Abstract
Purpose: In cytokine immunotherapy of cancer it is critical to deliver sufficiently high local cytokine concentrations in order to reach the therapeutic threshold needed for clinical efficacy. Simultaneously, for optimal clinical safety adverse effects caused by high systemic cytokine levels must be minimized. One of the most promising anti-cancer therapeutic cytokines, granulocyte-macrophage colony-stimulating factor (GM-CSF), has elicited anti-tumour immune responses in animal studies and clinical trials. However, the clinical efficacy has been limited, with local GM-CSF levels being therapeutically insufficient and systemic toxicity being a limiting factor.
Methods: To address these problems we have developed a novel GM-CSF expression vector, pAD-HotAmp-GM-CSF, which can provide high levels of GM-CSF expression, and induction of cytokine expression to limited tissue areas. This expression system combines inducible and amplifying elements in a single multi-genic construct. The first transcriptional unit contains the inducible element, the heat shock protein 70B (HSP70B) promoter that regulates expression of the transcription-activating factor tat.
Results: Upon the binding of tat to the second promoter, the HIV2 long terminal repeat amplifies downstream gene expression of the therapeutic cytokine GM-CSF. Moderate hyperthermia at 42°C for 30 min induced GM-CSF expression in pAD-HotAmp-GM-CSF that was over 2.5- and 2.8-fold higher than levels reached with HSP70B promoter alone and the prototypical human cytomegalovirus promoter.
Conclusions: Thus, the inducible amplifier vector, pAD-HotAmp-GM-CSF, represents a novel system for regulated and enhanced GM-CSF expression, which enables both greater efficacy and safety in cytokine immunotherapy of cancer.
Introduction
Strategies for the treatment of cancer include immunotherapy with recombinant cytokines, where the administration of single or combinations of cytokines can enhance a host's anti-tumour responses Citation[1]. Therefore, the composition of cytokines in the tumour micro-environment is critical Citation[2]. Systemic cytokine administration, in doses high enough to provide therapeutic tumour micro-environment levels, has proven toxic to the patient Citation[3], Citation[4]. Gene therapy with therapeutic cytokine genes provides an alternative strategy Citation[1], Citation[5–7] if the transfection or expression is local. Thus, systemic adverse effects can be circumvented. However, locally targeted in vivo transfection still remains a difficult task Citation[1], Citation[8]. Yet gene therapy with therapeutic cytokine genes shows great promise, as genetically engineered tumour cells secreting cytokines are often rejected quickly in mice that have additionally developed immune memory against subsequent challenges, even with poorly immunogenic tumours Citation[8]. Among the immunotherapy cytokines under investigation, granulocyte-macrophage colony-stimulating factor (GM-CSF) has demonstrated potential efficacy in modulating the immune system to combat cancer. GM-CSF is a factor with broad effects on progenitor cells of the myeloid lineage for proliferation and differentiation Citation[9].
In animal studies, GM-CSF cancer immunotherapy has been extensively researched. Vaccination with GM-CSF-secreting tumour cells has induced either complete rejection or development of tumours with smaller volumes compared to the parental tumours Citation[10], Citation[11], has induced significant decrease or inhibition of subcutaneous tumour growth Citation[7], Citation[10], has prolonged survival times Citation[7], Citation[11] and has induced protection from subsequent tumour challenges of the treated mice Citation[6]. In multiple murine models for head and neck squamous cell carcinoma Citation[12] and for metastatic melanoma Citation[13], potent and specific protection and in some cases long-lasting anti-tumour immunity against subsequent tumour challenges could be stimulated through this vaccination strategy. The response effect of tumour growth inhibition to GM-CSF strongly correlates with dose-dependence Citation[10], Citation[14]. Continuous high concentrations of the cytokine in the tumour micro-environment seem to be essential Citation[6], Citation[7], Citation[15].
Vaccination with GM-CSF-transfected tumour cells in a phase I clinical trial with patients having metastatic non-small-cell lung carcinomas elicited dendritic cell (DC), macrophage, granulocyte and lymphocyte infiltrates in the majority of patients and T lymphocyte and plasma cell infiltrates with tumour necrosis in some patients where anti-tumour immunity was enhanced Citation[13]. Lymphocyte infiltrates were also reported at the site of intra-dermal vaccination with GM-CSF-transfected melanoma and sarcoma cells in patients of a phase I study Citation[16]. Treatment with a GM-CSF-encoding vaccinia virus was found to increase the cytotoxic and phagocytic activity of peritoneal macrophages Citation[7]. Excised tissue sections from GM-CSF-producing tumour inoculation sites revealed, in contrast to the parental tumour, a dense inflammatory infiltrate Citation[10]. The infiltrate was composed of neutrophils, tissue macrophages, numerous CD4+ and CD8+ lymphocytes but few melanoma cells. Only within the site inoculated with clones secreting high GM-CSF levels were large numbers of DCs and cells expressing the B7.2 co-stimulatory molecule detected Citation[10]. High local and continuous cytokine concentrations in the tumour micro-environment seem to be essential for an increased macrophage and DC activation Citation[7], as well as an increased and significantly more effective tumour lysis mediated by blood lymphocytes Citation[6].
However, cytokine immunotherapy animal studies have generally yielded few successful clinical responses Citation[4], Citation[17–19], possibly because the required cytokine levels for humans were not achieved. A study using GM-CSF operating as an adjuvant in vaccination showed limited clinical benefits Citation[10]. Solitary T-cell responses may be incomplete to produce significant anti-cancer objective responses in patients with an advanced state of disease Citation[14]. In one study of intra-dermal injection of GM-CSF-transfected melanoma cells, one patient exhibited positive delayed-type hypersensitivity, a significant histological inflammatory response and developed clinically stable disease Citation[16]. In this patient GM-CSF production of greater than 30 ng per 106 cells per 24 h in vitro was observed, contrasted with the majority of the remaining patients who stayed below 3 ng of GM-CSF production. Promising clinical anti-tumour immune responses to vaccination with irradiated tumour cells, engineered to secrete GM-CSF, were detected in some of the treated patients suffering from kidney or prostate cancer Citation[20], non-small-cell lung cancer (NSCLC) Citation[13], Citation[21] and metastatic melanoma Citation[22], Citation[23]. A dose-related anti-tumour response to GM-CSF-secreting vaccines was observed Citation[20], Citation[21]. The sporadic potent clinical outcome in these patients supports the necessity of high local GM-CSF expression in order to reach the therapeutic threshold needed for anti-tumour immunotherapy.
In general the problem of expression of sufficient local levels of therapeutic anti-tumour cytokine has remained unsolved Citation[1], Citation[8]. Strategies to improve cytokine immunotherapy vectors have focused on improving cytokine expression using different promoters. It has been generally concluded that the cytomegalovirus (CMV) promoter is the strongest currently available promoter Citation[24–27]. Yet, further improvements in GM-CSF gene expression will be necessary in order to consistently meet the required therapeutic threshold Citation[28]. To achieve expression levels of GM-CSF higher than those possible using the CMV promoter, we have constructed a novel vector that is able to effectively ‘amplify’ the gene expression of any promoter, including CMV. The amplifier design applied in this study involves two interacting genes. The first gene, encoding the transcription-activator protein tat, derived from the human immunodeficiency virus (HIV), can activate the HIV2 long terminal repeat (LTR) promoter controlling the second gene, i.e. GM-CSF. To insure that the extremely high level of GM-CSF expression results in only high local tissue levels of GM-CSF, the expression of the tat gene in the construct is under the control of the well-characterized inducible human heat shock protein 70B (HSP70B) promoter Citation[29]. This promoter is induced by a set of heat shock factors that respond to diverse forms of physiological and environmental stress including hyperthermia, heavy metals, oxidative stress, anti-inflammatory drugs and some toxic agents Citation[30], Citation[31]. The tat-amplified expression of GM-CSF from this vector will, thus, be regulatable, with induction via a mild heat shock. The novel heat-inducible expression vector for amplified expression of GM-CSF addresses the need for high-level GM-CSF expression, as well as providing localized GM-CSF expression via locally applied hyperthermia.
Materials and methods
Cell line and culture conditions
B16 murine melanoma cells were obtained from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured in complete medium consisting of RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% (v/v) foetal bovine serum (Gemini Bioproducts, Calabasas, CA, USA), 100 U ml−1 penicillin, 100 µg ml−1 streptomycin, 5 ng ml−1 gentamycin sulphate, 1 mM non-essential amino acids, 1 mM sodium pyruvate, 0.001% (v/v) β-mercaptoethanol and 2 mM L-glutamine. Cells were maintained at 37°C in a 5% CO2 atmosphere. Adherent cells were detached for sub-culture (every 2–3 days) using 0.05% trypsin solution.
Lipid-mediated transfections
Cells were seeded in 6-well tissue culture plates (Falcon, Franklin Lakes, NJ, USA) at a concentration of 2 × 105 cells/well in 2 ml complete medium. Cells were incubated at 37°C in 5% CO2 for 24 h until they reached 60–80% confluency. Transfection of cells with plasmids was performed 24 h after sub-culture by lipid-mediated transfection (lipofection) using the lipid composition of GenePORTER2 Transfection Reagent (GTS Gene Therapy Systems Inc., San Diego, CA, USA). The lipid-DNA complex (lipoplex) preparation with GenePORTER2 was performed according to the manufacturer's protocol. Briefly, 2 µg of plasmid DNA were added to DNA diluent (GTS Gene Therapy Systems Inc.) to give a total volume of 50 µl. GenePORTER2 lipid formulation was diluted 1:5 in serum-free OptiMEM medium (GIBCO BRL, Gaithersburg, MD, USA) in a total volume of 50 µl. DNA solution and lipid mixture were combined and incubated for 5–10 min. That produced a DNA/lipid ratio of 2 µg of plasmid DNA to 5 µl of GenePORTER2 lipid. For each well, cells were washed with 2 ml PBS and subsequently incubated with 1 ml OptiMEM medium and 100 µl of the DNA/lipid mixture at 37°C in 5% CO2. After 4 h of exposure to the cells, the DNA-lipid-containing transfection medium was replaced by 2 ml complete medium. Supernatants were collected 24, 48 and 72 h after heat shock and stored at −80°C until analysed. After each harvest medium was renewed completely by fresh complete medium.
Transfection efficiency
Transfection efficiencies were assessed 24 h post-transfection by flow cytometric analysis of fluorescence from green fluorescence protein (GFP) after transfection with a reporter construct, expressing GFP under CMV promoter control. Cells were detached from their wells using a 0.05% trypsin solution, washed twice in PBS and then prepared in a 1% paraformaldehyde solution for fixation before analysis. Flow cytometry was performed using a FACStarPLUS flow cytometer (Becton Dickinson Immunocytometry Systems, Mountain View, CA, USA).
Heat shock treatment
Twenty-four hours post-transfection, cells were heat-treated in 6-well plates. Cells were washed with 2 ml PBS and 0.5 ml of complete medium was added to each well after removal of the wash buffer. This volume reduction served the purpose of minimizing heating-up periods of the suspension to favour heat shock conditions in contrast to gradual warming. Plates were sealed with parafilm and submersed in a Precision Dual Chamber water bath (Lehman Scientific, Wrightsville, PA, USA) at indicated pre-set temperatures and durations. Following heat exposure, wells were each filled up to 2 ml with complete medium and cells were returned to 37°C in a 5% CO2 atmosphere.
GM-CSF measurement
Cell culture supernatants were collected 24, 48 and 72 h after heat treatment as indicated. After each supernatant harvest, the remaining supernatant of each well was replaced with 2 ml fresh complete medium. GM-CSF levels were assayed in an ELISA using the Mouse GM-CSF BD OptEIA ELISA Set (BD Biosciences Pharmingen, San Diego, CA, USA) according to the manufacturer's protocol. Dilutions were performed in Assay Diluent for OptEIA ELISA Sets (BD Biosciences Pharmingen). Reactions were performed in 96-well flat-bottomed plates for ELISA (Falcon, Franklin Lakes, NJ, USA) with the TMB Substrate Reagent Set (BD Biosciences Pharmingen). Plates were read using a 96-well micro-plate reader (Bio-Rad, Hercules, CA, USA).
Heat shock survival measurement
B16 cells were seeded in 6-well plates at 2 × 105 cells/well in 2 ml complete medium and allowed to grow for 24 h prior to heat shock. Medium was then removed and 0.5 ml fresh medium was added to each well. Plates were sealed in parafilm and subjected to heat shock in a water bath at 40, 42 or 44°C for various time periods. After heating, 2 ml of fresh medium was added and cells were returned to 37°C incubator. After 24 h, medium was removed and saved. Wells were rinsed with PBS and treated with trypsin to detach cells. Detached cells were combined with the medium saved and all cells were counted. Viability was determined by trypan blue exclusion.
Luciferase assay
Cells were seeded at 2 × 105 cells/well in a 6-well tissue culture plate in 2 ml complete medium and incubated at 37°C until the cells were 40–60% confluent. DMRIE-C reagent (Invitrogen, Carlsbad, CA, USA) was used at a lipid:DNA ratio of 6:1. For each well, 1 µg of plasmid vector DNA was suspended in 500 µl OPTI Reduced Serum Medium (Gibco-BRL). Six microlitres of DMRIE-C lipid were suspended in 500 µl OPTI medium. The DNA and DMRIE-C solutions were mixed together and incubated at room temperature for 30 min. Cells were washed with OPTI medium and incubated with 1 ml DNA/lipid complex mixture at 37°C. Four hours later, the mixture was removed and 2 ml fresh medium was replaced in each well. Twenty-four hours after removal of the DNA/lipid transfection mixture, wells were washed twice with PBS. Cells were lysed with 100 µl lysing buffer as part of Luciferase Assay kit (Roche, Basel, Switzerland). Lysate was collected and stored at −80°C until assayed. As per kit protocol, lysate and luciferase reagent were mixed in opaque 96-well plates and read on a Perkins/Elmer Victor3 plate reader. Relative light units were measured from samples.
Plasmid construction
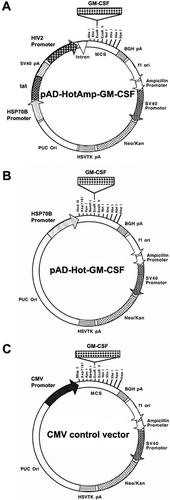
All restriction enzymes and their buffers were obtained from New England BioLabs (Beverly, MA, USA). The GFP reporter construct was created by inserting the green fluorescence protein (GFP) gene (BD Biosciences Clontech, Palo Alto, CA, USA) into the multiple cloning site (MCS) downstream of the cytomegalovirus (CMV) promoter in pcDNA3 (Invitrogen, San Diego, CA, USA). The reporter plasmid was used to estimate the transfection efficiency of the lipid-mediated transfection by fluorescence from GFP. pcDNA3 served also as the backbone for the CMV control vector. Replacing the CMV promoter of pcDNA3 (Invitrogen, San Diego, CA, USA) by the BamHI-HindIII fragment of the human HSP70B promoter from the p173OR plasmid (StressGen, Victoria, BC, USA) upstream of the MCS created the plasmid pH2. The functionally uncompromised BamHI-HindIII fragment of the promoter Citation[32], with its 451 bp in size, is significantly smaller than the full-length HSP70B promoter with 2.3 kb in size. This adaptation is advantageous for further cloning as it results in a smaller vector size. The construct pV3 originated from the pRSC plasmid, a mammalian expression vector with two multiple cloning sites (MCS) for the expression of two foreign genes, described elsewhere Citation[33]. The RSV promoter in pRSC was replaced with the HSP70B promoter fragment described earlier to generate the construct pV3. The gene for the transcription-activating factor, tat, was excised by XbaI digestion from pTAT, described elsewhere Citation[34], and ligated into the XbaI site of Sac-KiSS-λ Citation[35] following XbaI digestion to create Sac-KiSS-TAT. The tat gene was then cut back out with NotI and cloned into the NotI site downstream of the HSP70B promoter in pV3. The CMV promoter of the original pRSC construct was replaced by the HIV2 LTR that was excised from pGL2-HIV2, described elsewhere Citation[36], by BglII and HindIII digestion to produce pV3. The plasmid pV3 contains the bovine growth hormone (BGH) polyadenylation (pA) site following the MCS immediately downstream of the HIV2 promoter. The plasmids pcDNA3, pH2 and pV3 all carry Neomycin/Kanamycin (Neo/Kan) and Ampicillin selectable markers. Plasmids were isolated using a plasmid mini or maxi preparation (Qiagen, Valencia, CA, USA) according to the manufacturer's protocol. Each of the backbone constructs, pcDNA3, pH2 and pV3, were digested with EcoRI. The 5′ ends of the linearized plasmids were dephosphorylated utilizing a Shrimp Alkaline Phosphatase (SAP) kit (Roche Diagnostics GmbH, Mannheim, Germany) according to the manufacturer's protocol. Ligation reactions were performed with the Rapid DNA Ligation Kit (Roche Diagnostics GmbH). A 435 bp fragment encoding the murine GM-CSF gene was excised from pUMVC1-mGMCSF (Aldevron, Fargo, ND, USA) through a digestion with BamHI and EcoRI. The fragment was ligated into compatible sites of the MCS in the plasmid Sac-KiSS-λ, following a complete digestion of Sac-KiSS-λ with BamHI and a partial digestion with EcoRI. The GM-CSF gene was then excised through an EcoRI digestion and inserted into the EcoRI site of the different MCSs in pV3, pH2, and pcDNA3 backbones to generate the plasmids pAD-HotAmp-GM-CSF (), pAD-Hot-GM-CSF () and the CMV control vector (), respectively. Plasmids containing the luciferase gene were created by inserting the gene into the MCS of the HSP70B promoter-controlled pH2 and the CMV promoter-controlled pcDNA3 backbones.
Figure 1. Map of the GM-CSF expression constructs. (a) The first transcriptional unit consists of the inducible HSP70B promoter that drives the expression of the first gene, the transcription-activating factor tat. The second transcriptional unit contains the gene encoding GM-CSF driven by the HIV2 promoter (HIV2 LTR). (b) The HSP70B promoter directly controls the GM-CSF gene expression. (c) The constitutive CMV promoter directly drives the expression of the GM-CSF gene.
Statistical analysis
Values are given as means ± standard error of the mean (SEM). Results were compared using the Student's t-test. Statistical significance was considered at p < 0.05.
Results
Plasmid construction
Several GM-CSF expression plasmid vectors were constructed (described in Materials and methods): pAD-HotAmp-GM-CSF (), pAD-Hot-GM-CSF () and the CMV control vector (). The minimal HSP70B promoter fragment as described in Materials and methods is termed HSP70B promoter in the following text for simplicity. The novel vector, pAD-HotAmp-GM-CSF, contains inducible and amplifying features as two units in a single plasmid. The inducible unit is comprised of the transcription-activating factor tat located immediately downstream of the HSP70B promoter. A second promoter, the HIV2 LTR controls expression of the GM-CSF gene. The functional binding of the tat factor to the transactivation response RNA element contained in the HIV2 LTR enhances the processivity of RNA polymerase II and/or efficiency of the elongation and elevates gene expression from basal levels Citation[37].
Survival of B16 cells after heat shock
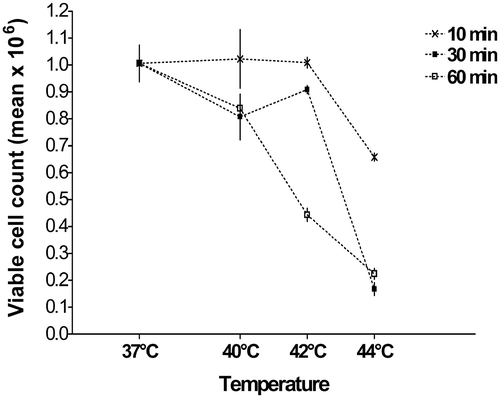
A heat shock at 42°C for up to 30 min had no significant impact on B16 cell viability (). Increasing the time period of heat shock to 60 min at 42°C diminished cell survival to a similar level as for 60 min at 44°C. Heat shock at 44°C had a significant negative impact on cell viability at shorter time periods, with a 30 min heat shock decreasing cell survival by over 50%.
Figure 2. Survival of B16 cells after heat shock. Cells were heat-shocked in a water bath at indicated times and temperatures 24 h after seeding. After another 24 h, cells were harvested and viable cells counted. Heating for 30 min at 42°C showed no significant difference from 37°C control in cell viability (p = 0.24). All conditions were repeated in triplicates with mean values shown (±SEM).
Inducible gene expression using HSP70B promoter
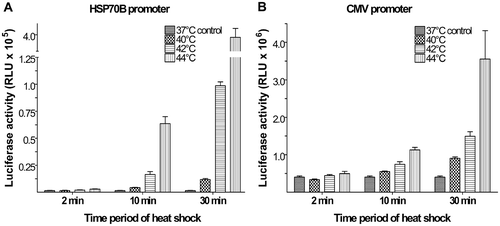
The luciferase system (where the expression of luciferase is proportional to the relative light units measured) was used as a reporter system to determine the inducibility of the HSP70B promoter () compared to the CMV promoter (). For both promoters, luciferase expression increased proportionally with temperature and time period of heat shock. The initial results indicated that the HSP70B promoter was highly inducible with low background expression levels at 37°C. However, its maximum expression was weak with expression levels that were an order of magnitude lower compared to the CMV promoter. It was determined that the heat shock condition of 42°C for 30 min was optimal due to the high increased expression induced concomitantly with preserved cell viability. In the next series of experiments, the inducibility of the HSP70B promoter was combined with the downstream expression of the transcription-activating factor tat and its amplifying effect on gene expression controlled by the HIV2 promoter.
Figure 3. Inducible gene expression using HSP70B promoter. Plasmids using the HSP70B promoter (a) showed low background activity at 37°C but large increases in luciferase expression upon heat shock compared to the CMV promoter (b). The maximum increase in HSP70B promoter-controlled luciferase expression of 250-fold was observed with heat shock of 44°C for 30 min. A heat shock of 42°C for 30 min provided an increase in luciferase activity by more than 60-fold over background. Values shown are means of relative light units (RLU) (±SEM) of triplicate experiments.
GM-CSF production levels
B16 mouse melanoma cells were seeded at a concentration of 0.2 × 106 cells in 2 ml complete medium per culture well of a 6-well plate. The vectors pAD-Hot-GM-CSF and pAD-HotAmp-GM-CSF and the CMV control vector were transiently transfected into B16 mouse melanoma cells. Negative controls were prepared as mock transfections lacking any plasmid DNA (data not shown). Cells were heat-shocked at 42°C for 30 min 24 h post-transfection and returned to 37°C afterwards. The overall lipid-mediated transfection efficiency during the experiments was determined using a GFP reporter construct. According to flow-cytometric analysis, GFP positive cells ranged from 39.1–49.8%, with a mean of 42.2% (data not shown).
If left at 37°C, basal GM-CSF production by pAD-HotAmp-GM-CSF stayed at ∼15% (2062 pg ml−1) of its heat shock-induced amplified levels whereas no basal activity of pAD-Hot-GM-CSF was detected (). The transient nature of GM-CSF production by the pAD-HotAmp-GM-CSF vector peaked at 24 h followed by an exponential decrease in production. Forty-eight hours after heat induction, GM-CSF production declined to similar levels for all constructs. At 72 h post-heat shock the GM-CSF production by pAD-HotAmp-GM-CSF decreases below 57% of the CMV promoter-controlled. If left at 37°C, the constitutive production by the CMV control vector and the basal production by pAD-HotAmp-GM-CSF reached similar levels during the first 24 h and vanished to undetectable levels at later time points. As expected, cells transfected with the backbone construct of pAD-HotAmp-, lacking the GM-CSF gene, as well as mock-transfected cells, lacking any DNA, did not produce measurable amounts of GM-CSF (data not shown).
Figure 4. GM-CSF production levels. Cells were heat-shocked at 42°C for 30 min 24 h post-transfection (▪) or kept at 37°C as controls (□). Supernatants were harvested 24, 48 and 72 h after transfection. GM-CSF levels in the supernatants were assayed using ELISA. Each bar represents the GM-CSF production within a 24 h period. Values shown are means of triplicate experiments (±SEM). The GM-CSF levels, 24 h after heat shock, produced by pAD-HotAmp-GM-CSF were compared to pAD-Hot-GM-CSF (p < 0.001) and the CMV control vector (p < 0.001) and to the non-heat-shocked control of pAD-HotAmp-GM-CSF (p < 0.001).
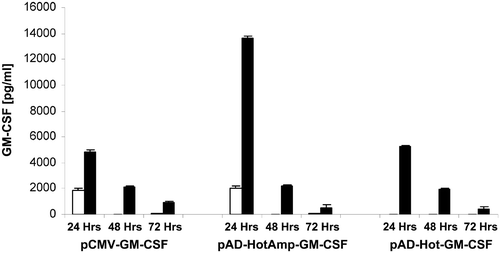
The highest GM-CSF production levels were achieved with the inducible and amplifying vector pAD-HotAmp-GM-CSF (). Twenty-four hours after heat shock at 42°C for 30 min, the mean GM-CSF production by pAD-HotAmp-GM-CSF (13634 pg ml−1) was over 2.5-fold higher than that of pAD-Hot-GM-CSF (5270 pg ml−1). In comparison to the GM-CSF production by the strong constitutively expressing CMV promoter (4788 pg ml−1), the amplified production by pAD-HotAmp-GM-CSF 24 h post-heat shock increased over 2.8-fold.
Discussion and conclusions
The heat-inducible and amplifying elements, combined into the single multi-genic construct, pAD-HotAmp-GM-CSF, responded to heat shock with the highest production of GM-CSF of all tested constructs, among which a prototypic CMV promoter construct. The inducible expression of the transactivator tat and its subsequent binding to the HIV2 LTR of the amplifying unit amplifies GM-CSF expression beyond the level that the HSP70B promoter alone could achieve. The heat-induced and amplified GM-CSF production by pAD-HotAmp-GM-CSF increased 6.6-fold over basal levels with a peak in the first 24 h after heat shock. The rather exponential decline of production thereafter underlines the inducible and amplifying properties. However, after a transient hyperthermic induction, production levels for all vectors were expected to weaken, in addition, due to plasmid loss over time in transient transfections. The transient activity of the HSP70B promoter alone also peaked during the first 24 h after single hyperthermic induction as previously shown by reporter gene expression Citation[32], Citation[38]. Interestingly, constitutive GM-CSF expression by the CMV promoter in the CMV control vector was also responsive to heat treatment with expression levels close to those achieved with the HSP70B promoter in pAD-Hot-GM-CSF. A similar stress-induced increase in transcription with the CMV promoter was previously observed Citation[39], Citation[40].
A one-plasmid system containing the HIV2 LTR with the tat-responsive elements and the HSP70B promoter-controlled tat gene allows for uncompromised gene expression and is more efficient than two-plasmid systems Citation[37]. A fixed ratio and co-ordinated expression of genes is assured in a one-plasmid system that is free from the problem of different transfection rates as occurs in co-transfection of multiple plasmids. Previously, it was shown that the incorporation of a second promoter on the same construct did not influence the temperature dependence of reporter gene expression and that in the absence of tat expression the HIV2 promoter demonstrated activity nearly independent of heat shock Citation[38]. A basal activity of the HIV2 LTR at 37°C (in the absence of tat) may be due to the influence of other intra-cellular factors such as NF-χB, which are capable of transactivating this promoter.
In addition to heat exposure, the minimal HSP70B promoter fragment can be induced by gamma-radiation and the chemotherapeutic drug geldanamycin Citation[32]. However, among them, hyperthermia remained the most effective. Hyperthermia may be applied together with the classical therapies, radiation and chemotherapy. Hyperthermia and/or certain chemotherapeutic agents may enhance the immune effects of genetically modified tumour vaccines Citation[17]. Therefore, a synergy effect can be achieved through the combination of such therapies.
The vector pAD-HotAmp-GM-CSF addresses the need for very high GM-CSF production to overcome limited clinical responses with sub-optimal doses. Moreover, GM-CSF production is inducible through the incorporation of the heat-responsive HSP70B promoter fragment. Thus, localized high levels of GM-CSF can be induced through localized hyperthermia. This strategy may provide an alternative to the difficult approach of locally targeted gene transfection Citation[41–43]. In an in vivo setting, administration of the vector may then be followed by local activation, e.g. locally applied hyperthermia. The pAD-HotAmp-GM-CSF expression vector may provide a feasible strategy for locally inducible, hence improving regulation and safety in, high-level expression of GM-CSF for anti-cancer gene therapy.
Acknowledgements
Special thanks belong to Debbie Sakiestewa and Dominic Titone for expert technical assistance.
References
- Tagawa M. Cytokine therapy for cancer. Curr Pharm Des 2000; 6: 681–699
- Dranoff G. GM-CSF-based cancer vaccines. Immunol Rev 2002; 188: 147–154
- Ravaud A, Delaunay M, Chevreau C, Coulon V, Debled M, Bret-Dibat C, Courbon F, Gualde N, Nguyen Bui B. Granulocyte-macrophage colony-stimulating factor alone or with dacarbazine in metastatic melanoma: A randomized phase II trial. Br J Cancer 2001; 85: 1467–1471
- Hast R, Hellstrom-Lindberg E, Ohm L, Bjorkholm M, Celsing F, Dahl IM, Dybedal I, Gahrton G, Lindberg G, Lerner R, Linder O, Lofvenberg E, Nilsson-Ehle H, Paul C, Samuelsson J, Tangen JM, Tidefelt U, Turesson I, Wahlin A, Wallvik J, Winquist I, Oberg G, Bernell P. No benefit from adding GM-CSF to induction chemotherapy in transforming myelodysplastic syndromes: Better outcome in patients with less proliferative disease. Leukemia 2003; 17: 1827–1833
- De Giovanni C, Nanni P, Forni G. The prospects for cancer gene therapy. Int J Immunopharmacol 2000; 22: 1025–1032
- Shi FS, Weber S, Gan J, Rakhmilevich AL, Mahvi DM. Granulocyte-macrophage colony-stimulating factor (GM-CSF) secreted by cDNA-transfected tumor cells induces a more potent antitumor response than exogenous GM-CSF. Cancer Gene Ther 1999; 6: 81–88
- Ju DW, Cao X, Acres B. Intratumoral injection of GM-CSF gene encoded recombinant vaccinia virus elicits potent antitumor response in a mixture melanoma model. Cancer Gene Ther 1997; 4: 139–144
- Schatzlein AG. Non-viral vectors in cancer gene therapy: Principles and progress. Anticancer Drugs 2001; 12: 275–304
- Buchsel PC, Forgey A, Grape FB, Hamann SS. Granulocyte macrophage colony-stimulating factor: Current practice and novel approaches. Clin J Oncol Nurs 2002; 6: 198–205
- Armstrong CA, Botella R, Galloway TH, Murray N, Kramp JM, Song IS, Ansel JC. Antitumor effects of granulocyte-macrophage colony-stimulating factor production by melanoma cells. Cancer Res 1996; 56: 2191–2198
- Chatterjee SK, Qin H, Manna S, Tripathi PK. Recombinant vaccinia virus expressing cytokine GM-CSF as tumor vaccine. Anticancer Res 1999; 19: 2869–2873
- Couch M, Saunders JK, O'Malley BW, Jr, Pardoll D, Jaffee E. Genetically engineered tumor cell vaccine in a head and neck cancer model. Laryngoscope 2003; 113: 552–556
- Salgia R, Lynch T, Skarin A, Lucca J, Lynch C, Jung K, Hodi FS, Jaklitsch M, Mentzer S, Swanson S, Lukanich J, Bueno R, Wain J, Mathisen D, Wright C, Fidias P, Donahue D, Clift S, Hardy S, Neuberg D, Mulligan R, Webb I, Sugarbaker D, Mihm M, Dranoff G. Vaccination with irradiated autologous tumor cells engineered to secrete granulocyte-macrophage colony-stimulating factor augments antitumor immunity in some patients with metastatic non-small-cell lung carcinoma. J Clin Oncol 2003; 21: 624–630
- Toda M, Martuza RL, Rabkin SD. Tumor growth inhibition by intratumoral inoculation of defective herpes simplex virus vectors expressing granulocyte-macrophage colony-stimulating factor. Mol Ther 2000; 2: 324–329
- Marshall E. Gene therapy's growing pains. Science 1995; 269: 1050, 1052–1055
- Mahvi DM, Shi FS, Yang NS, Weber S, Hank J, Albertini M, Schiller J, Schalch H, Larson M, Pharo L, Gan J, Heisey D, Warner T, Sondel PM. Immunization by particle-mediated transfer of the granulocyte-macrophage colony-stimulating factor gene into autologous tumor cells in melanoma or sarcoma patients: Report of a phase I/IB study. Hum Gene Ther 2002; 13: 1711–1721
- Armstrong TD, Jaffee EM. Cytokine modified tumor vaccines. Surg Oncol Clin N Am 2002; 11: 681–696
- Bohlius J, Reiser M, Schwarzer G, Engert A. Impact of granulocyte colony-stimulating factor (CSF) and granulocyte-macrophage CSF in patients with malignant lymphoma: A systematic review. Br J Haematol 2003; 122: 413–423
- Thomas X, Boiron JM, Huguet F, Reman O, Sutton L, Turlure P, Garban F, Gardin C, Espinouse D, Boulat O, Lheritier V, Fiere D. Efficacy of granulocyte and granulocyte-macrophage colony-stimulating factors in the induction treatment of adult acute lymphoblastic leukemia: A multicenter randomized study. Hematol J 2004; 5: 384–394
- Nelson WG, Simons JW, Mikhak B, Chang JF, DeMarzo AM, Carducci MA, Kim M, Weber CE, Baccala AA, Goeman MA, Clift SM, Ando DG, Levitsky HI, Cohen LK, Sanda MG, Mulligan RC, Partin AW, Carter HB, Piantadosi S, Marshall FF. Cancer cells engineered to secrete granulocyte-macrophage colony-stimulating factor using ex vivo gene transfer as vaccines for the treatment of genitourinary malignancies. Cancer Chemother Pharmacol 2000; 46(Suppl)S67–S72
- Nemunaitis J, Sterman D, Jablons D, Smith JW, 2nd, Fox B, Maples P, Hamilton S, Borellini F, Lin A, Morali S, Hege K. Granulocyte-macrophage colony-stimulating factor gene-modified autologous tumor vaccines in non-small-cell lung cancer. J Natl Cancer Inst 2004; 96: 326–331
- Dillman R, Selvan S, Schiltz P, Peterson C, Allen K, Depriest C, McClay E, Barth N, Sheehy P, de Leon C, Beutel L. Phase I/II trial of melanoma patient-specific vaccine of proliferating autologous tumor cells, dendritic cells, and GM-CSF: Planned interim analysis. Cancer Biother Radiopharm 2004; 19: 658–665
- Soiffer R, Hodi FS, Haluska F, Jung K, Gillessen S, Singer S, Tanabe K, Duda R, Mentzer S, Jaklitsch M, Bueno R, Clift S, Hardy S, Neuberg D, Mulligan R, Webb I, Mihm M, Dranoff G. Vaccination with irradiated, autologous melanoma cells engineered to secrete granulocyte-macrophage colony-stimulating factor by adenoviral-mediated gene transfer augments antitumor immunity in patients with metastatic melanoma. J Clin Oncol 2003; 21: 3343–3350
- Yew NS, Wysokenski DM, Wang KX, Ziegler RJ, Marshall J, McNeilly D, Cherry M, Osburn W, Cheng SH. Optimization of plasmid vectors for high-level expression in lung epithelial cells. Hum Gene Ther 1997; 8: 575–584
- Boshart M, Weber F, Jahn G, Dorsch-Hasler K, Fleckenstein B, Schaffner W. A very strong enhancer is located upstream of an immediate early gene of human cytomegalovirus. Cell 1985; 41: 521–530
- Hartikka J, Sawdey M, Cornefert-Jensen F, Margalith M, Barnhart K, Nolasco M, Vahlsing HL, Meek J, Marquet M, Hobart P, Norman J, Manthorpe M. An improved plasmid DNA expression vector for direct injection into skeletal muscle. Hum Gene Ther 1996; 7: 1205–1217
- Doll RF, Crandall JE, Dyer CA, Aucoin JM, Smith FI. Comparison of promoter strengths on gene delivery into mammalian brain cells using AAV vectors. Gene Ther 1996; 3: 437–447
- Tsang TC, Brailey JL, Vasanwala FH, Wu RS, Liu F, Clark PR, Meade-Tollin L, Luznick L, Stopeck AT, Akporiaye ET, Harris DT. Construction of new amplifier expression vectors for high levels of IL-2 gene expression. Int J Mol Med 2000; 5: 295–300
- Schiller P, Amin J, Ananthan J, Brown ME, Scott WA, Voellmy R. Cis-acting elements involved in the regulated expression of a human HSP70 gene. J Mol Biol 1988; 203: 97–105
- Morimoto RI, Kline MP, Bimston DN, Cotto JJ. The heat-shock response: Regulation and function of heat-shock proteins and molecular chaperones. Essays Biochem 1997; 32: 17–29
- Edwards MJ. Apoptosis, the heat shock response, hyperthermia, birth defects, disease and cancer. Where are the common links? Cell Stress Chaperones 1998; 3: 213–220
- Vasanwala F, Tsang T, Fellah A, Yorgin P, Harris D. A novel expression vector induced by heat, gamma-radiation and chemotherapy. Gene Ther Mol Biol 2000; 5: 1–7
- Tsang TC, Harris DT, Akporiaye ET, Chu RS, Brailey J, Liu F, Vasanwala FH, Schluter SF, Hersh EM. Mammalian expression vector with two multiple cloning sites for expression of two foreign genes. Biotechniques 1997; 22: 68
- Arya SK, Guo C, Josephs SF, Wong-Staal F. Trans-activator gene of human T-lymphotropic virus type III (HTLV-III). Science 1985; 229: 69–73
- Tsang TC, Harris DT, Akporiaye ET, Schluter SF, Bowden GT, Hersh EM. Simple method for adapting DNA fragments and PCR products to all of the commonly used restriction sites. Biotechniques 1996; 20: 51–52
- Xu J, Luznik L, Wong-Staal F, Gill GN. Hormone receptor regulation of the human immunodeficiency virus type 1 and type 2 long terminal repeats. J Biomed Sci 1996; 3: 323–331
- Zhang T, Tsang TC, Harris DT. Comparison of cis and trans tat gene expression in HIV LTR-based amplifier vectors. Biotechniques 2002; 33: 1146–1151
- Gerner EW, Hersh EM, Pennington M, Tsang TC, Harris D, Vasanwala F, Brailey J. Heat-inducible vectors for use in gene therapy. Int J Hyperthermia 2000; 16: 171–181
- Borrelli MJ, Schoenherr DM, Wong A, Bernock LJ, Corry PM. Heat-activated transgene expression from adenovirus vectors infected into human prostate cancer cells. Cancer Res 2001; 61: 1113–1121
- Andrews JM, Newbound GC, Lairmore MD. Transcriptional modulation of viral reporter gene constructs following induction of the cellular stress response. Nucleic Acids Res 1997; 25: 1082–1084
- Kaul G, Amiji M. Tumor-targeted gene delivery using poly(ethylene glycol)-modified gelatin nanoparticles: In vitro and in vivo studies. Pharm Res 2005; 22: 951–961
- Wolschek MF, Thallinger C, Kursa M, Rossler V, Allen M, Lichtenberger C, Kircheis R, Lucas T, Willheim M, Reinisch W, Gangl A, Wagner E, Jansen B. Specific systemic nonviral gene delivery to human hepatocellular carcinoma xenografts in SCID mice. Hepatology 2002; 36: 1106–1114
- Oshima Y, Sakamoto T, Yamanaka I, Nishi T, Ishibashi T, Inomata H. Targeted gene transfer to corneal endothelium in vivo by electric pulse. Gene Ther 1998; 5: 1347–1354