Abstract
Purpose: The aim of the study was to investigate the molecular mechanisms involved in apoptosis of human promyelocytic cells (HL60) induced by hyperthermia and to compare this to radiation-induced apoptosis as a reference model.
Materials and methods: Apoptosis of HL60 cells was induced by heat-treatment (43°C during 1 h) or by γ-radiation (8 Gy) and followed at increasing time periods after treatment with Annexin V binding to phosphatidylserine (PS). The transition of the mitochondrial membrane potential (Δψm) was estimated by the extent of mitochondrial JC-1 uptake. Bcl-2 and Bax protein expression levels were monitored using fluorescent-labelled antibodies. Caspase activation was studied using a fluorochrome-labelled pan-caspase inhibitor (FLICA), which also allowed one to study the kinetics of the apoptotic cascade.
Results: After heat-treatment or irradiation of HL60 cells, a decreased Δψm as well as PS membrane expression were detectable after 8 h. Bcl-2 and Bax protein expression levels were decreased and increased, respectively, 1 h after heat-treatment or irradiation. The apoptotic rate of HL60 cells, as measured by the FLICA binding, was faster with heat-treatment as compared to γ-irradiation. Addition of a pan-caspase inhibitor prevented PS externalization after heat-treatment but not after irradiation. The presence of a pan-caspase inhibitor did not influence the decrease of Δψm both after heat-treatment and γ-irradiation. However, the addition of the specific caspase-2 inhibitor zVDVAD-fmk prevented the mitochondrial breakdown after heat-treatment. Inhibition of caspase-2 had no effect on the γ-irradiation induced apoptosis.
Conclusion: These results suggest that the commitment to apoptosis in HL60 cells after heat-treatment is started by mitochondrial membrane transition involving the Bcl-2 family members and is mainly executed in a caspase-dependent pathway. The results suggest that caspase-2 plays a key role in the heat-induced apoptosis.
Introduction
Hyperthermia has been under investigation as an effective and useful tool in anti-cancer therapy Citation[1]. There are many reasons why hyperthermia should be a good anti-cancer treatment. First, hyperthermia enhances the effectiveness of other treatment modalities and, secondly, hyperthermia kills tumour cells that are normally resistant to other forms of treatment Citation[2]. Hyperthermia also induces apoptosis in a wide range of cancer cells Citation[3]. Although hyperthermia has been shown to induce apoptosis, the molecular mechanisms of heat-induced apoptosis are not fully understood Citation[4].
The aim of this study is to get more insight in the mechanisms of heat-induced apoptosis. γ-Irradiation was chosen as a reference treatment for inducing apoptosis since the molecular mechanisms involved have been well studied in vitro Citation[5–7].
For the initiation phase of apoptosis, the mitochondrial function was studied. Mitochondria play an essential role in commitment of cells to apoptosis via a decreased transmembrane potential resulting in an increased permeability of the outer mitochondrial membrane and finally the release of cytochrome-c in the cytosol Citation[8], Citation[9]. Anti-apoptotic Bcl-2 family members, such as Bcl-2 and Bcl-XL, can block these mitochondrial events, whereas pro-apoptotic Bcl-2 family members, including Bax, can trigger these changes Citation[10], Citation[11]. The effects of pro-apoptotic Bcl-2 family members are mediated by both caspase-dependent and caspase-independent mechanisms Citation[12], Citation[13].
In the effector phase of programmed cell death the cell has passed the point of no return and effector caspases are activated Citation[14]. Therefore, the role of caspases was studied in heat-induced and radiation-induced apoptosis of HL60 cells by inhibiting active caspases. With this method it is also possible to arrest cells in apoptosis and to label the arrested cells with fluorochrome to investigate the differences in apoptotic turnover in time between heat- and radiation-induced apoptosis Citation[15], Citation[16].
During the execution phase of the death programme, alterations of the plasma membrane occur, like exposure of phosphatidylserine (PS) due to transposition of PS from the inner to the outer leaflet of the plasma membrane (flip-flop mechanism) Citation[17], Citation[18]. These alterations in HL-60 cells were studied during heat- and γ-radiation-induced apoptosis by means of Annexin V binding to externalized PS.
Materials and methods
Cell cultures
Human promyelocytic leukemic HL60 cells were obtained from the German Collection of Micro-organisms (Braunschweig, Germany). Cells were cultured in RPMI-1640 medium (BioWhittaker, Verviers, Belgium) supplemented with 10% (v/v) heat-inactivated foetal calf serum, 100 units mL−1 penicillin, 100 µg mL−1 streptomycin and 2 mM L-glutamine (RPMI+ medium). Supplements and antibiotics were obtained from Life Technologies (Grand Island, NY). Cell cultures were maintained in a 5% CO2 humidified atmosphere at 37°C. The medium was refreshed every 3–4 days. For the experiments exponentially growing cells were used. Tissue culture disposables were supplied by Corning (Badhoevedorp, The Netherlands).
Apoptosis inducing treatments
Apoptosis was induced by heat using a thermostatically controlled water-bath filled with distilled water to heat the cell suspension at 43°C during 1 h. Tissue culture flasks containing 10 mL cell suspension (0.5 × 106 cells mL−1) were submerged into the water-bath. Temperature was monitored in all experiments with a flask containing only medium and a temperature probe to ensure a temperature of 43 ± 0.2°C. For radiation experiments, cells were exposed to γ-rays with a total dose of 8 Gy at a rate of 5 Gy min−1 with a Clinac600 6MV linear accelerator (Varian, Silicon Valley). To inhibit apoptosis a pan-caspase inhibitor zVAD-fmk (R&D systems, Minneapolis) was used at a final concentration of 20 µM. In addition, specific caspase-2 inhibitor zVDVAD-fmk (R&D systems, Minneapolis) was used 1 h before the experiments at a final concentration of 10 µM in the cell suspension according to the manufacturers protocol.
Annexin V assay
Binding of Annexin V- Fluorescein isothiocyanate (FITC; NeXins research BV, Hoeven, The Netherlands) to PS as a measure of PS externalization and the uptake of propidium iodide (PI; Sigma, St. Louis, MO) as a measure for membrane integrity were monitored by flow-cytometry Citation[19]. Cells (0.3 × 106) were washed twice with PBS and resuspended in freshly made HEPES buffer (Brunschwig, Amsterdam, The Netherlands) supplemented with 147 mM CaCl2.5H2O. Cells were incubated with 0.01 µg mL−1 Annexin V-FITC for 15 min at room temperature in the dark. Directly before measurements, cells were stained with 0.2 µg mL−1 PI. Samples were kept on ice until flow-cytometric analysis.
Mitochondrial membrane potential
To signal the breakdown of the mitochondrial membrane potential (Δψm), 0.3 × 106 cells were washed twice and resuspended in PBS, containing 0.2% BSA Citation[20]. Cells were incubated with the fluorescent probe JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl carbocyanine-iodide) at a concentration of 0.12 µM (Molecular Probes, Leiden, The Netherlands) for 20 min at 37°C. This cationic dye exhibits membrane potential-dependent accumulation in mitochondria. In vital cells JC-1 will accumulate and form aggregates (red fluorescence emission) in the mitochondrial matrix, whereas during apoptosis the mitochondrial membrane potential decreases resulting in the transition of JC-1 monomers (green fluorescence emission) to the cytoplasm. Cyclosporine-A (Sigma-Aldrich, Zwijndrecht, The Netherlands) was added to block further changes of the membrane potential, thereby stabilizing the mitochondrial membrane after harvesting the cells. Staining of the cells was quantified by flow-cytometry.
Caspase activation
To detect active caspases the Fluorochrome Inhibitor of Caspases (FLICA) method was used Citation[15]. Lyophilized fam-VAD-fmk fluorescent-labelled inhibitor of caspase was obtained from Chemicon International (ITK diagnostics, Uithoorn, The Netherlands). The solution of fam-VAD-fmk in PBS was mixed with a solution of the unlabelled inhibitor zVAD-fmk in a 1:4 molar ratio. The zVAD-fmk/fluorescent fam-VAD-fmk mixture was added to 0.5 × 106 cells at a 20 µM final concentration before apoptosis was induced. The cells were in the continuous presence of FLICA during the experiments. Before flow-cytometry, cells were washed twice in PBS and resuspended in PBS containing PI at a final concentration of 1 µg mL−1.
Quantification of Bcl-2 and Bax protein levels
Intra-cellular Bcl-2 and Bax protein levels were determined by flow-cytometry 1 and 2 h after induction of apoptosis Citation[20]. Cells (0.3 × 106) were fixed and permeabilized using the Intraprep kit (Immunotech, Marseille, France) and subsequently incubated with 1 µg anti-Bcl-2-FITC (IgG1, Dako, Glostrup, Denmark) or 1 µg anti-Bax-PE (IgG1, Brunschwig, Amsterdam, The Netherlands). Separately for each sample an additional negative control was performed by incubating cells with 1 µg mouse IgG1-FITC or IgG1-PE to measure non-specific fluorescent signals. All incubations with antibodies were performed at room temperature for 20 min. Cells were then washed twice with Isoton and resuspended in 500 µl PBS. The mean fluorescent ratio (MFR), defined as the ratio of the mean fluorescent intensity (MFI) of primary antibody and the isotype control stained cells, was used as a measure for Bcl-2 or Bax protein expression.
Flow cytometry
Fluorescence of individual cells was measured with a Coulter Epics XL flow-cytometer. Excitation was elicited at 488 nm with the Argon laser and emission was subsequently analysed by means of three fluorescence detectors; FL1 (530 nm band-pass filter), FL2 (585 nm band-pass filter) and FL3 (650 nm band-pass filter). In each sample, 10 000 events were measured. Annexin V-FITC and PI were detected in FL1 and in FL3, respectively. JC-1 fluorescence was analysed in FL1 and in FL2 for the detection of the dye monomer and aggregates, respectively. FLICA was also detected in FL1. Bcl-2 and Bax fluorescences were detected in FL1 and FL2, respectively, and the data are presented as percentage change in MFR of treated cells compared to the MFR of untreated cells. All other data are presented as percentage of cells, within the total population of 10 000 counted cells, showing fluorescence.
Light microscopy
Cell suspensions (300 µl) containing 0.5 × 106 cells mL−1 were centrifuged at 700 rpm for 10 min with low acceleration using a cytospin3 micro-centrifuge (Shandon, Pittsburg). The cells were fixed with methanol, stained with May-Grunwald-Giemsa and examined at 100 × magnification.
Results
Kinetics of Δψm transition; caspase activation; PS exposure and Bcl-2/Bax regulation
In the first set of experiments, cells were either heated at 43°C for 1 h or irradiated with 8 Gy and characteristic hallmarks of apoptosis were determined. The induction of apoptosis was associated with a decrease of Δψm, as determined by JC-1 staining shown in . Normal vital cells possess a high mitochondrial membrane potential. However, under apoptotic conditions the inner mitochondrial membrane potential decreases, resulting in an increased permeability of the outer mitochondrial membrane. Depolarization of the mitochondrial membrane occurred after 8 h for heat-induced HL60 cells and reached a maximum after 24 h when 70% of the cells showed Δψm transition. For radiation-induced cells JC-1 monomers were detected after 8 h at a lower rate compared to heat-induced cells, indicating slower Δψm transition kinetics. The activated proteolytic caspases were analysed using FLICA, which binds only to active caspases. As shown in , activated caspases were found 8 h after exposure to both heat and γ-radiation, closely associated with Δψm transition. The activation of effector caspases led to PS externalization, as can be seen in . Fifty per cent of the heat-induced cells showed Annexin V binding after 48 h. Since mitochondria are major targets for Bcl-2 family proteins, the expression levels of anti-apoptotic Bcl-2 and pro-apoptotic Bax was monitored directly after heat-treatment or γ-irradiation. In the percentage decrease of intra-cellular Bcl-2 fluorescence compared to that of untreated HL60 cells is shown. As soon as 1 h after treatment with heat or γ-radiation, Bcl-2 protein levels drop and continue to decrease for heat-treated cells (2 h), whereas for radiation-induced cells the Bcl-2 protein levels after 2 h are comparable to that of untreated cells. The pro-apoptotic protein Bax shows a significant up-regulation of expression levels up to 250% for heat-induced and 130% for radiation-induced HL60 cells compared to the expression levels of untreated cells ().
Figure 1. Kinetics of Δψm transition, caspase activation and PS exposure associated with heat- and radiation-induced apoptosis of HL60 cells. HL60 cells were exposed to 43°C during 1 h (HT) or to 8 Gy γ-radiation (RT). Figures show the percentage of cells, within the total population of 10 000 counted cells, showing fluorescence. (a) Reduction of Δψm determined by the extent of mitochondrial JC-1 uptake. JC-1 monomers represent a decreased Δψm, whereas with a normal Δψm, JC-1 aggregates in mitochondria to a crystalline structure. At the indicated time, the percentage of cells with JC-1 monomer fluorescence were determined with the flow-cytometer. (b) Percentage of cells with activated caspases determined using FLICA. (c) The percentage of cells with PS exposed to the outer leaflet of the cell membrane determined with Annexin V-FITC binding to PS. The results are the mean ± SD (n = 3).
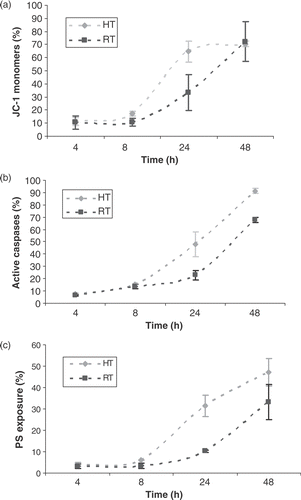
Figure 2. Intra-cellular Bcl-2 and Bax protein levels. (a) Percentage decrease in mean fluorescent ratio (MFR) of Bcl-2, compared to MFR of Bcl-2 for untreated HL60 cells, 1 and 2 h after heat-treatment (HT; 43°C during 1 h) or γ-irradiation (RT; 8 Gy). (b) Percentage increase in MFR of Bax, compared to MFR of Bax for untreated HL60 cells, 1 and 2 h after heat-treatment (HT) or γ-irradiation (RT). The MFR, defined as the ratio of the mean fluorescent intensity (MFI) of primary antibody and the MFI of the isotype control stained cells, was used as a measure for Bcl-2 or Bax protein expression. The results are the mean ± SD (n = 3).
Analysis of the apoptotic rate
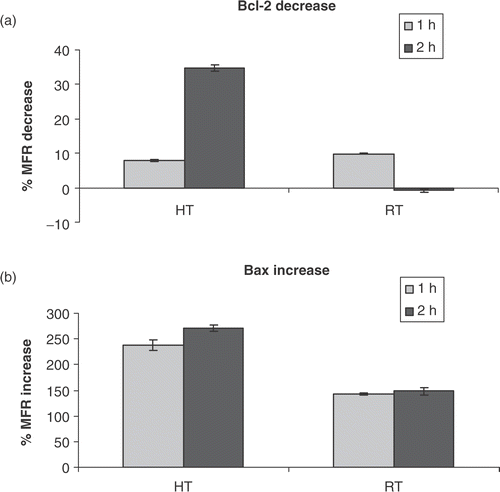
Studying the apoptotic rate is difficult due to loss of cells by cell disintegration in time. Exposure of cells to FLICA blocks the activation of caspases and arrests further progress of the apoptotic cascade and, therefore, prevents cellular disintegration. The fluorescent labelling of the cells that enter into apoptosis and the labelling of dead cells with PI offers the possibility to measure the cumulative apoptotic turnover in time Citation[16]. shows the different apoptotic stages of HL60 cells in time after treatment with heat (43°C during 1 h). After 8 h the first typical features of early apoptosis (FLICA+/PI−) start to emerge. The early apoptotic fraction reaches a maximum at 24 h, where 40% of the cells show caspase activity. Subsequently, HL60 cells lost their plasma membrane integrity and their ability to exclude PI (FLICA+/PI+). This late apoptotic fraction is visible after 24 h and increases up to 74% for heat-induced apoptosis at 48 h. The fraction of cells that are permeable to PI and lost the ability to bind FLICA (FLICA−PI+), also called the ‘necrotic stage’ was absent due to caspase inactivation (data not included). The viability (FLICA−/PI−) of the HL60 cells dropped from 92% to 9% within 48 h. The later onset of radiation-induced cell death observed by studying the kinetics of the apoptotic cascade () is in agreement with the findings that the breakdown of Δψm and the exposure of PS on the outer membrane leaflet occurred at a lower rate compared to heat-induced apoptosis in HL60 cells.
Figure 3. The apoptotic rate after heat-treatment (HT; 43°C during 1 h) and γ-radiation (RT; 8 Gy). The percentage of viable, early and late apoptotic cells are shown. Figures show the percentage of cells, within the total population of 10 000 counted cells, showing fluorescence. The different cell populations based on FLICA and PI uptake were plotted as a function of time after heat-treatment (a) or γ-irradiation (b) of HL60 cells in the continuous presence of FLICA. The fraction of viable cells is FLICA− and PI−, determined with the flow-cytometer as described in materials and methods. Early apoptotic cells are FLICA+ and PI−. In the late stage of apoptosis cells loose plasma membrane integrity and become FLICA+ and PI+. The results are the mean ± SD (n = 3).
Influence of caspase inhibition on Δψm and PS exposure
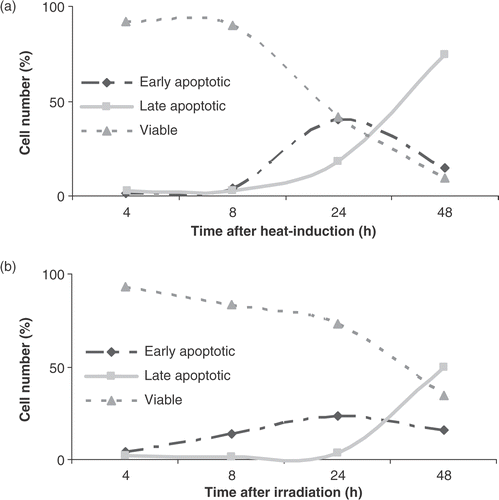
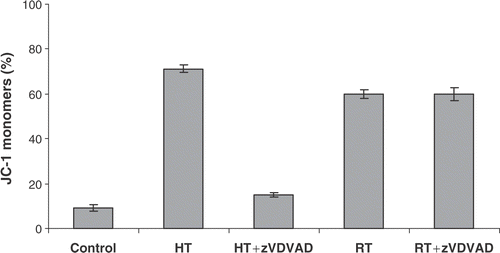
To investigate the role of caspases in Δψm transition and PS exposure, the effect was investigated of the pan-caspase inhibitor zVAD-fmk on heat- and radiation-induced apoptosis in HL60 cells. It was observed that the disruption of the mitochondrial membrane and subsequently the breakdown of Δψm does not require caspase activation, since zVAD-fmk was unable to block the mitochondrial membrane potential (MMP) shift (). However, a truly caspase-independent MMP pathway is hereby not proven since some caspases are insensitive to the caspase inhibitor zVAD-fmk. Caspase-2 is known to be insensitive to zVAD-fmk and can directly evoke the release of cytochrome c from isolated mitochondria. Therefore, the specific caspase-2 inhibitor zVDVAD-fmk was used to further investigate the role of caspases in MMP changes both after heat-treatment and irradiation of HL60 cells. The results presented in show that caspase-2 inhibition prevented the breakdown of the MMP after heat-treatment. In addition, inhibition of caspase-2 had no effect on γ-irradiation induced apoptosis. Although pan-caspase inhibition did not block mitochondrial events induced by hyperthermia, zVAD-fmk significantly reduced events downstream of mitochondrial changes such as PS externalization and cellular disintegration. PS exposure on the outer membrane leaflet of HL60 cells was not blocked by zVAD-fmk during radiation-induced apoptosis. As demonstrated in , PS externalization downstream Δψm transition is almost completely inhibited (from 47% to 10%, control is 5%) by zVAD-fmk in case of heat-induced but not for radiation-induced apoptosis.
Figure 4. Effect of pan-caspase inhibitor zVAD-fmk on Δψm disruption and PS exposure. HL60 cells were treated with heat (HT; 43°C during 1 h) or γ-radiation (RT; 8 Gy) in the presence (+) or absence (−) of 20 µM zVAD-fmk. Figures show the percentage of cells, within the total population of 10 000 counted cells, showing fluorescence. (a) Percentage of cells with JC-1 monomers after 24 h, correlating with a depolarized mitochondrial membrane potential. (b) Percentage of cells that show PS exposure 48 h after induction of apoptosis. zVAD-fmk was added before induction of apoptosis and was continuously present. The results are the mean ± SD (n = 3).
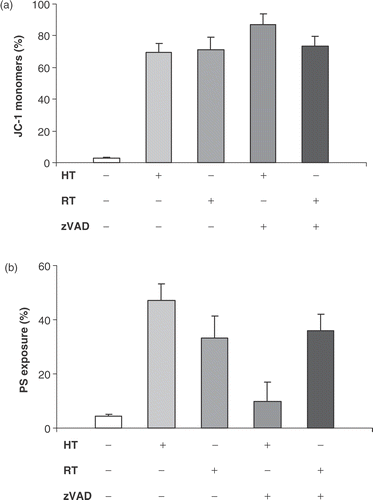
Figure 5. Effect of caspase-2 inhibitor zVDVAD-fmk on Δψm disruption. HL60 cells were treated with heat (HT; 43°C during 1 h) or γ-radiation (RT; 8 Gy) in the presence or absence of 10 µM zVDVAD-fmk. Figures show the percentage of cells, within the total population of 10 000 counted cells, showing fluorescence. Percentage of cells with JC-1 monomers after 24 h, correlating with a depolarized mitochondrial membrane potential. The results are the mean ± SD (n = 3).
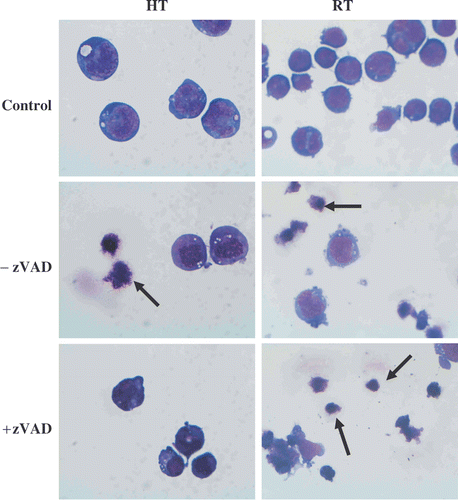
The light-microscopic images made 48 h after heat-treatment or irradiation of HL60 cells in the presence or absence of zVAD-fmk are shown in . The absence of apoptotic bodies after heat-induced apoptosis in the presence of caspase inhibitor is in agreement with the inhibition of PS exposure seen in . In the case of exposing HL60 cells to γ-radiation in the presence of zVAD-fmk there is no prevention of cellular disintegration.
Figure 6. Effects of heat-treatment (HT; 43°C during 1 h) and γ-radiation (RT; 8 Gy) on the morphology of HL60 cells in the presence or absence of 20 µM zVAD-fmk. The cells were fixed with methanol, stained with May-Grunwald-Giemsa and examined at 100 × magnification after 48 h. The images were taken with a light microscope (magnification × 100) and are representative of three independent experiments. Arrows indicate apoptotic cells and apoptotic bodies.
Discussion
This study shows that moderate hyperthermia (43°C during 1 h) of HL60 cells induces apoptosis by disruption of the mitochondrial function, resulting in an increased permeability of the outer mitochondrial membrane. Cellular changes, starting with Δψm disruption, occur as soon as 8 h after treatment of HL60 cells with heat. It has been documented that many different cell types die by apoptosis in response to mild hyperthermia. However, the rate and susceptibility for heat varies significantly with cell type Citation[21]. Comparing these data with similar studies using different cell types is therefore problematic. For γ-radiation-induced apoptosis the lag period before the onset of apoptosis is longer compared to heat-induced apoptosis. This finding is in agreement with the data obtained by determining the apoptotic turnover in time using FLICA, where the faster onset of heat-induced cell death based on the late apoptotic fraction was also visible. The increase of the late apoptotic fraction (AV+/PI+) seen in and , indicating permeabilization of the outer membrane, was unexpected in the presence of the caspase inhibitor FLICA. A possible explanation might be that, although the process of apoptosis was halted at the stage of caspase activation, caspase mediated events might occur, but at a much slower rate Citation[22].
Anti-apoptotic Bcl-2 protein levels decrease and pro-apoptotic Bax protein levels increase directly (compared to untreated HL60 cells) after induction of both heat- and radiation-induced apoptosis, suggesting the involvement of Bcl-2 family members in the mitochondrial membrane permeabilization.
Examination of events downstream of mitochondrial changes indicates that hyperthermia induces features of apoptosis associated with caspase activation such as PS exposure and the formation of apoptotic bodies. After the mitochondrial membrane permeabilization various soluble inter-membrane proteins, including cytochrome c, are released in the cytosol where they activate pro-caspases through the binding to Apaf-1 Citation[10]. The occurrence of the various apoptotic phases in time, presented in this study, is in agreement with the general conception involving the intra-cellular apoptotic pathway.
In both heat- and radiation-induced apoptosis, blockage of activated caspases by the pan-caspase inhibitor zVAD-fmk did not affect the decrease in mitochondrial membrane potential in HL60 cells. However, it has been shown that not all caspases show the same sensitivity to the pan-caspase inhibition Citation[23]. Not unexpectedly, it has been suggested recently that caspase-2 activation is an early initiating step in cell death induced by heat-treatm Citation[24], Citation[25]. Therefore, the effect of specific caspase-2 inhibitor zVDVAD-fmk was studied on the heat-induced changes in MMP and clearly it was showed that caspase-2 plays a key-role in the heat-induced apoptosis.
Since activated caspases were detected using FLICA and PS exposure was almost completely inhibited by a pan-caspase inhibitor, it can be concluded that the execution phase of heat-induced apoptosis in HL60 cells is mainly but not exclusively caspase-dependent. In contrast, the blockage of activated caspases during radiation-induced apoptosis did not result in a decrease of PS exposure on the cell membrane. This suggests that, in HL60 cells, radiation simultaneously activates two parallel pathways; one caspase-dependent, the other caspase-independent, that can lead to cell death. Other studies also indicated a role of caspase-independent pathways in radiation-induced apoptosis Citation[26], Citation[27]. This indicates that caspases act solely as executioners to facilitate heat- or radiation-induced apoptosis. The commitment of both heat- and radiation-induced apoptosis is caused by mitochondrial changes.
Conclusion
Hyperthermia mediates apoptosis of HL60 cells through mitochondrial signalling involving pro-apoptotic and anti-apoptotic Bcl-2 family members. Caspase-2 plays an important role in the breakdown of the mitochondrial membrane potential induced by heat-treatment. Downstream mitochondria caspases are activated and blockage of these caspases results in inhibition of PS exposure on the outer membrane and prevents cells from disintegration. Therefore, the execution phase of cell death during heat-induced apoptosis is mainly caspase-dependent. This insight might be useful for innovative strategies to increase heat-induced apoptosis by means of interfering with molecular processes, either stimulating pro-apoptotic pathways or inhibiting anti-apoptotic processes.
Acknowledgements
This work was financially supported by the University of Twente (Enschede, The Netherlands) as part of the strategic research project NIMTIK (Non-Invasive Molecular Tumour Imaging and Killing).
Related Research Data
References
- Watmough DJ, Ross WM. Hyperthermia. Blackie & Son, Glasgow 1986; 2–4
- Rong Y, Mack P. Apoptosis induced by hyperthermia in Dunn osteosarcoma cell line in vitro. Int J Hyperthermia 2000; 16: 19–27
- Falk MH, Issels RD. Hyperthermia in oncology. Int J Hyperthermia 2001; 17: 1–18
- Hildebrandt B, Wust P, Ahlers O, Dieing A, Sreenivasa G, Kerner T, Felix R, Riess H. The cellular and molecular basis of hyperthermia. Crit Rev Oncol Hematol 2002; 43: 33–56
- Olive PL, Durand RE. Apoptosis: An indicator of radiosensitivity in vitro?. Int J Radiat Biol 1997; 71: 695–707
- Dewey WC, Ling CC, Meyn RE. Radiation-induced apoptosis: Relevance to radiotherapy. Int J Radiat Biol 1995; 33: 781–796
- Mirkovic N, Meyn RE, Hunter NR, Milas L. Radiation-induced apoptosis in a murine lymphoma in vivo. Radiother Oncol 1994; 33: 11–16
- Tsujimoto Y, Shimizu S. The voltage-dependent anion channel: An essential player in apoptosis. Biochimie 2002; 84: 187–193
- Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 1999; 399: 483–487
- Desagher S, Osen-Sand A, Nichols A, Eshes R, Montessuit S, Lauper S, Maundrell K, Antonsson B, Martinou JC. Mitochondria as the central control point of apoptosis. Trends Cell Biol 2000; 10: 369–376
- Danial NN, Korsmeyer SJ. Cell death: Critical control points. Cell 2004; 116: 202–219
- Desagher S, Osen-Sand A, Nichols A, et al. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J Cell Biol 1999; 144: 891–901
- Fitch ME, Chang C-M, Parslow TG. The BH3 domain is required for caspase-independent cell death induced by Bax and oligomycin. Cell Death Differ 2000; 7: 338–349
- Green DR. Apoptotic pathways: Ten minutes to death. Cell 2005; 121: 671–674
- Wolbers F, Buijtenhuijs P, Haanen C, Vermes I. Apoptotic cell death kinetics in vitro depend on the cell types and the inducers used. Apoptosis 2004; 9: 385–392
- Smolewski P, Grabarek J, Phelps DJ, Darnzynkiewicz Z. Stathmo-apoptosis: Arresting apoptosis by fluorchrome-labeled inhibitor of caspases. Int J Oncol 2001; 19: 657–663
- Overbeeke R, Steffens-Nakken H, Vermes I, Reutelingsperger C, Haanen C. Early features of apoptosis detected by four different flow cytometry assays. Apoptosis 1998; 3: 115–121
- Overbeeke R, Yildirim M, Reutelingsperger C, Haanen C, Vermes I. Sequential occurrence of mitochondrial and plasma membrane alterations, fluctuations in cellular Ca2+ and pH during initial and late phases of cell death. Apoptosis 1999; 4: 455–460
- Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using Fluorescein labelled Annexin V. J Immunol Methods 1995; 184: 39–51
- Vermes I, Haanen C, Reutelingsperger C. Flow cytometry of apoptotic cell death. J Immunol Methods 2000; 243: 167–190
- O’Neill KL, Fairbairn DW, Smith MJ, Poe BS. Critical parameters influencing hyperthermia-induced apoptosis in human lymphoid cell lines. Apoptosis 1998; 3: 369–375
- Smolewski P, Grabarek J, Lee BW, Johnson GL, Darzynkiewics Z. Kinetics of HL-60 entry to apoptosis during treatment with TNF-α or camptothecin assayed by the stathmo-apoptosis method. Cytometry 2002; 74: 143–149
- Breckenridge DG, Xue D. Regulation of mitochondrial membrane permeabilization by BCL-2 family proteins and caspases. Curr Opin Cell Biol 2004; 16: 647–652
- Bonzon C, Bouchier-Hayes L, Pagliari LJ, Green DR, Newmeyer DD. Caspase-2-induced apoptosis requires bid cleavage: A physiological role for bid in heat shock-induced death. Mol Biol Cell 2006; 17: 2150–2157
- Tu S, McStay GP, Boucher LM, Mak T, Beere HM, Green DR. In situ trapping of activated initiator caspases reveals a role for caspase-2 in heat shock-induced apoptosis. Nat Cell Biol 2006; 8: 72–77
- Inagaki-Ohara K, Takamura N, Yada S, Alnadjim Z, Liu E, Yu X, Yoshida H, Lin T. Radiation-induced crypt intestinal epithelial cell apoptosis in vivo involves both caspase-3-dependent and -independent pathways. Dig Dis Sci 2002; 47: 2823–2830
- Cornelissen M, Vral A, Thierens H, Ridder L. The effect of caspase-inhibitors on radiation induced apoptosis in human peripheral blood lymphocytes: An electron microscope approach. Apoptosis 1999; 4: 449–495