL-12 induces complete tumor regression and protective immunity: response is correlated with a striking reversal of suppressed IFN-gamma production by anti-tumor T cells. International Immunology 1995; 7: 1135–1145
- Yu WG, Yamamoto N, Takenaka H, Mu J, Tai XG, Zou JP, Ogawa M, Tsutsui T, Wijesuriya R, Yoshida R, Hermann S, Fujiwara H, Hamaoka T. Molecular mechanisms underlying IFN-gamma-mediated tumor growth inhibition induced during tumor immunotherapy with rIL-12. International Immunology 1996; 8: 855–865
- Tsung K, Meko JB, Tsung YL, Peplinski GR, Norton JA. Immune response against large tumors eradicated by treatment with cyclophosphamide and IL-12. Journal of Immunology 1998; 160: 1369–1377
- Manetti R, Gerosa F, Giudizi MG, Biagiotti R, Parronchi P, Piccinni MP, Sampognaro S, Maggi E, Romagnani S, Trinchieri G. Interleukin 12 induces stable priming for interferon gamma (IFN-gamma) production during differentiation of human T helper (Th) cells and transient IFN-gamma production in established Th2 cell clones. Journal of Experimental Medicine 1994; 179: 1273–1283
- Brunda MJ, Luistro L, Warrier RR, Wright RB, Hubbard BR, Murphy M, Wolf SF, Gately MK. Antitumor and antimetastatic activity of interleukin 12 against murine tumors. Journal of Experimental Medicine 1993; 178: 1223–1230
- Tannenbaum CS, Wicker N, Armstrong D, Tubbs R, Finke J, Bukowski RM, Hamilton TA. Cytokine and chemokine expression in tumors of mice receiving systemic therapy with IL-12. Journal of Immunology 1996; 156: 693–699
- Cavallo F, Signorelli P, Giovarelli M, Musiani P, Modesti A, Brunda MJ, Colombo MP, Forni G. Antitumor efficacy of adenocarcinoma cells engineered to produce interleukin 12 (IL-12) or other cytokines compared with exogenous IL-12. Journal of the National Cancer Institute 1997; 89: 1049–1058
- Tahara H, Zeh HJ, 3rd, Storkus WJ, Pappo I, Watkins SC, Gubler U, Wolf SF, Robbins PD, Lotze MT. Fibroblasts genetically engineered to secrete interleukin 12 can suppress tumor growth and induce antitumor immunity to a murine melanoma in vivo. Cancer Research 1994; 54: 182–189
- Zitvogel L, Tahara H, Robbins PD, Storkus WJ, Clarke MR, Nalesnik MA, Lotze MT. Cancer immunotherapy of established tumors with IL-12. Effective delivery by genetically engineered fibroblasts. Journal of Immunology 1995; 155: 1393–1403
- Wu NZ, Klitzman B, Dodge R, Dewhirst MW. Diminished leukocyte-endothelium interaction in tumor microvessels. Cancer Research 1992; 52: 4265–4268
- Lohr F, Huang Q, Hu K, Dewhirst MW, Li CY. Systemic vector leakage and transgene expression by intratumorally injected recombinant adenovirus vectors. Clinical Cancer Research 2001; 7: 3625–3628
- Lohr F, Hu K, Huang Q, Zhang L, Samulski TV, Dewhirst MW, Li CY. Enhancement of radiotherapy by hyperthermia-regulated gene therapy. International Journal of Radiation Oncology, Biology & Physics 2000; 48: 1513–1518
- Lode HN, Dreier T, Xiang R, Varki NM, Kang AS, Reisfeld RA. Gene therapy with a single chain interleukin 12 fusion protein induces T cell-dependent protective immunity in a syngeneic model of murine neuroblastoma. Proceedings of the National Academy of Sciences (USA) 1998; 95: 2475–2480
- Rakhmilevich AL, Turner J, Ford MJ, McCabe D, Sun WH, Sondel PM, Grota K, Yang NS. Gene gun-mediated skin transfection with interleukin 12 gene results in regression of established primary and metastatic murine tumors. Proceedings of the National Academy of Sciences (USA) 1996; 93: 6291–6296
- Car BD, Eng VM, Lipman JM, Anderson TD. The toxicology of interleukin-12: A review. Toxicology & Pathology 1999; 27: 58–63
- Oppenheim JJ, Feldmann M, Durum SK. Cytokine reference: a compendium of cytokines and other mediators of host defense. Academic Press, San Diego 2001
- Cohen J. IL-12 deaths: Explanation and a puzzle. Science 1995; 270: 908
- Atkins MB, Robertson MJ, Gordon M, Lotze MT, DeCoste M, DuBois JS, Ritz J, Sandler AB, Edington HD, Garzonie PD, Mier JW, Canning CM, Battiato L, Tahara H, Sherman ML. Phase I evaluation of intravenous recombinant human interleukin 12 in patients with advanced malignancies. Clinical Cancer Research 1997; 3: 409–417
- Whitley D, Goldberg SP, Jordan WD. Heat shock proteins: a review of the molecular chaperones. Journal of Vascular Surgery 1999; 29: 748–751
- Parcellier A, Gurbuxani S, Schmitt E, Solary E, Garrido C. Heat shock proteins, cellular chaperones that modulate mitochondrial cell death pathways. Biochemistry & Biophysical Research Communications 2003; 304: 505–512
- Nollen EA, Morimoto RI. Chaperoning signaling pathways: molecular chaperones as stress-sensing ‘heat shock’ proteins. Journal of Cell Science 2002; 115: 2809–2816
- Morimoto RI, Tissiáeres A, Georgopoulos C. The biology of heat shock proteins and molecular chaperones. Cold Spring Harbor Laboratory Press, Plainview, NY 1994
- Borrelli MJ, Schoenherr DM, Wong A, Bernock LJ, Corry PM. Heat-activated transgene expression from adenovirus vectors infected into human prostate cancer cells. Cancer Research 2001; 61: 1113–1121
- Li CY, Dewhirst MW. Hyperthermia-regulated immunogene therapy. International Journal of Hyperthermia 2002; 18: 586–596
- Wang Q, Finer MH. Second-generation adenovirus vectors. Natural Medicine 1996; 2: 714–716
- Yang Y, Nunes FA, Berencsi K, Furth EE, Gonczol E, Wilson JM. Cellular immunity to viral antigens limits E1-deleted adenoviruses for gene therapy. Proceedings of the National Academy of Sciences (USA) 1994; 91: 4407–4411
- Zhang Y, Schneider RJ. Adenovirus inhibition of cell translation facilitates release of virus particles and enhances degradation of the cytokeratin network. Journal of Virology 1994; 68: 2544–2555
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proceedings of the National Academy of Sciences (USA) 1998; 95: 2509–2514
- Graham FL, Smiley J, Russell WC, Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. Journal of General Virology 1977; 36: 59–74
- Leutenegger CM, Mislin CN, Sigrist B, Ehrengruber MU, Hofmann-Lehmann R, Lutz H. Quantitative real-time PCR for the measurement of feline cytokine mRNA. Veterinary Immunology & Immunopathology 1999; 71: 291–305
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001; 25: 402–408
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Research 2001; 29: 45
- Mivechi NF, Ouyang H, Monson JM, Hahn GM. Correlation of heat resistance and HSP-70A mRNA levels in human tumor cells measured by competitive quantitative polymerase chain reaction. International Journal of Radiation Oncology, Biology & Physics 1994; 30: 141–149
- Li GC, Mivechi NF, Weitzel G. Heat shock proteins, thermotolerance, and their relevance to clinical hyperthermia. International Journal of Hyperthermia 1995; 11: 459–488
- Huang Q, Hu JK, Lohr F, Zhang L, Braun R, Lanzen J, Little JB, Dewhirst MW, Li CY. Heat-induced gene expression as a novel targeted cancer gene therapy strategy. Cancer Research 2000; 60: 3435–3439
- Yamada H, Chen D, Monstein HJ, Hakanson R. Effects of fasting on the expression of gastrin, cholecystokinin, and somatostatin genes and of various housekeeping genes in the pancreas and upper digestive tract of rats. Biochemical and Biophysics Research Communications 1997; 231: 835–838
- Foss DL, Baarsch MJ, Murtaugh MP. Regulation of hypoxanthine phosphoribosyltransferase, glyceraldehyde-3-phosphate dehydrogenase and beta-actin mRNA expression in porcine immune cells and tissues. Animals in Biotechnology 1998; 9: 67–78
- Bustin SA. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. Journal of Molecular Endocrinology 2000; 25: 169–193
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biology 2002; 3, RESEARCH0034
- Coss RA, Storck CW, Daskalakis C, Berd D, Wahl ML. Intracellular acidification abrogates the heat shock response and compromises survival of human melanoma cells. Molecular Cancer Therapy 2003; 2: 383–388
- Harvey BG, Maroni J, O’Donoghue KA, Chu KW, Muscat JC, Pippo AL, Wright CE, Hollmann C, Wisnivesky JP, Kessler PD, Rasmussen HS, Rosengart TK, Crystal RG. Safety of local delivery of low- and intermediate-dose adenovirus gene transfer vectors to individuals with a spectrum of morbid conditions. Human Gene Therapy 2002; 13: 15–63
- Gu W, Ogose A, Kawashima H, Ito M, Ito T, Matsuba A, Kitahara H, Hotta T, Tokunaga K, Hatano H, Morita T, Urakawa S, Yoshizawa T, Kuwano R, Endo N. High-level expression of the coxsackievirus and adenovirus receptor messenger RNA in osteosarcoma, Ewing's sarcoma, and benign neurogenic tumors among musculoskeletal tumors. Clinical Cancer Research 2004; 10: 3831–3838
- Witlox MA, Van Beusechem VW, Grill J, Haisma HJ, Schaap G, Bras J, Van Diest P, De Gast A, Curiel DT, Pinedo HM, Gerritsen WR, Wuisman PI. Epidermal growth factor receptor targeting enhances adenoviral vector based suicide gene therapy of osteosarcoma. Journal of Gene Medicine 2002; 4: 510–516
- Graat HC, Wuisman PI, van Beusechem VW, Carette JE, Gerritsen WR, Bras J, Schaap GR, Kaspers GJ, Ogose A, Gu W, Kawashima H, Hotta T. Coxsackievirus and adenovirus receptor expression on primary osteosarcoma specimens and implications for gene therapy with recombinant adenoviruses. Clinical Cancer Research 2005; 11: 2445–2447, author reply 2447–2448
- Rauen KA, Sudilovsky D, Le JL, Chew KL, Hann B, Weinberg V, Schmitt LD, McCormick F. Expression of the coxsackie adenovirus receptor in normal prostate and in primary and metastatic prostate carcinoma: potential relevance to gene therapy. Cancer Research 2002; 62: 3812–3818
- Okegawa T, Pong RC, Li Y, Bergelson JM, Sagalowsky AI, Hsieh JT. The mechanism of the growth-inhibitory effect of coxsackie and adenovirus receptor (CAR) on human bladder cancer: a functional analysis of car protein structure. Cancer Research 2001; 61: 6592–6600
- Dmitriev I, Krasnykh V, Miller CR, Wang M, Kashentseva E, Mikheeva G, Belousova N, Curiel DT. An adenovirus vector with genetically modified fibers demonstrates expanded tropism via utilization of a coxsackievirus and adenovirus receptor-independent cell entry mechanism. Journal of Virology 1998; 72: 9706–9713
- Miller CR, Buchsbaum DJ, Reynolds PN, Douglas JT, Gillespie GY, Mayo MS, Raben D, Curiel DT. Differential susceptibility of primary and established human glioma cells to adenovirus infection: targeting via the epidermal growth factor receptor achieves fiber receptor-independent gene transfer. Cancer Research 1998; 58: 5738–5748
- Li GC, He F, Shao X, Urano M, Shen L, Kim D, Borrrelli M, Leibel SA, Gutin PH, Ling CC. Adenovirus-mediated heat-activated antisense Ku70 expression radiosensitizes tumor cells in vitro and in vivo. Cancer Research 2003; 63: 3268–3274
- Sapareto SA, Dewey WC. Thermal dose determination in cancer therapy. International Journal of Radiation Oncology, Biology & Physics 1984; 10: 787–800
- Xu L, Zhao Y, Zhang Q, Li Y, Xu Y. Regulation of transgene expression in muscles by ultrasound-mediated hyperthermia. Gene Therapy 2004; 11: 894–900