Abstract
New knowledge of nuclear structure and DNA repair pathways has provided the basis for new insight into the effects of hyperthermia on the proteins involved in these processes. The nucleus is made up of mega protein-nucleic acid complexes that conduct various nuclear functions, including DNA packing, repair, replication and transcription. Heat shocks (41–50°C) cause unfolding of a number of nuclear proteins. Such unfolding changes protein associations within all of the intra-nuclear mega protein-nucleic acid complexes studied, with the exception that no alterations in the nucleosome-DNA bead and super bead complexes could be detected. This review will address heat effects on protein-nucleic acid complexes related to DNA replication and DNA repair. Heat-induced changes in DNA replication complexes can be related to the killing of S-phase cells by heat. The effects of heat on DNA repair foci, complexes involving MRE11, the nucleolus and on the complexes that anchor DNA to the nuclear matrix appear to contribute to radiosensitization as a function of increasing thermal dose. Thus, heat effects on these complexes can serve as molecular targets for the development of agents that can enhance the effectiveness of clinical thermal radiotherapy.
Introduction
Our first hyperthermia experiment was designed to use the effects of heat shock on DNA repair as a means to understand the proteins and mechanisms involved in DNA repair. The results from the first step of this study were so surprising that we found ourselves on a whole new study of the effects of heat shock on nuclear protein associations. Now after 30 years, numerous surprises and new horizons, we are able to study specific protein changes in specific steps of the DNA double strand break repair process. New knowledge of both nuclear structure and the DNA repair process has provided the basis for greater insight into the effects of hyperthermia on nuclear protein associations. It is now known that the nucleus is made up of mega protein-nucleic acid complexes that carry out various nuclear functions, including DNA packing, repair, replication and transcription. One long recognized mega protein-nucleic acid complex is the nucleolus. Recent results indicate that, in addition to its well established role in producing and exporting polysome components, the nucleolus is a repository of stress responsive proteins that are released in response to cellular stress. Heat shocks in the therapeutic range cause unfolding of a number of nuclear proteins (the exact number remains unknown). Such unfolding will change protein associations with and within all of the intra-nuclear mega protein-nucleic acid complexes studied, with the exception that no alterations in the nucleosome-DNA bead and super bead complexes could be detected. This review will address heat effects on several mega-protein-nucleic acid complexes and the subsequent role these effects play in cell killing by heat and the enhancement of the lethal effects of ionizing radiation. For example, heat-induced changes in DNA replication complexes can be related to the killing of S-phase cells by heat. Further, the effects of heat on DNA repair foci, complexes involving MRE11, the nucleolus and on the complexes that anchor DNA to the nuclear matrix all appear to contribute to radiosensitization as the thermal enhancement ratio (TER) increases as a function of increasing thermal dose. Given the clinical relevance of the latter endpoint, heat effects on these complexes can serve as molecular targets for the development of agents that can enhance the effectiveness of clinical thermal radiotherapy.
Early studies on the effects of hyperthermia on the nucleus
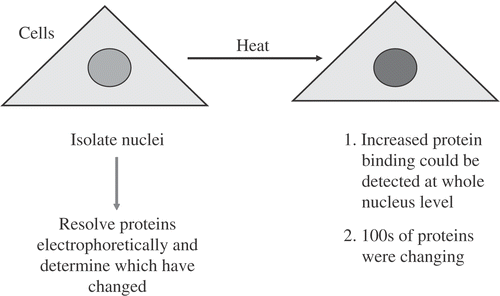
To start from the beginning, our first hyperthermia experiments were done for the wrong reasons. However, surprising results, launched on studies into the effects of hyperthermia on nuclear and eventually nuclear matrix proteins. Having been introduced to the radiosensitizing effects of hyperthermia by Bill Dewey and knowing about DNA repair, the logical direction was to study hyperthermia as an inhibitor of DNA repair. Thus, our original intent was to use hyperthermia as a way to get at the proteins critical for DNA repair. So I thought we would start defining DNA proteins by doing a very simple experiment (). Specifically, we heated cells, isolated nuclei and resolved proteins on 2D gels. The results were a big surprise. We found that the nuclei isolated from heat shocked cells had a whole lot more proteins than those from cells that had not been heated. We ran these proteins out on gels and the amounts of hundreds of proteins were altered. In fact, probably thousands of nuclear proteins had been affected by heat shock. We found that the proteins coisolating with the nucleus increased in a time–temperature dependent manner Citation[1–3]. So we stopped doing studies on individual proteins and instead focused on proteins and related changes in the nucleus and nuclear matrix. As will be seen, it took us several years to get back to studying individual proteins.
Figure 1. Initial experiment to Find Nuclear Proteins that were Sensitive to Heat Shock.
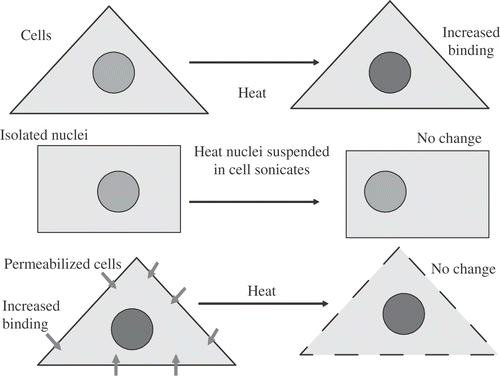
To determine the origin of the apparent increase in nuclear proteins, isolated nuclei were suspended in a whole cell sonicate, maintaining a high concentration of proteins approaching the protein concentration in a cell. The suspension of nuclei was heated. No change in the amount of proteins co-isolating with nuclei was detected (). Based on later work Citation[3–6], Citation[50] we know that a significant fraction of proteins binding with the nucleus after heat shock were soluble nuclear proteins prior to heat shock. If you isolate the nuclei in an aqueous environment, you lose these proteins. Once they are lost they cannot contribute to the increased amount of protein due to heat shock. This study along with later studies showed that the proteins were not binding during nuclear isolation Citation[7]. It should be noted that we were able to approximate the protein concentration of the cell but did not come close to the protein concentration of the nucleus. This result can be understood further if we consider the work of the late Dr James Lepock. His work showed that heat causes proteins to unfold and expose hydrophobic groups Citation[8]. These hydrophobic regions aggregate and the aggregated complex of proteins is what we observed as an increase in proteins co-isolating with the nucleus. When the nucleus was isolated prior to heat shock the protein-protein associations that lead to aggregation and/or altered binding were lost and no measurable aggregation was detected. Further, the mere presence of these proteins in the absence of the original structural associations did not restore the ability of heat shock to induce the aggregation of these proteins. One more early experiment was done, namely, to heat permeabilized cells and determine if we could observe the heat-induced increase in protein co-isolating with the nucleus. However, as soon as we permeabilized cells, the amount of proteins that were bound to the nucleus went up and heat shock did not further alter the extent of protein aggregation observed. Later studies by Dewey et al. Citation[9] showed that in S-phase cells, trauma to the cell membrane increased nuclear protein aggregation, cell killing and chromosome aberrations Citation[9]. Thus, changes in protein binding, leading to protein aggregation and its consequences at the cellular level, can occur following trauma to the nuclear membrane in an intact cell.
Figure 2. Increased Nuclear Protein Binding Induced by Heat Shock. Nuclear protein binding increased after heat shock or permeabilization of intact cells, but not after heat shock of isolated nuclei in whole cell sonicates or permeabilized cells.
Altered nuclear protein binding and cell killing
The enhanced protein binding within the nucleus should have a significant impact on nuclear function, because nuclear function is dependent upon a large number of proteins and nucleic acids interacting in a very small volume. For example the nucleus contains ∼2 meters of DNA, 5 × 109 base pairs, requiring a 40 000-fold compaction to fit within the nuclear volume Citation[10]. The protein mass is 3–6 times more than the amount of DNA in the nucleus. Thus, the nuclear volume is very tightly packed. Therefore, aberrant protein binding within that volume has the potential to disrupt a significant fraction of the dynamic interactions required to conduct what is normally a large number of nuclear functions. We knew from the above experiment that the de novo protein–protein associations and presumably some underlying structures are necessary for the heat induction of altered protein binding and aggregation within the nucleus based on Dr Coffey's work which had shown that one critical underlying nuclear structure is the nuclear matrix Citation[11–12]. Altered protein associations with the nuclear matrix have the potential to disrupt any and all nuclear matrix functions. Nuclear matrix functions include DNA replication, transcription of RNA, RNA processing Citation[13–14], molecular trafficking between the nucleus Citation[15] and cytoplasm and at least some DNA repair. All of these are known to be inhibited by heat shock Citation[16]. Thus, the aberrant binding of proteins to the nuclear matrix could contribute to a number of hyperthermic effects at the cellular level. What is the evidence for heat shock causing protein unfolding within the nuclear matrix? and show the results of differential scanning calorimetry curves obtained by the late Dr James Lepock, which detect protein unfolding as a function of temperature Citation[8]. The various peaks, A–D, represent endothermic denaturation of the proteins of various cellular components. The lower the temperature that a peak is detected, e.g., peak A, the more sensitive the proteins of that cellular component are to unfolding at lower temperatures. is the scanning curve for isolated nuclear matricies, which corresponds to the peak A, in , showing that the nuclear matrix is (at least one of) the most heat sensitive parts of the cell Citation[8]. This result leads us and other workers to consider if the aberrant binding of proteins to the nuclear matrix could play a role in cell killing.
Figure 3. Thermal Denaturation of Cellular Components. DSC profiles of excess Cp vs. temperature for V79 cells (solid line) an nuclei isolated from V79 cells (dotted line). Reprinted from Ref. 8.
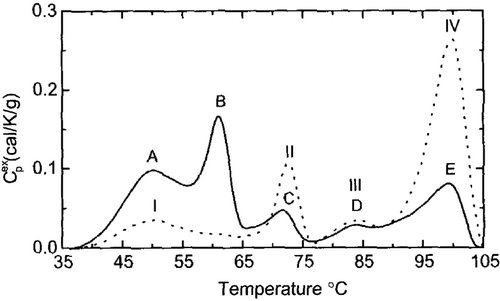
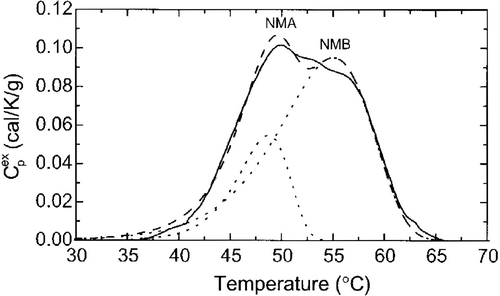
Figure 4. The Nuclear Matrix is the Most Temperature Sensitive Subcellular Components Identified so Far. DSC profile of V79 nuclear matrices(solid line), the best two components fits (dashed line) and the individual components: NMA and NMB (dotted lines). This profile corresponds to peak A in . Reprinted from Ref. 8.
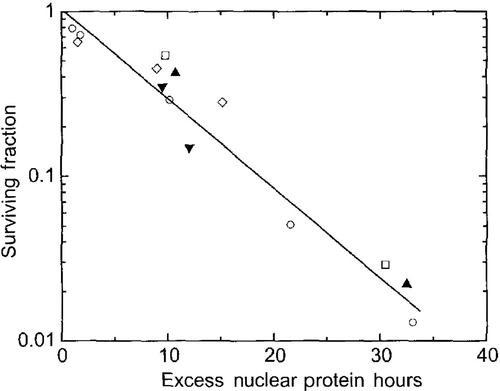
The temperature dependence of aberrant protein binding to the nuclear matrix has two components: (1) the time- and temperature-dependent increase in protein binding to the nuclear matrix and (2) the time-dependent, post-heat shock resolution of the aberrant nuclear matrix-protein binding to pre-heat shock levels. The first order time course of the latter is dependent on the amount of post heat nuclear matrix-protein binding and the cellular levels of molecular chaperones (i.e., heat shock proteins). If both components of the time course are taken into account, the binding of proteins in excess of the amount bound to the nuclear matrix in unheated cells, correlates linearly with cell killing () Citation[17]. We developed (or modified) three additional measures of heat effects on the nuclear matrix: (1) limit DNA digests by DNase I, RNase stability assay, and divalent cation stability assay Citation[18–19]. The first assay showed that at the limit DNase I digest in nuclei from heated cells, a larger fraction of DNA associated with the nuclear matrix was inaccessible to the enzyme than when the nuclei were isolated from control cells Citation[18]. The two stability assays showed that nuclear matrices from heated cells cannot be disintegrated by RNase digestion or the removal of divalent cations, showing the residual protein–protein interactions have become stabilized, in other words less functional Citation[19]. These are clearly changes that would alter the functional/structural role of the nuclear matrix in DNA metabolism. At this point, even though we started to address the effects of heat on DNA repair we were now thinking about how alterations in DNA metabolism could lead to cell killing.
Figure 5. Nuclear Matrix as a Target for Cell Killing. Cell survival after heat shock is plotted as a function of excess nuclear protein hours for various heating conditions, including normal and thermotolerant cells and the presence of sensitizers (ethanol and procaine) or protectors (glycerol). Each symbol represents a different treatment. Excess nuclear protein hours is calculated by summing the amount of protein coisolating with the nucleus (above control amounts) times the time (in hours) that there is an excess. Reprinted from 8 originally published in 17.
The killing of S-phase cells
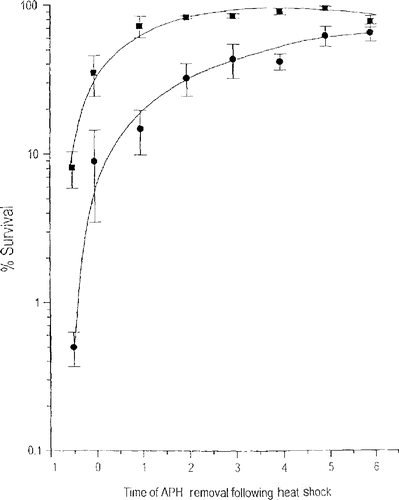
When the rate constants for cell killing are subjected to Arrhenius analysis one finds that both the first and second order rate constants are linear with the reciprocal of the absolute temperature Citation[20]. While the results of this analysis suggested that heat-induced cell killing was a combination of first and second order mechanisms, the strong temperature dependence of the second order rate constant caught our attention because it suggested that there is a repairable, heat-induced lesion that is interacting either with another heat-induced lesion or a heat-modulated cellular process. Considering what cellular process could be interacting with a heat-induced lesion, the first possibility that came to mind was DNA synthesis. Westra, Dewey and coworkers showed that S phase cells are highly sensitive to heat shock Citation[21]. In addition, chromosome aberrations correlate with the killing of S-phase cells, suggesting a role for DNA damage. In our opinion, the DNA damage is secondary to attempted DNA replication during the time that heat shock-induced aberrant protein binding with the nuclear matrix is ongoing. This idea is supported by the observation that when cells are heated in G1, they are less sensitive to heat-induced cell killing and no chromosome aberrations are induced Citation[21]. Thus, we began to look at the role of DNA replication and nuclear matrix protein binding following heat shock in the hypersensitivity of S-phase cells to hyperthermia. This work was done by Robert vanderWaal Citation[22] who used a reversible DNA synthesis inhibitor, aphidicolin, to first obtain a population of cells in the first 30 min of S-phase and then after heat shock either allow the cells to synthesize DNA as best they could or to block DNA synthesis and allow the cells to recover from the effects of heat shock for various time intervals prior to attempting DNA synthesis. When cells synthesized DNA as best they could, the survival levels were 0.08 and 0.005 after the 15 min and 30 min at 45°C heat shocks, respectively (). When DNA synthesis was blocked, there was an increase in cell survival that was dependent on time that DNA synthesis was blocked until survival equaled that of a non-S phase cell. For example, if DNA synthesis was blocked for 5 hr the survival levels were 0.9 and 0.55 after the 15 min and 30 min at 45°C. This result means that all of the S phase hypersensitivity to hyperthermia is due to the cells attempting to make DNA while some repairable heat-induced damage is present. Our next step was to determine if aberrant binding of proteins to the nuclear matrix is the repairable lesion.
Figure 6. Delaying DNA Synthesis Reduces the Thermal Sensitivity of S-phase Cells. S-phase Cells were heated at 45°C for 15 (squares) or 39 (circles) min and DNA synthesis was blocked for the indicated time with aphidicolin. Reprinted from Ref. 22.
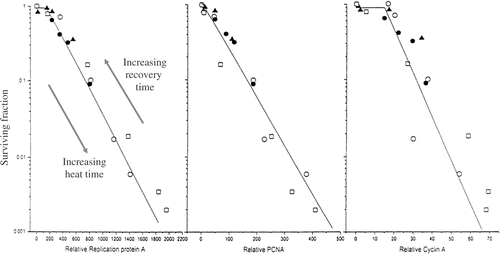
To test the possibility that aberrant binding of proteins to the nuclear matrix is repaired during post hyperthermia recovery in a manner that relates to the loss of S-phase hypersensitivity, we measured the binding of three replication proteins: RPA, PCNA and cyclin A to the nuclear matrix as an indirect measure of their binding to DNA replication complexes which are associated with the nuclear matrix. The amount of excess DNA replication proteins binding to the nuclear matrix at the time cells attempted DNA synthesis demonstrated a linear relationship with the logarithm of the fraction of cells surviving, regardless of whether the heat dose or the recovery time prior to the resumption of DNA synthesis was varying (). In contrast there was no correlation between proteins that demonstrated heat-induced binding to the nuclear matrix, without binding to DNA replication complexes Citation[22]. This result suggests that altered protein binding within the DNA replication complexes is the potentially lethal damage that can be repaired if DNA synthesis is halted, but will be fixed to lethal damage if the cells attempt to synthesize DNA when the enhanced protein binding is present. Further evidence for the tighter binding of proteins within DNA replication complexes is the observation that larger complexes are isolated effectively from heated cells compared to unheated cells Citation[22–23]. The question then becomes, why would enhanced binding of proteins in DNA replication complexes be a potentially lethal condition. To understand this point it is first important to recognize that functionality of large protein complexes such as DNA replication factories requires a certain level of protein mobility within the complex to successfully conduct that complex's normal function. When heat shock induces the aggregation of two or more protein members within a complex, its function will be impaired, in this case inhibiting DNA synthesis. Inhibition of DNA synthesis can lead to lethal lesions by either or both of two mechanisms: stalled replication DNA replication forks Citation[24], and imbalanced timing of DNA replication in S-phase due to epigenetically inhibited check points.
Figure 7. The Abundance of Unextractable DNA Replication Proteins Relates to Cell Survival after Heat Shock. The surviving fraction of S-phase Cells following various heat shocks of 7.5 to 45 min at 45°C and after blocking DNA synthesis for 0-6h after the heat shocks was plotted as a function of RPA, PCNA or Cyclin A insolubly bound to the nuclear matrix. The different symbols relate to different experiments were the heating time or the DNA synthesis blocking time was varied. Reprinted from Ref. 22.
To understand the importance of the latter mechanism one must note the observations by Berezeny Citation[25] that different DNA replication patterns occur as cells progress through S–phase and that the timing of the transition between these patterns is critical for the segregation of replication and transcription, which must be coordinated in time and space, as cells progress through S-phase. To observe these DNA replication patterns, cells are labelled with the thymidine analog, BrdU. DNA replication foci fall into three distinct patterns: (1) a pattern characteristic of early S-phase, called type I; (2) a pattern characteristic for middle S-phase, called type II; and (3) a pattern characteristic for late S-phase, called type III Citation[24]. Heat shock disrupts the timing of these replication patterns, as can be seen by a split labelling experiment. A split labelling experiment is done by labelling first with chloro-deoxyuridine, which is detected, for example, by green immunofluorescence, followed after a time interval by labelling with iodo-deoxyuridine, which is detected by red immunofluorescence. This approach allows one to detect the replication factories that are synthesizing DNA at the time of each label and those that are replicating DNA at both times, detected as yellow fluorescence. When there is a 4 hr time interval between labels in normal S-phase progression you can see either all green, Type I replication foci, or all red, Type II replication foci, and very little yellow. Therefore, essentially all of the factories making DNA in early S-phase had completed synthesis 4 hr later. In contrast, after a 30 min at 45°C heat shock, with a 6.5 hr interval between the labels, all of the Type I foci, fluoresced yellow, while the Type II foci fluoresced red. This result means that all of the early S, Type I replication factories are still making DNA 6.5 hr later, a time interval that is long enough that most unheated cells would have completed S-phase. Thus, heat shock disrupts the spatial and temporal coordination of S phase progression, which appears to be critical for proper cellular function Citation[26].
The second consequence of inhibited DNA replication is stalled replication forks. Stalled replication forks can lead DNA double strand breaks via longer lived single strand regions as reported by Wong et al. Citation[24], Citation[27–28] which can collapse into ‘chicken foot’ structures Citation[27]. DNA double strand breaks secondary to stalled replication forks require homologous recombination for their repair. Ironically heat shock inhibits homologous recombination, as we will see below. The cell death pathways secondary to DNA double strand breaks are well established Citation[29–31]. Therefore, if heat-induced disruption of DNA synthesis that leads to DNA double strand breaks through replication fork stalling and the homologous recombination (HR) repair pathway is inhibited by heat shock, we have two heat dependent interacting alterations that can lead to cell death, which could explain the second order cell death mechanism inferred from the Arrhenius analysis described above.
Effects of heat shock on DNA repair
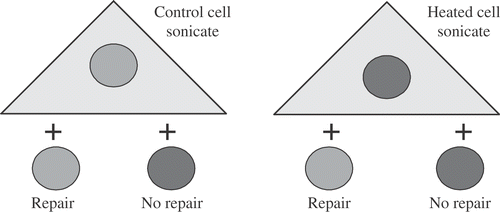
The foregoing considerations have led us back to the need to understand the effects of heat shock on DNA repair. Given that hyperthermia causes protein unfolding, an early question was whether hyperthermia was directly altering DNA repair enzymes or if altered protein associations with the nuclear matrix blocked access of the repair system to the DNA lesions within chromatin. The following experiment supports the latter possibility at least for base excision repair. This experiment is based on the fact that isolated nuclei are able to excise DNA base damage if suspended in whole cell lysates Citation[32–33]. The specific experiment was to isolate nuclei from heated or unheated cells and prepare lysates from heated or unheated cells. We then found that lysates from control cells were unable to excise damaged nucleotide bases from nuclei isolated from heated cells (). In contrast, lysates from heated cells were able to excise damaged bases from nuclei isolated from control cells. Thus, the heat-induced alteration that inhibits the excision of damaged bases is in the nucleus, not in the enzymes that are released when a whole cell lysate is prepared. A lot more is known about DNA repair today than when we did the experiments. Specifically, there are three phases of any repair pathway to be considered: (1) sensing the DNA damage in the chromatin, (2) transduction which mediates the transition between sensing and actual repair pathway, and (3) the effector steps in which the removal of damage and restoration of the native DNA and chromatin structure is accomplished. We now know that the removal of damaged bases from DNA is accomplished by glycosylases, the first step in the effector portion of the base excision repair process. Thus, the results of the repair reconstitution experiment, suggests that the heat-induced nuclear (matrix) alteration would have inhibited a step in the repair process at or before the very first step in the effector process.
Figure 8. First Experiment to Determine the Effects of Hyperthermia on Base Excision Repair (BER). Nuclei isolated from γ-irradiated cells that were either heat-shocked 30 min at 45°C or not heated were mixed with whole cell sonicates from either heat-shocked 30 min at 45°C or control cells and assayed for the ability to excise altered thymine bases. The result shows that there is an alteration in nuclei from heated cells that prevents excision of damaged bases.
This conclusion appears to apply also for the repair of DNA double stand breaks. DNA double strand breaks can be repaired by either of two pathways: HR or non-homologous end joining (NHEJ), which are thought to compensate for each other Citation[34]. The work recently reviewed by Kampinga et al. Citation[35] showed that mutants defective in the effector steps of the HR or the NHEJ pathways do not have a reduced TER Citation[35]. Given the extensive prior evidence that heat-induced radiosensitization was due to the inhibition of DNA double strand break repair Citation[35], the opinion of this author is that both pathways are impacted by inhibition of steps prior to the effector portions of these pathways. Heat-induced alterations within the nucleus must, therefore, inhibit the sensing and/or transducing steps of DNA repair. This can occur either by heat effects on the sensing/transducing molecules or by heat-induced aggregation of proteins at the nuclear matrix sterically preventing the sensing or transducing molecules from accessing the damage. We are finding evidence that both are occurring after heat shock if the temperature is high enough. We will consider the latter first, which has been described as the masking of DNA damage from DNA repair pathways Citation[36].
If DNA damage is masked by proteins bound to the nuclear matrix, one would expect that such binding would affect the DNA that is associated with the nuclear matrix or MAR DNA. Changes in DNA nuclear matrix anchoring should be detectable through changes in the supercoiling ability of the anchored DNA loops (). In addition, the strand breaks induced by ionizing radiation are known to alter the ability of DNA loops to supercoil by disrupting the integrity of the DNA strand Citation[37–38]. Thus, assaying DNA supercoiling ability appeared to be a good approach to studying the effects of heat plus radiation on DNA repair and, thereby, heat-induced radiosensitization. The first question was when cells are heat shocked and then irradiated, do the effects of heat on proteins binding to MAR DNA, which should tighten loop anchoring and increase supercoiling ability, dominate or are dominated by the effects of ionizing radiation which through strand breaks inhibit the ability of loops to supercoil. A textbook DNA supercoiling experiment uses a closed circular plasmid which can be supercoiled, relaxed or supercoiled in the opposite sense. However, Vogelstein and Coffey developed a method, which uses fluorescent intercalating compounds to visualize DNA loops, with their endogenous nuclear matrix anchoring, as they unwind and rewind Citation[39]. Our modification of this technique allows us to measure DNA unwinding and rewinding in cells that can be suspended after various treatments or even isolated from in vivo tissues Citation[38]. In this is a simple assay, as the DNA loops unwind, we can see the maximum DNA loop diameter and as they rewind are returned to a diameter that is just a bit larger then the original nuclear diameter. As an additional approach, we took advantage of a recently discovered anchoring protein, protein disulfide isomerase (PDI), which under normal redox or oxidative conditions, binds DNA more tightly, enhancing DNA loop rewinding Citation[40]. However, under reducing conditions the disulfide bridges unlock and PDI loosens its grip on the DNA and the rewinding of DNA loops are inhibited. Thus, the sensitivity of nuclear matrix DNA anchoring to reducing conditions allows us to ask the question of whether the tightening effects of heat shock on DNA anchoring will dominate the loosening effects of reducing conditions. In fact, the heat-induced binding of proteins to the MAR DNA dominates both the inhibition of DNA loop rewinding by DNA strand breaks induced by ionizing radiation and that due to the loosening of PDI-MAR DNA binding. We have termed this phenomenon, ‘the masking effect’ Citation[36], Citation[41–42].
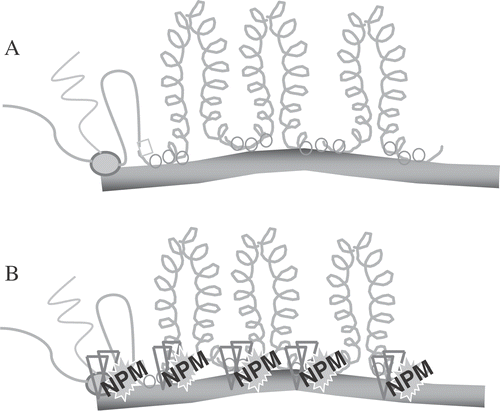
Figure 9. An Illustration of Nuclear Matrix DNA Anchoring. DNA loops are periodically attached to the nuclear matrix creating domains for DNA supercoiling changes (shown in A for unheated cells), which occur secondarily to transcription (shown on the left side) and replication (not shown). After heat shock (B) proteins, such as NPM (see text) bind to the attachment points.
Two studies demonstrate the importance of the masking effect in heat-induced radiosensitization and in the inhibition of DNA repair. The first is an experiment done by Andrei Laszlo in collaboration with us. We have here three CHO cell lines, which vary in their resistance to heat-induced radiosensitization. In fact, these are the only mutants we know of that are heat resistant to heat-induced radiosensitization Citation[42]. The wild type (HA1) has a TER of around 2.12–2.16 after either a 60 min at 43°C or a 15 min at 45°C heat shock. In contrast, the heat resistant variant (HR1) has TERs of 1.65 or 1.83 after these two heat shocks, respectively. The third cell line (OC14) is interesting in that it is resistant to 43°C hyperthermia but not 45°C hyperthermia Citation[42]. After a 60 min at 43°C heat shock, the TER for OC14 cells was 1.65, but after 15 min at 45°C, the TER was 2.11, even though these two heat shocks are considered to be equivalent thermal doses. After both heat shocks in wild type cells a significant masking of DNA damage (as measured by the inhibition of DNA supercoiling) is observed Citation[42]. The extent of masking can be quantified by defining a quantity, called the excess halo diameter, which quantifies the difference between the DNA loop rewindability in nucleoids from control cells and that observed in nucleoids from treated cells. The excess halo diameter is illustrated in . Thus, the reduction in excess halo diameter becomes a measure of the masking effect. Wild type cells showed a significant making effect after both the 60 min at 43°C and 15 min at 45°C heat shocks. In contrast, in the heat-resistant variant the masking effect was greatly reduced after both the 60 min at 43°C and 15 min at 45°C heat shocks. Interestingly, the OC14 cells showed a reduction in the masking effect after the 60 min at 43°C heat shock, but not after the 15 min at 45°C heat shock, even though the two heat shocks tested were isothermal doses Citation[42]. The reduction in excess halo diameter showed a linear correlation with TER for these cell lines and heat shocks (). This result is consistent with the conclusion that the masking effect contributes to the increase in radiation sensitivity induced by heat shock. However, it should be noted that this correlation extrapolated to zero reduction of excess halo diameter at a TER of 1.5. Thus, the masking effect can only contribute to a higher TER value at higher temperatures and there must be additional targets for heat-induced radiosensitization at moderate temperatures (see below for alternative targets). The second study was done by Jim Wynstra, who showed that the masking effect linearly correlates with both the reduction in the rate of repair of radiation damage and the residual unrepaired DNA damage () Citation[41]. We also found that both the masking effect and the inhibition of DNA repair were reduced to their pre-heat shock levels faster in thermotolerant cells than in normal cells Citation[41].
Figure 10. Graphic Definition of the Excess Halo Diameter (EHD).
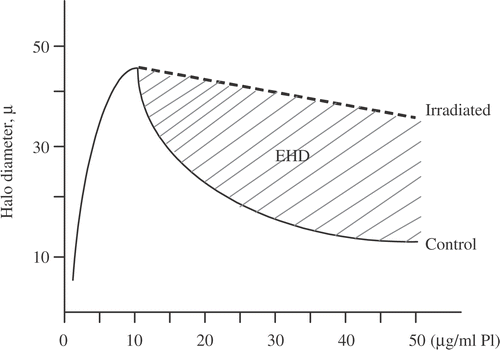
Figure 11. Relationship between masking of DNA damage and HIR multiple molecular targets must be affected by hyperthermia to achieve TERs > 1.5
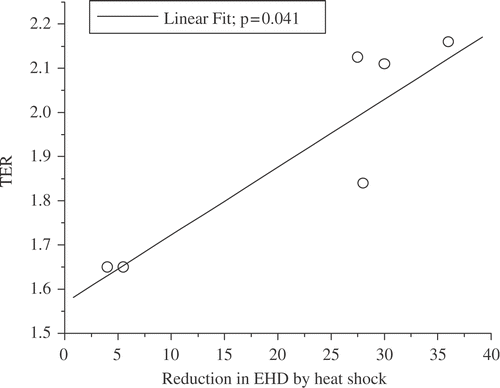
Figure 12. The Effects of MAR DNA Masking on the Repair of DNA Damage. For various time intervals (1–6 hr) between 30’ at 45°C and 5 Gy, the rate of repair and the percent of unrepaired DNA damage were measured and plotted vs. the reduction in EHD (41).
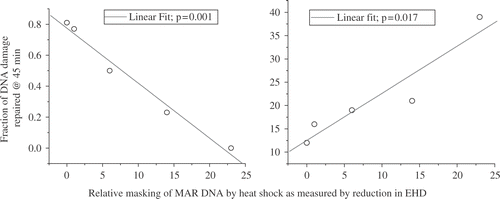
These two studies show that the heat-induced masking of DNA damage at or near MAR regions keeps a fraction of DNA strand breaks from being repaired (i.e., increases the residual unrepaired DNA damage) and at the same keeping the strand breaks from the repair. Further, these results imply that when we have protein binding to that MAR region and we have a strand break in this DNA, that protein blocks the repair of that strand break. Once the proteins responsible for the masking effect are removed, repair can go forward to completion. The question becomes what proteins are binding to MAR DNA and the nuclear matrix and causing the masking effect.
Protein candidates for the masking of DNA damage
Using a proteomics screening approach, we have found a number of proteins that increase their binding with DNA after heat shock Citation[43]. One of these is a nucleolar protein nucleophosmin (NPM), which binds DNA and increases its DNA binding after heat shock Citation[43]. In the unheated cell NPM cooperates with nucleolin and escorts the polysome component, L5, from the nucleus to the cytoplasm Citation[44]. NPM is essential for polysome assembly and rate limiting for protein synthesis Citation[44]. In the heated cell, these two proteins appear to act independently instead of together. Nucleophosmin binds p53 and DNA. George Iliakis has shown that nucleolin binds RPA, escorts RPA away from the nucleus, preventing its role in DNA replication Citation[45]. We have found that when NPM is associated with DNA, reducing conditions cannot reduce the association of PDI with DNA Citation[43], suggesting that NPM is contributing to the masking effect. Ongoing work is verifying the role of NPM in the masking effect.
Effects of heat shock on MRE11
The foregoing discussion suggested the need to consider a moderate temperature target. Based on the work of Kampinga et al. Citation[35] we decided to look upstream of the effector portions of both HR and NHEJ DNA double strand break repair pathways. One molecular complex that is thought to participate in the sensing/transducing steps of DNA double strand break repair is the MRN complex consisting of MRE11, RAD50 and NBS1. Further work by Dynlacht and coworkers had shown that acute heat shock can affect the nuclear localization of MRE11 Citation[46]. Thus, we began to evaluate the MRN complex as our moderate temperature target by measuring the effects of hyperthermia on MRE11. Moderate hyperthermia has several distinct effects on MRE11. Moderate hyperthermia (41°C) delocalizes a significant fraction of MRE11 from the nucleus. In a study of MRE11 localization by immunofluorescence one can observe a time (at 41°C) dependent increase in cytoplasmic MRE11 and a corresponding decrease in nuclear associated MRE11 Citation[47]. This observation was confirmed using cell fractionation experiments. Cells were split into a cytoplasmic fraction and nuclear fraction using Triton X100 and analysing the amount of MRE11 by Western blotting. Prior to hyperthermia most (80–95%) of MRE11 was recovered in the nuclear fraction. Depending upon the tumor cell line, the fraction of MRE11 in the nucleus, decreases to 60% or less in 2 to 6 hr Citation[47]. In the NSY human tumor cell line the reduction in nuclear MRE11 correlates with both the 2 hr at 41°C required to radiosensitize the cells (when irradiation is done after hyperthermia) and the extent of radiosensitization measured. Note that after 8 hr at 41°C cell cycle redistribution also modulates radiation sensitivity Citation[47]. The functional relationship between TER and MRE 11 nuclear delocalization is being tested to other human tumor cell lines (work in progress). The observation that TER correlates with MRE11 nuclear delocalization in NSY cells suggests that a reduction in availability of MRE11 is responsible for the increased radiation sensitivity. This possibility was tested using siRNA technology.
To test the possibility that reduced MRE11 could radiosensitize cells in a manner similar to that by hyperthermia, we transfected NSY cells with a siRNA specific for MRE11. Twenty-four hours after transfection, MRE11 levels were reduced 60% below normal Citation[48]. The results showed that in the cells with ∼60%, MRE11 knockdown had a dose modifying factor of 1.39, only slightly higher than that for cells heated for 2 hr at 41°C (a TER of 1.28). Further, when the cells with 60% reduced MRE11 cells were treated with 2 hr at 41°C and then X-irradiated, the TER was not different than that for the unheated knocked down cells Citation[48]. Thus, the RNAi knockdown of MRE11 caused about the same radiosensitization as did 2 hr at 41°C and hyperthermia did not increase the radiation sensitivity over the knockdown alone. These results suggest that the unavailability of MRE11 for DNA repair due to heat-induced nuclear delocalization of the molecule is a contributing factor to radiosensitization.
The question then becomes what could be causing MRE11 to delocalize from the nucleus? If the hyperthermia is causing the MRE11 molecule to unfold, then one would expect an increase in the interaction between MRE11 and HSP70. MRE11 and its partner, RAD50, both interact with HSP70 a minimum basal level in unheated cells. The associations between Hsp70 and RAD 50 and MRE11 increase up to between 10- (for RAD50) and 100-fold (for MRE11) as a function of time at 41°C Citation[49]. The associations of both MRE11 and RAD50 with HSP70 are correlated with radiosensitization as measured by TER (). Interestingly, these correlations were clearer than those between TER and MRE11 delocalization. Thus, there could be more heat effects on MRE11 than just nuclear delocalization, which require HSPs for recovery. As described below, another effect of heat is the aggregation of the residual nuclear MRE11. The astute reader may have noticed that the nuclear delocalization of MRE11 required 2 hr at 41°C, but significant radiosensitization occurs after 1 hr at 41°C if the tumor cells are irradiated during the heating time, i.e., 45 min at 41°C X-irradiated at 41°C, followed by 15 min at 41°C. We, therefore, decided to determine if hyperthermia had effects on MRE11 in addition to nuclear delocalization. It is known that X or γ-irradiation induces foci containing MRE11 at sites of DNA double strand breaks as part of the repair process Citation[50]. In heated cells you can see MRE11 foci or aggregates, at the nuclear periphery after 2 hr at 41°C and are different morphologically from the foci formed after radiation () Citation[49]. At a non-radiosensitizing heat dose (1 hr at 41°C) a small amount of aggregation at the nuclear periphery can be observed Citation[49]. However, if cells are X-irradiated during the 1 hr of heating at 41°C, the foci are different from either heat or radiation alone Citation[49]. In this case the aggregates are large and distributed throughout the nucleus Citation[49]. The enhanced aggregation of MRE11 when heat and radiation are given simultaneously could explain how 1 hr at 41°C can effectively radiosensitize these cells for simultaneous heat and radiation when there is less nuclear delocalization of MRE11 than is required for radiosensitization when radiation is given after heating. In summary, after heat shock, MRE11 increases its association with HSP70, a fraction of it is delocalized from the nucleus, and much of the remaining nuclear MRE11 is aggregated. Correlation analysis suggests that for TER values up to 1.5–1.9 targets in addition to the MRN complex must be affected. The results described in this review are consistent with the idea that multiple molecular targets are affected by hyperthermia to achieve TER values on the order of 2.5 or more (see ).
Figure 13. The Associations Between MRE11/Rad50 and Hsp70 Relate to the TER Obtained from a Heat Treatment. The association between the indicated molecules was measured by immunoprecipitation and Western blotting.
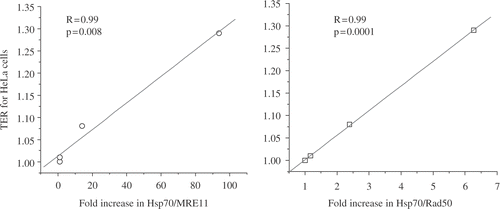
Figure 14. Hyperthermia appears to induce the formation of aggregates containing MRE11, associated with the nuclear matrix. Reprinted from Ref. 49.

Table I. TERs, targets, and temperature.
Strategies to enhance the thermal effectiveness of moderate hyperthermia
Given that there are molecular changes in DNA repair-related functions, it is attractive to consider hyperthermia as one of the best enhancers of radiotherapy. However, as we are all aware, the results of several clinical studies have not provided the impetus to make hyperthermia a major contributor to radiation therapy Citation[51]. This appears to be due to the difficulty of heating human tumors in a clinical setting. Thus, there is a need to develop methods to improve the delivery and/or efficacy of hyperthermia. While improvement due to the simultaneous delivery of heat and radiation in a clinical setting has been reported Citation[52] further improvement is needed. Pioneering studies by Gius and coworkers demonstrated that indomethacin is an effective enhancer for thermal radiosensitization Citation[53]. Further, at higher concentrations, indomethacin can act as an apparent mimic of the radiosensitizing effects of hyperthermia. However, at Indomethacin concentrations which either enhance or mimic the radiosensitizing effects of hyperthermia, there are unacceptable toxic side effects. Therefore, an effort to develop agents which can enhance the effective thermal dose of a given time–temperature combination is both feasible and would address a clear need for the enhancement of thermal radiotherapy.
Conclusion
Nuclear activity and nuclear metabolism, e.g. transcription, DNA replication, DNA repair require proper functioning of large dynamic protein complexes. These numerous complexes form, function and dissociate in a very small volume. Heat-induced protein unfolding and aggregation in these concentrated molecular conditions, in which critical functions (for the cell) are being conducted, will have catastrophic effects on cellular processes. Disruption of cellular processes such as DNA repair should significantly sensitize tumor cells to a significant variety of cellular stresses. Examples of such stresses include those resulting from radiotherapy or chemotherapy, and those associated with tumor microenvironments, such as hypoxic or oxidative stress. The foregoing review supports the points raised in the recent commentary by Coffey et al. Citation[54] that stresses the role of heat-induced changes in nuclear protein associations as an enhancer of several cancer therapies. Thus, the heat-induced changes in nuclear protein associations and functions represent a road map to molecular targets which can be impacted to enhance the efficacy of a number of cancer therapies.
Acknowledgements
It is a great honour for me to have this recognition from the Society for Thermal Medicine. For more than 25 years, our laboratory group has been presenting results on the effects of hyperthermia on the nuclear matrix and on the DNA functions that are dependent on the nuclear matrix. Thus, it is a welcome coincidence that Dr Donald S. Coffey chose to attend as his first Society for Thermal Medicine meeting, the very meeting where you decided that I should give this lecture. Dr Donald Coffey is one of the two people, whose work forms the basis for my own experiments. Thus, his comments on this lecture cannot be more welcomed. The other inspiration for my studies is William Dewey, whom most of you know, and with whom many of you here have trained. I should also acknowledge the excellent training I received from Dr S. Okada who was my PhD advisor and also the people who really taught me to do lab work: Dr Y. Doida and Dr S. Sawada. Dr P. Cerutti was my post-doc mentor and I learned a lot about radiation chemistry from him. Dr L. Dethlefsen was my first boss, Dr C. Perez was the second and the third would be Dr S. Powell. I thank them for their support of my career. It should be noted that Lyle Dethlefsen at the University of Utah has made an impact on the Society for Thermal Medicine by designing the symbol for the Society, which has lasted longer than the several names of the Society. Many graduate students, post-doctoral trainees, and support staff have contributed much to the work being presented and will be acknowledged in the cited work.
Related Research Data
References
- Roti Roti JL, Winward RT. The effects of hyperthermia on the protein to DNA ratio of isolated HeLa chromatin. Radiat Res 1978; 74: 159–169
- Tomasovic SP, Turner GN, Dewey WC. Effects of hyperthermia on non-histone proteins isolated with DNA. Radiat Res 1978; 73: 535–552
- Blair OC, Winward RT, Roti Roti JL. The effect of hyperthermia on the protein content of HeLa nuclei: A flow cytometric analysis. Radiat Res 1979; 78: 474–484
- Kampinga HH, Jorritsma JB, Konings AW. Heat-induced alterations in DNA polymerase activity of HeLa cells and of isolated nuclei. Relation to cell survival. Int J Radiat Biol Relat Stud Phys Chem Med 1985; 47: 29–40
- Chu GL, Ross G, Wong WSL, Warters R, Dewey WC. Content of nonhistone protein in nuclei after hyperthermic treatment. J Cell Phys 1993; 154: 217–221
- Borrelli MJ, Lepock JR, Frey HE, Lee YJ, Corry PM. Excess protein in nuclei isolated from heat-shocked cells results from a reduced extractability of nuclear proteins. J Cell Physiol 1996; 167: 369–379
- Roti Roti JL, Higashikubo R, Mace M. Protein cross-migration during isolation of nuclei from mixtures of heated and unheated HeLa cells. Radiat Res 1984; 98: 107–114
- Lepock JR. Role of nuclear protein denaturation and aggregation in the thermal radiosensitization. Int J Hyperthermia 2004; 20: 115–130
- Dewey WC, Li X, Wong RSL. Cell killing, chromosomal aberrations, and division delay as thermal sensitivity is modified during the cell cycle. Radiat Res 1990; 122: 268–274
- Pienta KJ, Getzenberg RH, Coffey DS. Cell structure and DNA organization. Crit. Rev Eukaryot Gene Expr 1991; 1: 355–385
- Berezney R, Coffey DS. Identification of a nuclear protein matrix. Biochem Biophys Res Commun 1974; 60: 1410–1417
- Berezney R, Mortillaro MJ, Ma H, Wei X, Samarabandu J. The nuclear matrix: A structural milieu for nuclear genomic function. Nuclear matrix: Structural and functional organization, R Berezney, K Jeon. Academic Press, California 1995; 1–63
- Nickerson JA, Blencowe BJ, Penman S. The architectural organization of nuclear metabolism. Nuclear matrix: Structural and functional organization, R Berezney, K Jeon. Academic Press, California 1995; 67–124
- Jackson DA, Cook PR. The structural basis of nuclear function. Nuclear matrix: Structural and functional organization, R Berezney, K Jeon. Academic Press, California 1995; 125–160
- Agutter PS. Intracellular structure and nucleocytoplasmic transport. Nuclear matrix: Structural and functional organization, R Berezney, K Jeon. Academic Press, California 1995; 183–224
- Roti Roti JL, Laszlo A. The effects of hyperthermia on cellular macromolecules. Hyperthermia and Oncology, Thermal effects on cells and tissues, M Urano, E Douple. VSP, The Netherlands 1988; 1: 13–56
- Kampinga HH, Turkel-Uygur N, Roti Roti JL, Konings AWT. The relationship of increased nuclear protein content induced by hyperthermia to killing of HeLa S3 cells. Radiat Res 1989; 117: 511–522
- Roti Roti JL, Wright WD, Higashikubo R, Dethlefsen LA. DNase I sensitivity of nuclear DNA measured by flow cytometry. Cytometry 1985; 6: 101–108
- Wright WD, Higashikubo R, Roti Roti JL. Flow cytometric studies of the nuclear matrix. Cytometry 1989; 10: 303–311
- Roti Roti JL, Henle KJ. Comparison of two mathematical models for describing heat-induced cell killing. Radiat Res 1980; 81: 374–383
- Dewey WC, Westra A, Miller HH, Nagasawa H. Heat-induced lethality and chromosomal damage in synchronized Chinese hamster cells treated with 5-bromodeoxyuridine. Int J Radiat Biol 1971; 20: 505–520
- vanderWaal RP, Higashikubo R, Xu M, Roti Roti JL. Cytometric methods to analyze thermal effects. Methods in Cell Biology 2001; 64: 269–286
- vanderWaal RP, Wright WD, Roti Roti JL. The effects of heat-shock on nuclear-matrix-associated DNA-replication complexes. Crit Rev Eukaryotic Gene Expression 1999; 9: 363–371
- Wong RSL, Kapp LN, Dewey WC. DNA for displacement rate measurements in heated Chinese hamster ovary cells. Biochim Biophys Acta 1989; 1007: 224–227
- Berezney R. Visualizing DNA replication sites in the cell nucleus. Sem Cell Biol 1991; 2: 103–115
- Wei X, Samarabandu J, Devdhar RS, Siegel AJ, Acharya R, Berezney R. Segregation of transcription and replication sites into higher order domains. Science 1998; 281: 1502–1506
- Wong RSL, Kapp LN, Krishnaswamy G, Dewey WC. Critical steps for induction of chromosomal aberrations in CHO cells heated in S phase. Radiat Res 1003; 133: 52–59
- Wong RSL, Thompson LL, Dewey WC. Recovery from effects of heat on DNA synthesis in Chinese hamster ovary cells. Radiat Res 1988; 114: 125–137
- Carney JP, Maser RS, Olivares H, Davis EM, Le Beau M, Yates JR, Hays L, Morgan WF, Petrini JH. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: Linkage of double-strand break repair to the cellular DNA damage response. Cell 1998; 93: 477–486
- De la Torre C, Pincheira J, Lopez-Saez JF. Human syndromes with genomic instability and multiprotein machines that repair DNA double-strand breaks. Histol Histopathol 2003; 18: 225–243
- Thompson LH, Schild D. Recombinational DNA repair and human disease. Mutat Res 2002; 509: 49–78
- Warters RL, Roti Roti JL. Production and excision of 5’,6’-dihydroxydihydrothymine type products in the DNA of preheated cells. Int J Radiat Biol 1978; 34: 381–384
- Warters RL, Roti Roti JL. Excision of X-ray induced thymine damage in chromatin from heated cells. Radiat Res 1979; 79: 113–121
- Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, Utsumi H, Yamaguchi-Iwai M, Shinohara A, Takeda S. Homologous recombination and non-homologous en-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J 1998; 17: 5497–5508
- Kampinga HH, Dynlacht JR, Dikomey E. Mechanism of radiosensitization by hyperthermia (> or = 43) as derived from studies with DNA repair defective mutant cell lines. Int J Hyperthermia 2004; 20: 131–139
- Kampinga HH, Wright WD, Konings AWT, Roti Roti JL. The interaction of heat and radiation affecting the ability of nuclear DNA to undergo supercoiling changes. Radiat Res 1988; 116: 114–123
- Roti Roti JL, Painter RB. Effects of hyperthermia on the sedimentation of nucleoids from HeLa cells in sucrose gradients. Radiat Res 1981; 89: 166–175
- Roti Roti JL, Wright WD. Visualization of DNA loops in nucleoids from HeLa cells: Assays for DNA damage and repair. Cytometry 1987; 8: 461–467
- Vogelstein B, Pardoll DM, Coffey BS. Supercoiled loops and eukaryotic DNA replication. Cell 1980; 22: 79–85
- VanderWaal RP, Spitz DR, Griffith CL, Higashikubo R, Roti Roti JL. Evidence that protein disulfide isomerase (PDI) is involved in DNA-nuclear matrix anchoring. J Cell Biochem 2002; 85: 689–702
- Wynstra JH, Wright WD, Roti Roti JL. Repair of radiation-induced DNA damage in thermotolerant and non-thermotolerant HeLa cells. Radiat Res 1990; 124: 85–89
- Laszlo A, Davidson T, Harvey A, Sim J, Malyapa RS, Spitz DR, Roti Roti JL. Alterations in heat-induced radiosensitization accompanied by nuclear structure alterations in Chinese hamster cells. Int J Hyperthermia 2006; 22: 43–60
- Vanderwaal RP, Roti Roti JL. Heat induced “masking” of redox sensitive component(s) of the DNA-nuclear matrix anchoring complex. Int J Hyperthermia 2004; 20: 234–239
- Yu Y, Maggi LB, Brady SN, Apicelli A, Dai M-S, Lu H, Weber JD. Nucleophosmin is essential for ribosomal protein L5 nuclear export. Mol Cell Biol 2006; 26: 3798–3809
- Wang Y, Guan J, Wang H, Wang Y, Leeper D, Illiakis G. Regulation of DNA replication after heat shock by replication protein A-nucleolin interactions. J Biol Chem 2001; 267: 20579–20588
- Zhu WG, Seno J, Beck BD, Dynlacht JR. Translocation of Mre11 from the nucleus to the cytoplasm as a mechanism of radiosensitization by heat. Radiat Res 2001; 156: 92–102
- Xu M, Myerson RJ, Straube WL, Moros EG, LaGroye I, Wang LL, Lee JT, Roti Roti JL. Radiosensitization of heat resistant human tumor cells by one hour at 41.1. Int J Hyperthermia 2002; 18: 385–403
- Xu M, Myerson RJ, Hunt C, Kumar S, Moros EG, Straube WL, Roti Roti JL. Transfection of human tumor cells with Mre11 siRNA increases radiation sensitivity and reduces heat induced radiosensitization. Int J Hyperthermia 2004; 20: 157–162
- Dynlacht JR, Xu M, Pandita RK, Roti Roti JL. Effects of heat shock on the Mre11/Rad50/Nbs1 complex in irradiated or unirradiated cells. Int J Hyperthermia 2004; 20: 144–156
- Mirzoeva OK, Petrini JH. DNA damage-dependent nuclear dynamics of the Mre11 complexes. Mol Cell Biol 2001; 21: 281–288
- Roti Roti JL. Radiosensitization by Hyperthermia: Mechanisms and clinical implications. Int J Hyperthermia 2004; 20: 109–114
- Myerson RJ, Roti Roti JL, Moros EG, Straube WL, Xu M. Modelling heat-induced radiosensitization: Clinical implications. Int J Hyperthermia 2004; 20: 201–212
- Bradbury CM, Markovina S, Wei SJ, Rene LM, Zoberi I, Horikoshi N, Gius D. Indomethacin-induced radiosensitization and inhibition of ionizing radiation-induced NFκB activation in HeLa cells occur via a mechanism involving p38 MAP kinase. Cancer Res 2001; 61: 7689–7696
- Coffey DS, Getzenberg RH, DeWeese TL. Hyperthermic biology and cancer therapies: A hypothesis for the ‘Lance Armstrong effect’. JAMA 2006; 296: 445–448