Abstract
Immune protection from microbial invaders or malignant progression is dependent on the ability of lymphocytes to efficiently traffic across morphologically and biochemically distinct vascular sites throughout the body. Lymphocyte trafficking to target tissues is orchestrated by adhesion molecules and chemokines that stabilize dynamic interactions between circulating lymphocytes and endothelial cells lining blood vessels. While the molecular mechanisms that regulate the efficient migration of lymphocytes across specialized high endothelial venules (HEVs) in secondary lymphoid organs have been extensively characterized, there is a paucity of information available regarding the mechanisms that dictate the rate of lymphocyte entry into tumor tissues. This article summarizes recent evidence that inflammatory cues associated with fever-range thermal stress promote lymphocyte extravasation across HEVs of lymphoid organs through a highly regulated lymphocyte–endothelial–interleukin-6 (IL-6) biological axis. The potential for using thermally-based strategies to improve lymphocyte delivery to the tumor microenvironment during T cell-based immunotherapy will also be discussed.
Advantages and limitations of T-cell based cancer immunotherapy
Significant progress has been achieved in the development of T cell-based immunotherapeutic modalities for the treatment of cancer Citation[1–6]. These strategies are based on the notion that the adaptive immune system can be harnessed to mount a specific attack on tumor cell targets. Target-specific immune approaches are attractive as they avoid the serious side-effects of other conventional treatments such as chemotherapy and radiation that have relatively non-specific mechanisms of action. CD8 T cell subsets are potent effectors of adaptive immunity by virtue of their ability to initiate cell death following receptor-mediated engagement by antigens expressed on the surface of target cells. T cells mediate tumor destruction by the release of pore-forming lytic enzymes (granzyme, perforin) as well as initiation of apoptotic pathways following ligation of death-receptors on tumor cells (TRAIL receptor, Fas receptor) Citation[2], Citation[7]. T cell cytotoxicity requires direct contact with target cells, thereby limiting damage to bystander cells. An inherent feature of the immune response, unlike conventional cancer therapies, is that it can elicit durable long-term protection from recurring disease.
An additional advantage of T cell-based anti-tumor immunity is that this treatment strategy does not depend on knowledge of tumor location. T cells, through the expression of trafficking molecules, are equipped with the potential to ‘search out and destroy’ widely disseminated tumor cell targets. However, the prevailing paradigm is that the ratio of T cells to tumor targets within tissues must be high in order to cause tumor regression. This assumption is based on in vitro studies where effective killing requires relatively high effector-to-target ratios (e.g., ranging from 20 : 1 to 1 : 1) that are difficult to achieve during immunotherapy. Thus, CD8 T cell-based anti-tumor responses are likely to be most effective when the tumor burden is limited such as in micrometastases or in a setting of minimal residual disease after tumor debulking by surgery, chemotherapy, or radiation.
The generation of CD8 T cell-mediated anti-tumor immunity involves two integrated arms of the immune system. In the first arm, tumor-derived antigens are captured in peripheral organs by resident antigen-presenting cells (i.e., dendritic cells) and carried via the afferent lymphatics to tumor-draining lymph nodes () Citation[2–4], Citation[8], Citation[9]. The capture and delivery of tumor antigens to draining lymph nodes follows the same sequence of events as for microbial-derived antigens. Antigen-loaded dendritic cells localize in close proximity to specialized blood vessels, termed high endothelial venules (HEVs), which are the major portals of entry of blood-borne naïve and central memory CD8 T cells into lymphoid organs Citation[10], Citation[11]. Thus, newly arrived T cells have the opportunity to sample a vast array of antigens presented by dendritic cells upon their entry into lymph nodes.
Figure 1. Tumor immunity depends on the generation of tumor-reactive T cells in the lymph node compartment and subsequent trafficking to the tumor microenvironment. Antigens derived from microbial pathogens or tumor cells in peripheral tissues are carried by dendritic cells to draining lymph nodes via the afferent lymphatic network. Naïve T cells enter lymph nodes across high endothelial venules (HEVs) and proliferate in response to antigen presentation by dendritic cells. Following exit of effector T cells via the efferent lymphatics, these cells circulate throughout the body in search of cognate target cells. The inset illustrates how fever-range thermal stress improves immune surveillance by increasing naïve T cell trafficking across HEVs. Thermal stress modifies the intravascular landscape in HEVs by increasing the density of trafficking molecules (CCL21 and ICAM-1) that support lymphocyte extravasation. Vessels within tumor sites are hypothesized to be non-permissive to effector T lymphocyte trafficking. Thus, effector T cells are physically segregated from tumor cell targets, providing one potential tumor escape mechanism. The role of thermal stress in regulating endothelial adhesion in tumor vessels remains to be determined.
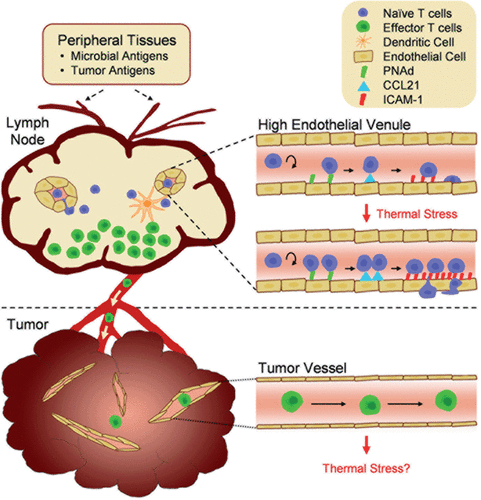
HEVs lined by cuboidal endothelial cells are distinguished from ubiquitous flat-walled vascular beds found throughout the body by their ability to support efficient extravasation of lymphocytes into the underlying parenchymal tissue Citation[10], Citation[12], Citation[13]. HEVs are found exclusively in lymphoid organs (i.e., lymph nodes and Peyer patches) located in close proximity to the skin or the respiratory, gastrointestinal or genitourinary tracts. These organs are strategically positioned to provide the first line of defense against invading pathogens. Lymph nodes are also a major site of immune detection of endogenous antigens derived from transformed tumor cells. It is estimated in mice that lymphocytes continuously cross the total HEVs within a single lymph node at a rate of ∼1.25 × 105 cells per hour Citation[14]. As a result, the lymphocyte content of an individual murine lymph node comprised of ∼106 cells turns over ∼3 times a day. This high throughput screening process is critical for maintaining immune homeostasis and ensures that rare antigen-specific T cells present at a frequency of only 1 in 105 − 106 T cells Citation[15], Citation[16] will encounter dendritic cells carrying cognate antigens from the periphery.
Antigen recognition leads to expansion and differentiation of CD8 T cells with potent lytic potential within the interior of lymph nodes () Citation[2], Citation[4], Citation[8]. Tumors can interfere with the development of tumor-reactive CD8 T cells by multiple mechanisms including preventing dendritic cell maturation, promoting the development of regulatory T cells (Treg), and limiting the rate of entry of naïve and central memory CD8 T cells across HEVs in tumor-draining lymph nodes Citation[3], Citation[8] Citation[17–20]. These limitations can be overcome clinically by hyper-vaccination with purified tumor antigens or by adoptive T cell transfer regimens in which large numbers of patient-derived tumor-reactive T cells are infused back into cancer patients Citation[1–6]. Despite these advances, clinical trials of T cell-based immunotherapy have been disappointing, thus far, with objective response rates achieved in only a low proportion of patients Citation[1–4], Citation[21].
The second arm of the anti-tumor immune response depends on the delivery of blood-borne CD8 T cells to peripheral sites of primary and metastatic tumor lesions. ‘Armed to kill’ CD8 T cells exit the lymph nodes via the efferent lymphatics and ultimately return to the bloodstream where they patrol the body in search of antigen depots within tissues (). CD8 T cell entry at all tissue sites is dependent on a highly ordered sequence of lymphocyte-endothelial adhesion events mediated by matching pairs of ‘Velcro-like’ receptors on T cells and venular walls as well as by interactions between chemokine receptors on T cells with chemoattractant molecules displayed on vascular endothelial cells Citation[10], Citation[11], Citation[22], Citation[23]. While the success of T cell-based cancer immunotherapy clearly depends on the ability of blood-borne T cells to gain access to malignant tissues in order to spearhead tumor-target destruction, remarkably little is known about the molecular mechanisms that control this dynamic process.
A landmark paper by Galon et al. has reinvigorated interest in this topic Citation[24]. This retrospective study in colorectal cancer patients demonstrated that the extent of infiltration in tumor lesions by CD3 T cells as well as by CD8 effector T cell subsets is a better prognostic indicator of patient outcome than conventional TNM (tumor, node, metastasis) staging. Thus, stage III patients with high tumoral infiltration by CD3 and CD8 T cells have a greater probability of disease-free survival and overall survival than stage I patients with low CD3 or CD8 T cell infiltration. Similar observations have been reported in melanoma, colon, lung, and ovarian cancer Citation[23], Citation[25], Citation[26]. Moreover, CD8 T cells have been identified in close proximity to tumor cells undergoing apoptosis in melanoma patients manifesting spontaneous tumor regression Citation[27], Citation[28]. Taken together, these observations suggest that T cell infiltration is causally linked to tumor control and improved patient outcome. Consequently, the mechanisms controlling T cell trafficking to the tumor microenvironment are likely to be a significant determinant of patient responses during T cell-based immunotherapy.
A frequently overlooked consideration is that the extent of CD8 T cell infiltration depicted in a histologic ‘snapshot’ reflects a balance between the rate of entry across a vascular barrier and subsequent events that affect the fate of T cells in situ including survival, proliferation, retention, and egress from tumor tissues Citation[23], Citation[26], Citation[29]. Despite the obvious importance of the initial entry step, surprisingly little is known about the molecular events controlling adhesive interactions between CD8 T cells and the vascular endothelium lining tumor vessel walls. Studies correlating the extent of vascular expression of specific adhesion molecules or chemokines with intratumoral accumulation of T cells over a period of days, while informative, do not provide precise information about entry mechanisms, per se, since many of these same molecules also regulate T cell survival, proliferation, and retention within tissues.
Limited leukocyte interactions with tumor vessels under steady-state conditions
Tumor microvessels are differentiated from vessels of normal organs with respect to their structure, organization, and ability to support leukocyte interactions Citation[30], Citation[31]. Normal vessels are typified by a hierarchical organization in which blood flow is unidirectional through arterioles, capillaries, venules, and subsequently collecting veins. Leukocyte trafficking occurs preferentially across postcapillary venules which respond to acute inflammatory cues because of expression of histamine receptors that trigger vasodilation and increased vascular permeability as well as expressions of adhesion molecules that support leukocyte-endothelial interactions Citation[30], Citation[32–37]. Tumor vessels lack the sequential hierarchy of normal vessels such that arterioles, capillaries, and venules typically cannot be discriminated within tumor tissues Citation[30], Citation[37], Citation[38].
The postcapillary venules of secondary lymphoid organs (lymph nodes, Peyer patches) have additional refinements with respect to structure and function which allow them to support efficient homeostatic trafficking of naïve and central memory lymphocytes. Within the venular tree, successive increases in diameter and branch points provide landmarks for the discrimination of high-order level V through level III venules (with level V being most proximal to capillaries) which comprise the specialized HEVs Citation[14]. Immediately downstream are flat-walled, low-order level II–I venules which are not supportive of lymphocyte interactions under non-inflammatory conditions. Postcapillary HEVs (high-order level V–III venules) of lymphoid organs constitutively express a high density of adhesion molecules and chemokines that support the multistep adhesion cascade required for lymphocyte extravasation that includes: (1) primary tethering and rolling of lymphocytes along vessel walls, (2) activation of chemokine receptors on lymphocytes by chemokines presented on the glycocalyx of vascular endothelium, (3) firm arrest of lymphocytes on vessels, and (4) migration of lymphocytes across the vascular endothelial barrier () Citation[10], Citation[11], Citation[22], Citation[39]. The efficiency of these interactions has been demonstrated by intravital microscopy. For example, in high-order level V venules of inguinal lymph nodes, ∼50% of blood-borne lymphocytes undergo tethering and rolling, and ∼40% of these cells transition to firm arrest Citation[40]. This is in sharp contrast to low-order level I or level II venules where ≤10% of lymphocytes initiate tethering and rolling while ≤5% of these cells undergo firm arrest Citation[40]. Lymphocyte-endothelial interactions in HEVs occur over seconds (tethering/rolling and chemokine activation) and minutes (firm arrest), while it takes approximately 20–30 minutes for lymphocytes to fully complete the extravasation process resulting in entry into the underlying parenchyma Citation[13], Citation[40].
This four-step adhesion cascade is required for trafficking of leukocytes throughout the body although the precise adhesion molecules and chemokines can differ under various inflammatory conditions and at different anatomical sites Citation[36]. Postcapillary venules within non-lymphoid organs normally support only low levels of trafficking of leukocytes under homeostatic conditions but are rapidly transformed to active recruitment sites in response to acute inflammatory signals provided by local cytokines (e.g., tumor necrosis factor (TNF), interleukin-1β (IL-1β), IL-6, interferon-γ (IFN-γ), and lymphotoxin) released by resident mast cells and macrophages in response to infectious agents or inflammatory stimuli Citation[26], Citation[36], Citation[37]. Lymphocyte trafficking can also occur across modified ‘HEV-like’ capillaries and arterioles during chronic inflammation Citation[37], Citation[41].
The highly disordered organization of flat-walled tumor vessels is exemplified by irregular diameters, aberrant branching patterns, abnormal blood flow rates, and anastomotic structures Citation[30], Citation[31], Citation[35], Citation[37], Citation[38], all of which could influence leukocyte trafficking. Moreover, intratumoral vessels in murine tumor models (e.g., RIP-Tag5 pancreatic tumors, EMT6 mammary tumors, CT26 colon tumors, B16 melanoma) poorly express primary adhesion molecules (e.g., E-selectin), chemokines (e.g., CXCL9, CXCL10) or prototypical arrest molecules such as intercellular adhesion molecule-1 (ICAM-1) or vascular adhesion molecule-1 (VCAM-1) Citation[23], Citation[25], Citation[26], Citation[42–48]. These observations in murine tumors correspond with histologic studies showing limited expression of adhesion molecules and chemokines in the intratumoral region of human tumors Citation[23], Citation[25], Citation[26], Citation[42], Citation[45], Citation[49–51].
Intravital microscopy has been used to evaluate the interaction of circulating leukocytes with tumor vessels Citation[26], Citation[31], Citation[52–55]. These studies are primarily based on the analysis of leukocytes labeled in situ with fluorescent tracking dyes (e.g., rhodamine-6-G). Thus, neutrophils are likely to be the major leukocyte population under observation since they comprise the predominant circulating leukocyte pool. These studies provide insight into the intrinsic adhesive properties of tumor microvasculature, although intravital microscopy has yet to be applied for the direct analysis of interactions between CD8 effector T cells and tumor microvessels. In contrast to the efficient interactions of lymphocyte observed in HEVs of lymphoid organs or inflamed vessels in normal organs Citation[10], Citation[11], Citation[14], Citation[25], Citation[35], Citation[36], Citation[40], Citation[56], leukocytes interact poorly with tumor vessels, at a level comparable to or less than what is detected in normal vessels under homeostatic conditions Citation[52–55], Citation[57], Citation[58]. Angiogenic factors present at high concentrations in the tumor microenvironment such as basic fibroblast growth factor (bFGF) and vascular endothelial growth factor (VEGF) C and D have been implicated in repressing adhesion in tumor vessels by in vitro and intravital studies Citation[54], Citation[58–61].
Short-term (1 hour) homing studies further suggest that CD8 T cell entry across tumor vessels is limited. This time point is relevant to the 20–30 minute time frame required for lymphocyte entry into tissues across vascular endothelial barriers. In this regard, tracking of CD8 T cells during the first hour after adoptive transfer into mice bearing MCA-205 tumor nodules in the lung showed that CD8 T cells interacted with capillaries but not venules in normal lung tissue and interacting cells were rarely found in the intratumoral region Citation[62]. Moreover, in murine studies examining the fate of tumor-specific T cells within the first 24 hours after adoptive transfer, it has been shown that CD8 T cells fail to accumulate in large numbers in tumor tissues while these cells are readily detected in other organs such as the spleen Citation[29], Citation[48], Citation[62–64].
While direct experimentation is required to elucidate CD8 T cell extravasation mechanisms in tumor vessels, the overall picture to emerge is that the lymphocyte-endothelial interface in tumor tissues is an important determinant controlling access of CD8 effector T cells to tumor targets. Restrictions imposed by the endothelial barrier provide a plausible explanation for observations in melanoma patients that tumor progression occurs despite high levels of tumor-reactive T cells in the peripheral blood after repeated vaccination or adoptive T cell therapy Citation[3], Citation[21], Citation[65–67]. Tumor growth is similarly unimpeded in a B16 melanoma mouse model even though >90% of the CD8 T cell population expresses a T cell receptor transgene capable of recognizing the gp100 antigen (pmel) on melanoma cells Citation[64], Citation[68].
Identification of the lymphocyte-HEV axis as a thermally sensitive alert system that heightens immune surveillance
There is an accumulating body of evidence indicating that fever-range thermal stress actively promotes egress of blood-borne lymphocytes across HEVs of secondary lymphoid organs where the probability of encountering cognate antigens or pathogens is enhanced () Citation[13], Citation[23], Citation[26], Citation[42], Citation[69]. The notion that thermal stress improves immune surveillance by augmenting lymphocyte trafficking has primarily been studied within the context of the relevance to physiological fever. These findings have provided insight into the beneficial mechanism of action of the thermal component of fever that has generally been relegated to a bystander function during infection and inflammation. Studies of the dynamics of lymphocyte trafficking across HEVs also have broader relevance to cancer immunotherapy since lymph nodes are the major site of priming of CD8 T cells by tumor antigens as well as a common destination of early metastatic spread. Moreover, there is strong speculation that optimal antitumor immunity depends on the ability of a subset of tumor-reactive T cells to gain access to secondary lymphoid organs during adoptive T cell transfer immunotherapy Citation[1], Citation[70–72].
The molecular mechanisms underlying thermal control of lymphocyte trafficking are highly integrated, involving all four steps of the adhesion cascade. In addition, these mechanisms represent independent, but complementary responses in both lymphocytes and high endothelial cells that line HEVs. Temperatures that mimic febrile episodes (38–40°C) were shown to act directly on T and B lymphocytes to enhance the avidity and/or affinity of two lymphocyte homing molecules, L-selectin and α4β7 integrin Citation[69], Citation[73–78]. These molecules are obligatory for the initial tethering and rolling of lymphocytes along the luminal surface of HEVs in lymph nodes () or Peyer patches, a requisite first step in the adhesion cascade that ultimately directs lymphocytes into the underlying parenchymal tissue. Fever-range thermal stress augments the binding activity of L-selectin and α4β7 integrin without altering the surface density of these molecules on lymphocytes Citation[74–76], Citation[78]. The effects of thermal stress are highly specific such that no change in the binding activity of leukocyte function-associated molecule-1 (LFA-1) for its endothelial ligand, ICAM-1, has been detected Citation[74].
Studies focusing on L-selectin further determined that thermal stress does not alter the intrinsic lectin activity of the N-terminal extracellular domain of L-selectin Citation[74]. Nor does it alter the topographical distribution of L-selectin on microvillous processes where it must reside in order to initiate transient tethering and rolling interactions Citation[74]. Instead, thermal stress causes L-selectin to become stably associated with the detergent-insoluble cytoskeletal matrix prior to engagement by its cognate HEV ligand, peripheral lymph node addressin (PNAd) Citation[75], Citation[78]. The C-terminal 11 amino acids of the cytoplasmic tail of L-selectin are necessary for the thermal response Citation[75]. Notably, this region contains a binding site for the cytoskeletal linker protein α-actinin Citation[79]. On the basis of these studies, it has been proposed that stable associations with the cytoskeletal scaffold induced by fever-range thermal stress enhance L-selectin tensile strength and the ability to withstand hemodynamic shear within HEVs Citation[75], Citation[78].
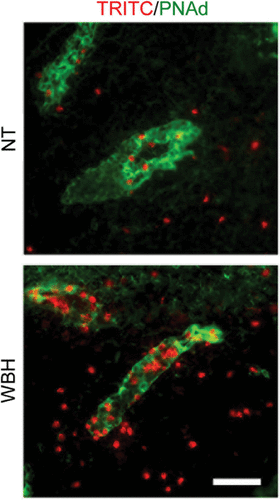
Fever-range thermal stress has also been found to improve immune surveillance by augmenting the binding function of HEVs Citation[13], Citation[26], Citation[40], Citation[77]. These studies employed the administration of fever-range whole body hyperthermia (WBH) in order to elevate core body temperatures to the range of physiological fever (39–40°C for 3–6 hours) Citation[76], Citation[80–84]. Systemic thermal therapy using fever-range WBH causes a transient decrease in the number of circulating lymphocytes in mice as well as in advanced cancer patients Citation[77], Citation[81], Citation[83]. In mice it was shown that the loss of lymphocytes from the peripheral blood pool is accompanied by increased trafficking of lymphocytes across HEVs of lymphoid organs Citation[13], Citation[23], Citation[40], Citation[77]. This process is illustrated in where increased accumulation of lymphocytes labeled with a fluorescent tracking dye (TRITC, red) is detected within the walls of HEVs (demarked by staining for the HEV-specific adhesion molecule, PNAd; green) as well as in the parenchyma of lymph nodes following WBH-treatment of recipient mice when compared with normothermic (NT) controls Citation[13], Citation[23], Citation[40]. An important aspect of the experimental design in these studies is that the evaluation of the adhesive properties of HEVs is performed after the cessation of heat treatment Citation[13], Citation[40]. This allows for the segregation of the endothelial response to thermal stress from the effects on lymphocyte adhesion (as detailed above) or hemodynamic parameters such as vasodilation, vascular permeability, and blood flow that could influence trafficking.
Figure 2. Fever-range thermal stress increases lymphocyte recruitment across HEVs. Photomicrographs show typical images of TRITC-labeled lymphocytes (red) associated with PNAd-positive HEVs (green) or infiltrated into the stroma of PLN tissue. NT, normothermic control; WBH, whole body hyperthermia. Bar = 50 µm.
Fever-range thermal therapy was further found to improve homeostatic trafficking of lymphocytes across HEVs Citation[40]. In this regard, thermal stress increases the absolute number of blood-borne lymphocytes that migrate across HEVs without preferentially affecting the composition of cells that traffic to peripheral lymph nodes. Thus, naïve and central memory T lymphocytes and B lymphocytes represent the major population of cells trafficking under both normothermic and hyperthermic conditions while neutrophils, monocytes, and activated effector/memory T cells are essentially excluded from crossing HEVs. This is a key difference from the response in inflamed nodes driven by the administration of potent inflammatory cytokines (e.g., TNF) or antigenic challenge which ‘open the HEV gateways’ for cells normally excluded from trafficking at this vascular site including NK cells, monocytes, and effector CD8 T cells Citation[11], Citation[85–91]. Based on these findings, it is tempting to speculate that heightened immune surveillance by fever in draining lymph nodes as well as in distal secondary lymphoid organs would provide a substantial advantage to the host by protecting against widespread dissemination of rapidly multiplying infectious agents.
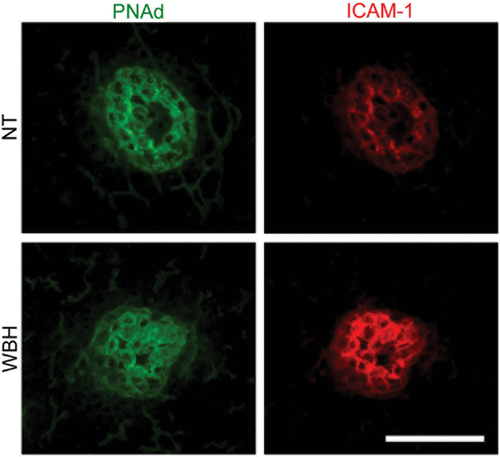
Several approaches have been taken to pinpoint the adhesion molecules or chemokines that mediate the thermal response in HEVs. Intravital microscopy revealed that fever-range temperatures do not affect the ability of HEVs to support primary tethering and rolling interactions of lymphocytes Citation[40]. Instead, thermal stress causes a profound increase in the transition of lymphocytes from rolling interactions to firm arrest in high-order level III–V postcapillary venules. The remarkable site-specificity of the response is evidenced by findings that adhesion is not altered in low-order level I–II venules, i.e., the vascular segments immediately proximal to the HEVs. Consistent with these observations, thermal stress does not change the expression of trafficking molecules that mediate primary adhesion (tethering and rolling) such as PNAd in lymph node HEVs () or mucosal addressin cell adhesion molecule-1 (MAdCAM-1) in Peyer patch HEVs Citation[13], Citation[26], Citation[40], Citation[63], Citation[77]. In contrast, fever-range thermal stress increases the intravascular display of two gatekeeper homing molecules, ICAM-1 () and the CCL21 chemokine, exclusively in HEVs Citation[40], Citation[63], Citation[69]. Moreover, enhanced endothelial expression of ICAM-1 and CCL21 is causally linked to improved lymphocyte trafficking across HEVs. While elevated ICAM-1 expression is generally considered a signature of inflammation, the findings for CCL21 are particularly unexpected since this chemokine was previously considered to be a homeostatic chemokine that is not susceptible to inducible expression on blood vessel walls.
Figure 3. Fever-range thermal stress increases ICAM-1 display on HEVs of peripheral lymph nodes. Intravascular staining of ICAM-1 (red) and PNAd counterstaining (green). Bar = 50 µm.
The site-specific nature of the fever-range thermal response in vascular endothelium would be predicted to be a benefit during physiologic fever since it would maintain the focus of the immune response in lymphoid organs, where lymphocytes have the greatest opportunity to encounter antigens. The failure of flat-walled non-HEVs to augment adhesion in response to fever-range thermal stress in vivo Citation[40] is paralleled by results of in vitro studies which examined the effects of fever-range hyperthermia (39–40°C for 6 hours) on endothelial expression of adhesion molecules and the ability to support leukocyte adhesion Citation[92], Citation[93]. Fever-range thermal stress has no effect on baseline endothelial expression of adhesion molecules (ICAM-1, E-selectin, VCAM-1) by endothelial cells in vitro, nor does it improve the ability of cultured endothelial cells to support adhesion of lymphocytes or neutrophils Citation[92], Citation[93]. Febrile temperatures further do not affect TNF-induced upregulation of ICAM-1, E-selectin, or VCAM-1 Citation[93]. Flat-walled endothelial cells are not entirely refractory to fever-range thermal stress however, since heat-treated endothelial cells release proadhesive factors in vitro that can act in trans to stimulate L-selectin or α4β7 integrin adhesion in lymphocytes Citation[78], Citation[93].
Contradictory results have been observed in endothelial cells in vitro in response to high temperature (≥41°C), short-duration (>60 minutes) heat shock. Differences among the various studies may reflect the types of endothelial cells and the temperature conditions under investigation. Several studies have reported a moderate induction of ICAM-1 in endothelial cells in response to high temperature heat shock alone Citation[93], Citation[94]. Other studies have reported that heat shock does not influence induction of adhesion molecules (ICAM-1, VCAM-1) in cultured endothelial cells either in the absence or presence of inflammatory stimuli such as TNF Citation[93], Citation[95–97]. Finally, several studies have shown that high temperature heat shock actually inhibits cytokine-induced upregulation of ICAM-1, E-selectin, and VCAM-1 by preventing NFκB activation Citation[98], Citation[99]. While heat shock temperatures are not achieved in vivo during physiologic febrile responses, these temperatures could be relevant during local-regional clinical hyperthermia regimens.
Conserved role of IL-6 trans-signaling in mediating thermal control of lymphocyte-endothelial adhesion
An unexpected unifying aspect of the fever-range thermal response is that the same proinflammatory cytokine, IL-6, is required for increased adhesion in both lymphocytes and HEVs. Thus, IL-6 is obligatory for thermally-induced increases in L-selectin binding activity in lymphocytes as well as enhanced intravascular display of ICAM-1 in HEVs Citation[40], Citation[69], Citation[73], Citation[78]. Other proinflammatory cytokines that are more often recognized for the regulation of lymphocyte-endothelial adhesion such as TNF, IL-1β, IFN-γ are not necessary for the response to fever-range thermal stress Citation[40], Citation[78]. Thermal regulation of lymphocyte-HEV adhesion further involves a tightly regulated IL-6 trans-signaling mechanism whereby the membrane-anchored gp130 signal transduction molecule is engaged by a complex comprised of IL-6 and a soluble form of the IL-6 receptor α (sIL-6Rα) binding subunit () Citation[40], Citation[73], Citation[78]. The dual requirement for both IL-6 and sIL-6Rα provides an additional level of control which is typical of potent proinflammatory cytokines, where multiple checks and balances prevent adverse bystander effects during inflammation Citation[69], Citation[100], Citation[101]. The majority of cell types throughout the body are refractory to IL-6 despite ubiquitous expression of membrane-associated gp130 because of their limited expression of membrane IL-6Rα. These cells are rapidly converted to IL-6–responsive cells during acute inflammation as a result of IL-6 trans-signaling due to an increase in local availability of sIL-6Rα Citation[100], Citation[101].
Figure 4. Fever-range thermal stress regulates lymphocyte and HEV adhesion through a common IL-6 trans-signaling mechanism. IL-6 trans-signaling is mediated by ligation of the membrane-anchored gp130 signal transducing subunit by a heterodimeric complex comprised of IL-6 and a soluble form of the IL-6 receptor α (sIL-6Rα) binding subunit. Downstream activation of ERK1/2 mediates thermal enhancement of L-selectin binding activity in lymphocytes. The signal transduction pathways underlying thermal induction of ICAM-1 density in HEVs are currently unknown.
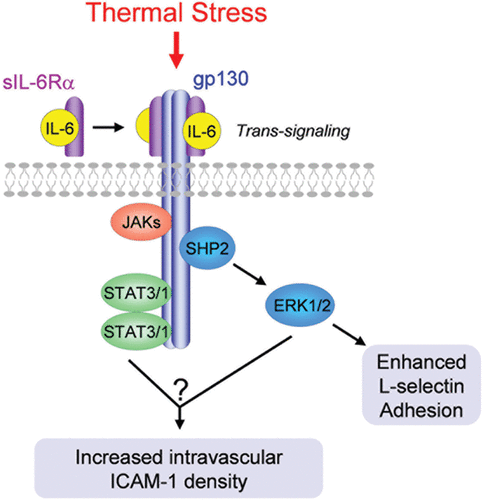
Perhaps one of the most intriguing findings relates to evidence for the evolutionary conservation of the IL-6 trans-signaling mechanism governing lymphocyte adhesion during thermal stress. Elevated temperatures in the range of physiological fever stimulate L-selectin adhesion via an endogenous IL-6 trans-signaling mechanism in leukocytes of species representing four major taxa of jawed vertebrates (mammals, avians, amphibians, teleosts) Citation[39], Citation[73–78]. The evolutionary conservation of a cytokine-dependent, thermally-regulated adhesion response in species that evolved from a node of common ancestry ∼450 million years ago suggests that this process is beneficial since conserved functions point to preservation of organismal integrity. MEK1-ERK1/2, but not p38 MAPK or JNK, has been identified in the IL-6/sIL-6Rα/gp130 signaling pathway upstream of activation of L-selectin adhesion by fever-range thermal stress () Citation[78]. The canonical IL-6 signaling molecule, STAT3, is also activated by a gp130-dependent mechanism during lymphocyte stimulation by fever-range temperature Citation[78] although it remains to be determined if a STAT3 pathway acts cooperatively with MEK1-ERK1/2 to promote L-selectin binding activity. The downstream signal transduction pathway(s) responsible for mediating ICAM-1 induction in HEVs during thermal stress is under investigation.
Several lines of evidence suggest that fever-range thermal regulation of lymphocyte trafficking involves local rather than systemic action of IL-6. In the case of lymphocytes, the IL-6 trans-signaling response necessary for thermal enhancement of L-selectin adhesion in vivo is fully recapitulated during heat treatment of lymphocytes in vitro Citation[69], Citation[73], Citation[78] In this regard, IL-6 and sIL-6Rα present in the conditioned medium of heat-treated leukocytes are capable of acting in trans to stimulate L-selectin adhesion in non-heat treated lymphocytes Citation[78]. Thus, an intact hormonal-neuronal-cytokine axis is not required to induce thermal responses in lymphocytes. Further support for the notion that IL-6/sIL-6Rα acts locally is provided by evidence that thermal stimulation of ICAM-1–dependent firm arrest of lymphocytes is restricted to high-order level III–V venules whereas adhesion in endothelial cells lining low-order venules (levels I–II), immediately adjacent to HEVs, is unaltered Citation[40]. Conversely, systemic administration of a recombinant fusion protein comprised of IL-6 linked to sIL-6Rα indiscriminately induces ICAM-1 on both HEVs and non-HEVs via a trans-signaling mechanism Citation[40].
Perspectives
Insight into the potential protective mechanism of action of fever is provided by emerging evidence that a thermally sensitive lymphocyte–HEV–IL-6 axis positively regulates lymphocyte trafficking (). These observations may also have clinical relevance during fever-range thermal therapy in cancer, since lymphocyte trafficking into tumor-draining lymph nodes would be predicted to be improved. Enhanced migration of naïve CD8 T cells into tumor-draining nodes could result in the generation of greater numbers of tumor-reactive CD8 T cells that are licensed to kill tumor cell targets in peripheral organs. Systemic fever-range thermal therapy is well tolerated in cancer patients Citation[81], Citation[84], suggesting that this modality could be used in an adjuvant setting together with T cell-based immunotherapies.
It remains to be determined if the site-restricted specificity of the vascular thermal response reflects intrinsic properties of specialized high endothelial cells that line HEVs or extrinsic influences of the surrounding cytokine-rich microenvironment within lymphoid organs. A related question is whether tumor microvessels will respond to clinical thermal therapy in a manner analogous to HEVs, or whether they will be non-responsive like non-HEVs. There are early indications that the tumor microenvironment could be converted to a site that actively recruits immune cells based on findings that systemic thermal therapy increases the intratumoral accumulation of leukocytes including natural killer cells, granulocytes, and lymphocytes Citation[25], Citation[80], Citation[102]. The tumor microenvironment is known to be rich in inflammatory cytokines and chemokines such as TNF, IL-1β, IL-6 and CCL2 Citation[103–106]. Moreover, studies in murine tumor models indicate that tumor cells are capable of releasing sIL-6Rα into the local tissue environment Citation[101], Citation[107–109]. The inflammatory milieu associated with these factors drives tumor progression by promoting angiogenesis, tumor cell proliferation and survival. Thus, it is feasible that thermal therapy can target the tumor vasculature by exploiting this pre-existing inflammatory environment, thereby opening the gateways for trafficking of cytolytic effector lymphocytes ().
Acknowledgements
We thank E.A. Repasky, H. Baumann, and J. Subjeck for valuable discussions during the development of this work and M.M. Appenheimer, J.B. Muhitch, W.C. Wang, and T.D. Vardam for critical review of the manuscript. Supported by the US National Institutes of Health (CA79765 and CA094045) and the Department of Defense (W81XWH-04-1-0354).
References
- Gattinoni L, Powell Jr DJ, Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: Building on success. Nat Rev Immunol 2006; 6: 383–393
- Dudley ME, Rosenberg SA. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat Rev Cancer 2003; 3: 666–675
- Boon T, Coulie PG, Eynde BJ, Bruggen PV. Human T cell responses against melanoma. Annu Rev Immunol 2006; 24: 175–208
- Pardoll DM. Spinning molecular immunology into successful immunotherapy. Nat Rev Immunol 2002; 2: 227–238
- Ho WY, Blattman JN, Dossett ML, Yee C, Greenberg PD. Adoptive immunotherapy: Engineering T cell responses as biologic weapons for tumor mass destruction. Cancer Cell 2003; 3: 431–437
- June CH. Adoptive T cell therapy for cancer in the clinic. J Clin Invest 2007; 117: 1466–1476
- Harty JT, Tvinnereim AR, White DW. CD8+ T cell effector mechanisms in resistance to infection. Annu Rev Immunol 2000; 18: 275–308
- Gajewski TF, Meng Y, Blank C, Brown I, Kacha A, Kline J, Harlin H. Immune resistance orchestrated by the tumor microenvironment. Immunol Rev 2006; 213: 131–145
- Cochran AJ, Huang RR, Lee J, Itakura E, Leong SP, Essner R. Tumour-induced immune modulation of sentinel lymph nodes. Nat Rev Immunol 2006; 6: 659–670
- Butcher EC, Picker LJ. Lymphocyte homing and homeostasis. Science 1996; 272(5258)60–66
- von Andrian UH, Mempel TR. Homing and cellular traffic in lymph nodes. Nat Rev Immunol 2003; 3: 867–878
- Engelhardt B, Wolburg H. Mini-review: Transendothelial migration of leukocytes: Through the front door or around the side of the house?. Eur J Immunol 2004; 34: 2955–2963
- Chen Q, Clancy KA, Wang WC, Evans SS. Inflammatory cues controlling lymphocyte-endothelial interactions during fever-range thermal stress. Endothelial Biomedicine, W Aird. Cambridge University Press, Cambridge 2007
- von Andrian UH. Intravital microscopy of the peripheral lymph node microcirculation in mice. Microcirculation 1996; 3: 287–300
- Pabst R, Binns RM. Heterogeneity of lymphocyte homing physiology: Several mechanisms operate in the control of migration to lymphoid and non-lymphoid organs in vivo. Immunol Rev 1989; 108: 83–109
- Blattman JN, Antia R, Sourdive DJ, Wang X, Kaech SM, Murali-Krishna K, Altman JD, Ahmed R. Estimating the precursor frequency of naive antigen-specific CD8 T cells. J Exp Med 2002; 195: 657–664
- Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer 2005; 5: 263–274
- Gabrilovich D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol 2004; 4: 941–952
- Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest 2007; 117: 1155–1166
- Carriere V, Colisson R, Jiguet-Jiglaire C, Bellard E, Bouche G, Al Saati T, Amalric F, Girard JP, M'Rini C. Cancer cells regulate lymphocyte recruitment and leukocyte-endothelium interactions in the tumor-draining lymph node. Cancer Res 2005; 65: 11639–1148
- Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: Moving beyond current vaccines. Nat Med 2004; 10(9)909–915
- Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: The multistep paradigm. Cell 1994; 76: 301–314
- Chen Q, Fisher DT, Kucinska SA, Wang WC, Evans SS. Dynamic control of lymphocyte trafficking by fever-range thermal stress. Cancer Immunol Immunother 2006; 55: 299–311
- Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006; 313: 1960–1964
- Chen Q, Wang WC, Evans SS. Tumor microvasculature as a barrier to antitumor immunity. Cancer Immunol Immunother 2003; 52: 670–679
- Fisher DT, Chen Q, Appenheimer MM, Skitzki J, Wang WC, Odunsi K, Evans SS. Hurdles to lymphocyte trafficking in the tumor microenvironment: Implications for effective immunotherapy. Immunol Invest 2006; 35: 251–277
- Yamshchikov GV, Mullins DW, Chang CC, Ogino T, Thompson L, Presley J, Galavotti H, Aquila W, Deacon D, Ross W, et al. Sequential immune escape and shifting of T cell responses in a long-term survivor of melanoma. J Immunol 2005; 174: 6863–6871
- Yamshchikov G, Thompson L, Ross WG, Galavotti H, Aquila W, Deacon D, Caldwell J, Patterson JW, Hunt DF, Slingluff CL, Jr. Analysis of a natural immune response against tumor antigens in a melanoma survivor: Lessons applicable to clinical trial evaluations. Clin Cancer Res 2001; 7(Suppl 3)S909–S916
- Skitzki J, Muhitch J, Evans SS. Tracking the elusive lymphocyte: Methods of detection during adoptive immunotherapy. Immunol Invest in press
- McDonald DM, Choyke PL. Imaging of angiogenesis: From microscope to clinic. Nat Med 2003; 9: 713–725
- Jain RK, Munn LL, Fukumura D. Dissecting tumour pathophysiology using intravital microscopy. Nat Rev Cancer 2002; 2: 266–276
- Springer TA. Traffic signals on endothelium for lymphocyte recirculation and leukocyte emigration. Annu Rev Physiol 1995; 57: 827–872
- Kubes P, Kanwar S. Histamine induces leukocyte rolling in post-capillary venules. A P-selectin-mediated event. J Immunol 1994; 152: 3570–3577
- McDonald DM, Thurston G, Baluk P. Endothelial gaps as sites for plasma leakage in inflammation. Microcirculation 1999; 6: 7–22
- Ley K. Integration of inflammatory signals by rolling neutrophils. Immunol Rev 2002; 186: 8–18
- Luster AD, Alon R, von Andrian UH. Immune cell migration in inflammation: Present and future therapeutic targets. Nat Immunol 2005; 6: 1182–1190
- McDonald DM. Imaging of angiogenesis in inflammation and cancer: Lessons for novel treatment of allergic rhinitis. Clin Exp Allergy Rev 2007; 7: 11–18
- McDonald DM, Foss AJ. Endothelial cells of tumor vessels: Abnormal but not absent. Cancer Metastasis Rev 2000; 19: 109–120
- Appenheimer MM, Chen Q, Girard RA, Wang WC, Evans SS. Impact of fever-range thermal stress on lymphocyte-endothelial adhesion and lymphocyte trafficking. Immunol Invest 2005; 34: 295–323
- Chen Q, Fisher DT, Clancy KA, Gauguet JM, Wang WC, Unger E, Rose-John S, von Andrian UH, Baumann H, Evans SS. Fever-range thermal stress promotes lymphocyte trafficking across high endothelial venules via an interleukin 6 trans-signaling mechanism. Nat Immunol 2006; 7: 1299–1308
- Hansson GK. Immune mechanisms in atherosclerosis. Arterioscler Thromb Vasc Biol 2001; 21: 1876–1890
- Chen Q, Evans SS. Thermal regulation of lymphocyte trafficking: Hot spots of the immune response. Int J Hyperthermia 2005; 21(8)723–729
- Ganss R, Ryschich E, Klar E, Arnold B, Hammerling GJ. Combination of T-Cell therapy and trigger of inflammation induces remodeling of the vasculature and tumor eradication. Cancer Res 2002; 62: 1462–1470
- Onrust SV, Hartl PM, Rosen SD, Hanahan D. Modulation of L-selectin ligand expression during an immune response accompanying tumorigenesis in transgenic mice. J Clin Invest 1996; 97: 54–64
- Carlos TM. Leukocyte recruitment at sites of tumor: Dissonant orchestration. J Leukoc Biol 2001; 70: 171–184
- Ganss R, Arnold B, Hammerling GJ. Mini-review: Overcoming tumor-intrinsic resistance to immune effector function. Eur J Immunol 2004; 34: 2635–2641
- Gollnick SO, Evans SS, Baumann H, Owczarczak B, Maier P, Vaughan L, Wang WC, Unger E, Henderson BW. Role of cytokines in photodynamic therapy-induced local and systemic inflammation. Br J Cancer 2003; 88: 1772–1779
- Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol 2005; 174: 7516–7523
- Weishaupt C, Munoz KN, Buzney E, Kupper TS, Fuhlbrigge RC. T-cell distribution and adhesion receptor expression in metastatic melanoma. Clin Cancer Res 2007; 13: 2549–2556
- Piali L, Fichtel A, Terpe HJ, Imhof BA, Gisler RH. Endothelial vascular cell adhesion molecule 1 expression is suppressed by melanoma and carcinoma. J Exp Med 1995; 181: 811–816
- Kunkel EJ, Butcher EC. Chemokines and the tissue-specific migration of lymphocytes. Immunity 2002; 16: 1–4
- Wu NZ, Klitzman B, Dodge R, Dewhirst MW. Diminished leukocyte-endothelium interaction in tumor microvessels. Cancer Res 1992; 52: 4265–4268
- Ryschich E, Schmidt J, Hammerling GJ, Klar E, Ganss R. Transformation of the microvascular system during multistage tumorigenesis. Int J Cancer 2002; 97: 719–725
- Hellebrekers DM, Castermans K, Vire E, Dings RP, Hoebers NT, Mayo KH, Oude Egbrink MG, Molema G, Fuks F, van Engeland M, Griffioen AW. Epigenetic regulation of tumor endothelial cell anergy: Silencing of intercellular adhesion molecule-1 by histone modifications. Cancer Res 2006; 66: 10770–10777
- Bessa X, Elizalde JI, Mitjans F, Pinol V, Miquel R, Panes J, Piulats J, Pique JM, Castells A. Leukocyte recruitment in colon cancer: Role of cell adhesion molecules, nitric oxide, and transforming growth factor beta1. Gastroenterology 2002; 122: 1122–1132
- Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat Rev Immunol 2007; 7: 678–689
- Fukumura D, Salehi HA, Witwer B, Tuma RF, Melder RJ, Jain RK. Tumor necrosis factor alpha-induced leukocyte adhesion in normal and tumor vessels: Effect of tumor type, transplantation site, and host strain. Cancer Res 1995; 55: 4824–4829
- Melder RJ, Koenig GC, Witwer BP, Safabakhsh N, Munn LL, Jain RK. During angiogenesis, vascular endothelial growth factor and basic fibroblast growth factor regulate natural killer cell adhesion to tumor endothelium. Nat Med 1996; 2: 992–997
- Griffioen AW, Damen CA, Martinotti S, Blijham GH, Groenewegen G. Endothelial intercellular adhesion molecule-1 expression is suppressed in human malignancies: The role of angiogenic factors. Cancer Res 1996; 56: 1111–1117
- Dirkx AE, Oude Egbrink MG, Kuijpers MJ, van der Niet ST, Heijnen VV, Bouma-ter Steege JC, Wagstaff J, Griffioen AW. Tumor angiogenesis modulates leukocyte-vessel wall interactions in vivo by reducing endothelial adhesion molecule expression. Cancer Res 2003; 63: 2322–2329
- Bouma-ter Steege JC, Baeten CI, Thijssen VL, Satijn SA, Verhoeven IC, Hillen HF, Wagstaff J, Griffioen AW. Angiogenic profile of breast carcinoma determines leukocyte infiltration. Clin Cancer Res 2004; 10: 7171–7178
- Skitzki J, Craig RA, Okuyama R, Knibbs RN, McDonagh K, Chang AE, Stoolman LM. Donor cell cycling, trafficking, and accumulation during adoptive immunotherapy for murine lung metastases. Cancer Res 2004; 64: 2183–2191
- Skitzki JJ, Chen Q, Wang WC, Evans SS. Primary immune surveillance: Some like it hot. J Mol Med 2007; 85: 1361–1367
- Palmer DC, Balasubramaniam S, Hanada K, Wrzesinski C, Yu Z, Farid S, Theoret MR, Hwang LN, Klebanoff CA, Gattinoni L, et al. Vaccine-stimulated, adoptively transferred CD8+ T cells traffic indiscriminately and ubiquitously while mediating specific tumor destruction. J Immunol 2004; 173: 7209–7216
- Rosenberg SA, Sherry RM, Morton KE, Scharfman WJ, Yang JC, Topalian SL, Royal RE, Kammula U, Restifo NP, Hughes MS, et al. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J Immunol 2005; 175: 6169–6176
- Nestle FO, Alijagic S, Gilliet M, Sun Y, Grabbe S, Dummer R, Burg G, Schadendorf D. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat Med 1998; 4: 328–332
- Banchereau J, Palucka AK, Dhodapkar M, Burkeholder S, Taquet N, Rolland A, Taquet S, Coquery S, Wittkowski KM, Bhardwaj N, et al. Immune and clinical responses in patients with metastatic melanoma to CD34(+) progenitor-derived dendritic cell vaccine. Cancer Res 2001; 61: 6451–6458
- Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med 2003; 198: 569–580
- Vardam TD, Zhou L, Appenheimer MM, Chen Q, Wang WC, Baumann H, Evans SS. Regulation of the lymphocyte–endothelial–IL-6 axis by fever-range thermal stress: Hot spot of immune surveillance cytokine. 2007; 39: 84–96
- Wherry EJ, Teichgraber V, Becker TC, Masopust D, Kaech SM, Antia R, von Andrian UH, Ahmed R. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol 2003; 4: 225–234
- Huang J, Khong HT, Dudley ME, El-Gamil M, Li YF, Rosenberg SA, Robbins PF. Survival, persistence, and progressive differentiation of adoptively transferred tumor-reactive T cells associated with tumor regression. J Immunother (1997) 2005; 28: 258–267
- Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, Finkelstein SE, Theoret MR, Rosenberg SA, Restifo NP. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest 2005; 115: 1616–1626
- Appenheimer MM, Girard RA, Chen Q, Wang WC, Bankert KC, Hardison J, Bain MD, Ridgley F, Sarcione EJ, S. B, Kaspers B, et al. Conservation of IL-6 trans-signaling mechanisms controlling L-selectin adhesion by fever-range thermal stress. Eur J Immunol 2007; 37: 2856–2867
- Wang WC, Goldman LM, Schleider DM, Appenheimer MM, Subjeck JR, Repasky EA, Evans SS. Fever-range hyperthermia enhances L-selectin-dependent adhesion of lymphocytes to vascular endothelium. J Immunol 1998; 160: 961–969
- Evans SS, Schleider DM, Bowman LA, Francis ML, Kansas GS, Black JD. Dynamic association of L-selectin with the lymphocyte cytoskeletal matrix. J Immunol 1999; 162: 3615–3624
- Evans SS, Bain MD, Wang WC. Fever-range hyperthermia stimulates alpha4beta7 integrin-dependent lymphocyte-endothelial adhesion. Int J Hyperthermia 2000; 16: 45–59
- Evans SS, Wang WC, Bain MD, Burd R, Ostberg JR, Repasky EA. Fever-range hyperthermia dynamically regulates lymphocyte delivery to high endothelial venules. Blood 2001; 97: 2727–2733
- Chen Q, Wang WC, Bruce R, Li H, Schleider DM, Mulbury MJ, Bain MD, Wallace PK, Baumann H, Evans SS. Central role of IL-6 receptor signal-transducing chain gp130 in activation of L-selectin adhesion by fever-range thermal stress. Immunity 2004; 20: 59–70
- Pavalko FM, Walker DM, Graham L, Goheen M, Doerschuk CM, Kansas GS. The cytoplasmic domain of L-selectin interacts with cytoskeletal proteins via alpha-actinin: Receptor positioning in microvilli does not require interaction with alpha-actinin. J Cell Biol 1995; 129: 1155–1164
- Burd R, Dziedzic TS, Xu Y, Caligiuri MA, Subjeck JR, Repasky EA. Tumor cell apoptosis, lymphocyte recruitment and tumor vascular changes are induced by low temperature, long duration (fever-like) whole body hyperthermia. J Cell Physiol 1998; 177: 137–147
- Kraybill WG, Olenki T, Evans SS, Ostberg JR, O'Leary KA, Gibbs JF, Repasky EA. A Phase I study of fever-range whole body hyperthermia (FR-WBH) in patients with advanced solid tumours: Correlation with mouse models. Int J Hyperthermia 2002; 18: 253–266
- Ostberg JR, Gellin C, Patel R, Repasky EA. Regulatory potential of fever-range whole body hyperthermia on Langerhans cells and lymphocytes in an antigen-dependent cellular immune response. J Immunol 2001; 167: 2666–2670
- Ostberg JR, Repasky EA. Comparison of the effects of two different whole body hyperthermia protocols on the distribution of murine leukocyte populations. Int J Hyperthermia 2000; 16: 29–43
- Pritchard MT, Ostberg JR, Evans SS, Burd R, Kraybill W, Bull JM, Repasky EA. Protocols for simulating the thermal component of fever: Preclinical and clinical experience. Methods 2004; 32: 54–62
- Tedla N, Wang HW, McNeil HP, Di Girolamo N, Hampartzoumian T, Wakefield D, Lloyd A. Regulation of T lymphocyte trafficking into lymph nodes during an immune response by the chemokines macrophage inflammatory protein (Mip)-1α alpha and Mip-1β beta. J Immunol 1998; 161: 5663–5672
- Mackay CR, Marston W, Dudler L. Altered patterns of T cell migration through lymph nodes and skin following antigen challenge. Eur J Immunol 1992; 22: 2205–2210
- Guarda G, Hons M, Soriano SF, Huang AY, Polley R, Martin-Fontecha A, Stein JV, Germain RN, Lanzavecchia A, Sallusto F. L-selectin-negative CCR7− effector and memory CD8(+) T cells enter reactive lymph nodes and kill dendritic cells. Nat Immunol 2007; 8: 743–752
- Sallusto F, Mackay CR. Chemoattractants and their receptors in homeostasis and inflammation. Curr Opin Immunol 2004; 16: 724–731
- Martin-Fontecha A, Thomsen LL, Brett S, Gerard C, Lipp M, Lanzavecchia A, Sallusto F. Induced recruitment of NK cells to lymph nodes provides IFN-y for TH 1 priming. Nat Immunol 2004; 5(12)1260–1265
- McEvoy LM, Jutila MA, Tsao PS, Cooke JP, Butcher EC. Anti-CD43 inhibits monocyte-endothelial adhesion in inflammation and atherogenesis. Blood 1997; 90: 3587–3594
- Janatpour MJ, Hudak S, Sathe M, Sedgwick JD, McEvoy LM. Tumor necrosis factor-dependent segmental control of MIG expression by high endothelial venules in inflamed lymph nodes regulates monocyte recruitment. J Exp Med 2001; 194: 1375–1384
- Hasday JD, Bannerman D, Sakarya S, Cross AS, Singh IS, Howard D, Drysdale BE, Goldblum SE. Exposure to febrile temperature modifies endothelial cell response to tumor necrosis factor-alpha. J Appl Physiol 2001; 90: 90–98
- Shah A, Unger E, Bain MD, Bruce R, Bodkin J, Ginnetti J, Wang WC, Seon B, Stewart CC, Evans SS. Cytokine and adhesion molecule expression in primary human endothelial cells stimulated with fever-range hyperthermia. Int J Hyperthermia 2002; 18: 534–551
- Lefor AT, Foster III CE, Sartor W, Engbrecht B, Fabian DF, Silverman D. Hyperthermia increases intercellular adhesion molecule-1 expression and lymphocyte adhesion to endothelial cells. Surgery 1994; 116: 214–220
- Gnant MF, Turner EM, Alexander Jr HR. Effects of hyperthermia and tumour necrosis factor on inflammatory cytokine secretion and procoagulant activity in endothelial cells. Cytokine 2000; 12: 339–347
- Nakayama J, Terao H, Koga T, Furue M. Induction of CD54 and CD58 Expression in cultured human endothelial cells by beta-interferon with or without hyperthermia in vitro. J Dermatol Sci 2001; 26: 19–24
- Ito A, Shinkai M, Honda H, Wakabayashi T, Yoshida J, Kobayashi T. Augmentation of MHC class I antigen presentation via heat shock protein expression by hyperthermia. Cancer Immunol Immunother 2001; 50: 515–522
- Nakabe N, Kokura S, Shimozawa M, Katada K, Sakamoto N, Ishikawa T, Handa O, Takagi T, Naito Y, Yoshida N, Yoshikawa T. Hyperthermia attenuates TNF-alpha-induced up regulation of endothelial cell adhesion molecules in human arterial endothelial cells. Int J Hyperthermia 2007; 23: 217–224
- Frossard JL, Pastor CM, Hadengue A. Effect of hyperthermia on NF-κB binding activity in cerulein-induced acute pancreatitis. Am J Physiol Gastrointest Liver Physiol 2001; 280: G1157–1162
- Jones SA. Directing transition from innate to acquired immunity: Defining a role for IL-6. J Immunol 2005; 175: 3463–3468
- Rose-John S, Scheller J, Elson G, Jones SA. Interleukin-6 biology is coordinated by membrane-bound and soluble receptors: Role in inflammation and cancer. J Leukoc Biol 2006; 80: 227–236
- Ostberg JR, Ertel BR, Lanphere JA. An important role for granulocytes in the thermal regulation of colon tumor growth. Immunol Invest 2005; 34: 259–272
- Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest 2007; 117: 1175–1183
- Coussens LM, Werb Z. Inflammation and cancer. Nature 2002; 420: 860–867
- Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 2005; 7: 211–217
- Szlosarek P, Charles KA, Balkwill FR. Tumour necrosis factor-alpha as a tumour promoter. Eur J Cancer 2006; 42: 745–750
- Becker C, Fantini MC, Schramm C, Lehr HA, Wirtz S, Nikolaev A, Burg J, Strand S, Kiesslich R, Huber S, et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity 2004; 21: 491–501
- Mitsuyama K, Sata M, Rose-John S. Interleukin-6 trans-signaling in inflammatory bowel disease. Cytokine Growth Factor Rev 2006; 17: 451–461
- Atreya R, Neurath MF. Involvement of IL-6 in the pathogenesis of inflammatory bowel disease and colon cancer. Clin Rev Allergy Immunol 2005; 28: 187–196