Abstract
Purpose: Quercetin (QCT), an important flavonol, is known to sensitize tumour cells to hyperthermia by suppressing heat shock protein 72 (Hsp72) induction, and is also reported to inhibit p53 accumulation. This study was conducted to examine the effects of QCT on the heat sensitivities of human tumour cell lines with different p53 statuses.
Material and methods: Cell lines derived from human cancers and p53-inducible cells were used. After heat treatment at 43°C for 2 h with or without QCT, cell survival was determined in a clonogenic assay. The cellular and nuclear content of Hsp72 as well as that of p53 was determined by Western blotting analysis.
Results: Treatment of cells with 150 µM QCT, which completely abolished Hsp72 induction, potentiated the lethal effects of hyperthermia in all tumour cell lines. Particularly, remarkable enhancement of cell death was observed in tumour cell lines having little or no p53 proteins. Although nuclear translocation of Hsp72 is induced by hyperthermia, it was significantly compromised in p53-deficient cells.
Conclusions: These results indicate that p53 is a component for nuclear accumulation of Hsp72; therefore, p53 status is an important determinant of the sensitization of human tumour cells to hyperthermia by QCT.
Introduction
Cells or organisms respond to heat shock by inducing the synthesis of heat shock proteins (HSPs). Among HSP groups, the Hsp70 family is one of the most abundant and is highly conserved among organisms Citation[1], Citation[2]. In mammalian cells, two major members of the Hsp70 family are found: a constitutively expressed Hsp73 and a stress-inducible Hsp72 Citation[2], Citation[3]. Like other stress proteins, Hsp70 functions as a molecular chaperone, which binds to unfolded proteins under stressed and physiological conditions. This may protect proteins from misfolding, and it facilitates protein maturation by assisting their correct assembly Citation[2–4]. The activation of heat shock genes is transcriptionally regulated by heat shock transcription factor 1 (HSF1), which binds to the heat shock element (HSE) in the promoter region of the heat shock genes. In response to heat shock or other stresses, HSF1 is thought to be liberated from its association or interaction with Hsp72; the released HSF1 then assembles into a trimer, accumulates within the nucleus, binds to HSE, becomes phosphorylated, and activates the transcription of the heat shock genes Citation[5–8]. Hsp72 after heat shock migrates from the cytoplasm to the nucleus Citation[9], Citation[10], which has been shown to accelerate the recovery of heat-induced alterations of nuclei and nucleoli Citation[11]. Although the molecular mechanism of heat shock-induced nuclear accumulation of Hsp72 has not been fully understood, it has recently been reported that 20 amino acid residues of nuclear localization-related signals (NLRS) in the protein-binding domain are necessary for proper nuclear localization of HSC70 Citation[12], and this sequence also exists in Hsp72.
Heat shock, in addition to DNA-damaging agents including radiation, ultraviolet light (UV), and chemicals, induces p53 stabilization Citation[13–19]. The gene encoding p53 is a tumour suppressor gene, whose mutations are found in more than 50% of all human cancers Citation[20]. In response to cellular stresses, activated p53 protects the genome from destabilization by transactivating the downstream genes whose products regulate cell cycle arrest, apoptosis, and DNA repair Citation[21–23]. Stabilization of p53 appears to be primarily regulated post-transcriptionally Citation[24], and p53 stabilization has been shown to result from an inhibition of its degradation. Stabilized p53 is translocated into nuclei, and nuclear accumulation is essential for its function. Interestingly, interaction between p53 and heat shock proteins has been reported by several studies. For example, association of wild-type p53 (wtp53) and mutant p53 (mp53) with Hsp72/73 has been demonstrated not only under normal conditions Citation[25–27] and after UV and γ-ray irradiation Citation[28] but also after heat shock Citation[13], Citation[25], Citation[29]. Thus, p53 protein could be a component that is involved in the nuclear accumulation of Hsp72 after hyperthermia.
Quercetin (QCT; 3, 3′, 4′, 5, 7-pentahydroxyflavone) is an important member of the flavonols, which are the most ubiquitous flavonoids in dietary plants Citation[30]. QCT has a wide range of biological activities, including cancer inhibition. For this function, many mechanisms of QCT's anticancer action have been identified, such as estrogenic/anti-estrogenic activity, anti-proliferation, cell cycle arrest and apoptosis, and antioxidation (reviewed in Citation[31]). QCT is also known to be a hyperthermic sensitizer Citation[32–34] by suppressing heat shock response. In heat-shocked tumour cell lines, QCT inhibits the induction of Hsp72 Citation[35], Citation[36] by inhibiting the post-transcriptional activation of HSF1; this occurs by reducing HSF1 phosphorylation without affecting HSF1 trimer formation Citation[37], Citation[38]. QCT is further reported to inhibit p53 accumulation Citation[39].
Thus, QCT treatment simultaneously inhibits activation of HSF1 and p53 in response to heat shock. However, the effects of QCT on heat sensitivities of human cancer cell lines with different p53 statuses have not been examined. In this study, we investigated the effects of QCT on the levels of Hsp72 and p53 as well as on the heat sensitivities of human cancer cell lines showing different p53 statuses.
Materials and methods
Cells and cultures
We used cell lines derived from human cancers. Their origins and p53 statuses are summarized in .
Table I. List of human tumour cells used in this study.
HT1080, MCF-7, A431, and HeLa cells were obtained from the Japanese Cancer Research Resources Bank (JCRB), while the other cancer cells were purchased from American Type Culture Collection (ATCC). All cells were cultured in Eagle's MEM (Nissui Pharmaceutical, Tokyo) supplemented with 10% fetal bovine serum (Trace Bioscience PTY, Perth, Australia) at 37°C in a humidified atmosphere with 5% CO2.
Establishment of 99v9 cells and 99v9-derived p53-inducible (99v9–p53) cells has been reported elsewhere Citation[40].
QCT and heat treatment
QCT (Sigma-Aldrich, St Louis, MO) was dissolved in dimethylsulfoxide (DMSO; Sigma-Aldrich) and stored at a concentration of 100 mM at −20°C. When the stock solution was needed, it was thawed at room temperature and then added to culture medium to give the required concentration. The final concentration of DMSO in the culture medium was adjusted to 0.15% (v/v). Exponentially growing cells in the culture flasks were heated at 43°C for 2 h in a water-bath controlled to ± 0.1°C in the presence or absence of QCT.
Colony formation assay
Cell survival after the treatments was determined in a clonogenic assay, and was expressed in terms of the plating efficiency by inoculating an appropriate number of cells into 100 mm dishes. The cells were incubated at 37°C for 10–12 days, fixed with 99.5% ethanol, and stained with a 3% solution of Giemsa (Wako Pure Chemical Industries, Osaka). The colonies composed of more than 50 cells were counted as having grown from surviving cells. All data points were tested in triplicate dishes. The surviving fraction was calculated by dividing the plating efficiency of each treatment by the plating efficiency of the non-treated control.
Western blotting analysis
After the treatments, cells were trypsinized and lysed in RIPA buffer (50 mM Tris-HCl, pH 7.2, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) containing 1 mM 4-(2-amino-ethyl)-benzenesulfonyl fluoride hydrochloride. After freezing at −20°C, the cell lysates were thawed and centrifuged at 15 000 rpm for 10 min at 4°C. The supernatant was harvested and the protein content was measured by the bicinchoninic acid (BCA) method (Pierce Chemical, Rockford, IL).
For extraction of the nuclear protein, cells were lysed in Lysis buffer (10 mM HEPES, pH 8.0, 50 mM NaCl, 0.5 M sucrose, 0.1 mM EDTA, 0.5% Triton X-100, 1 mM dithiothreitol, 5 mM MgCl2). The lysate was centrifuged at 5, 000 rpm for 1 min. After the supernatant was removed, the resultant pellet was washed repeatedly with Lysis buffer, suspended in Lysis buffer containing 0.5 M NaCl and 5 mM spermidine, and incubated for 30 min at 4°C. The lysate was purified by centrifugation at 12,000 rpm for 30 min at 4°C, and the supernatant was used as nuclear protein. Protein concentration was determined with a protein assay kit (Bio-Rad, Tokyo).
Eight to 16 µg of total or nuclear proteins were subjected to SDS-polyacrylamide gel electrophoresis (PAGE) as described by Laemmli Citation[41]. The proteins were transferred electrophoretically to polyvinyl difluoride (PVDF) filters in transfer buffer (0.1 M Tris, 0.192 M glycine). The filters were blocked with 10% skim milk overnight and incubated with anti-Hsp72/73 monoclonal antibody (Ab-1, clone W27, Oncogene Research Products, San Diego, CA) for detection of Hsp72 or anti-p53 monoclonal antibody (clone BP53–12, NeoMarkers, Fremont, CA) to detect p53. For visualization of the bound primary antibodies, the filters were incubated with biotinylated secondary antibody and streptavidin/biotinylated alkaline phosphatase conjugate (Amersham Japan, Tokyo, code RPN 1234). Then the membranes were incubated with 5-bromo-4-chloro-3-indolylphosphate p-toluidine salt (BCIP) and nitroblue tetrazolium chloride (NBT) (Gibco BRL, Rockville, MD) for color development. The band intensities were measured densitometrically with a scanning imager.
Statistical analysis
Differences between groups were determined by ANOVA with post-hoc Scheffe analysis. Synergism was determined by two-way ANOVA. All statistical procedures were performed using the statistics program SPSS (version 11.0; SPSS, Inc., Chicago, IL). A probability level of p < 0.05 was considered to be statistically significant.
Results
Effects of 0.15% DMSO on cellular Hsp72 and p53 levels in human cancer cell lines
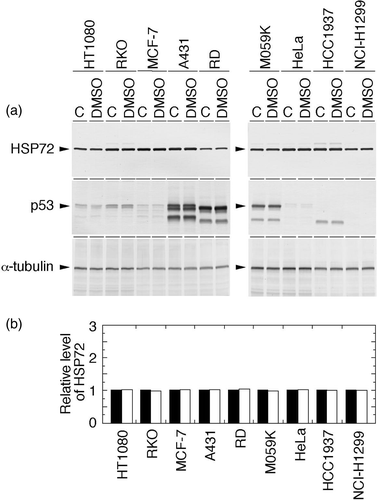
The effects of 0.15% DMSO, the vehicle for QCT, on cellular Hsp72 and p53 levels were examined in all tumour cell lines used in this study. As shown in , we confirmed that DMSO alone has no effect on Hsp72 and p53 levels under the concentration used in this study. In addition, 0.15% DMSO had little or no effect on cell survival in all cases.
Figure 1. Effects of 0.15% DMSO on the levels of Hsp72 and p53 in human cancer cell lines. Exponentially growing cells were treated with or without 0.15% DMSO for 2 h. Whole-cell proteins were extracted and analysed with Western blotting. (a) Hsp72 and p53 were visualized using anti-Hsp72/73 antibody and anti-p53 antibody, respectively. C, control (without any treatment). (b) The intensity of the band indicated in (a) was measured by densitometry and the relative level of Hsp72 protein was calculated. The protein level detected in the control cells was determined as 1.0. Filled column, control; open column, DMSO.
QCT's effects on cellular Hsp72 and p53 levels in heat-shocked human cancer cell lines
Human cancer cell lines with different p53 statuses were heat-shocked at 43°C for 2 h, and the effects of QCT (150 µM) on cellular Hsp72 and p53 levels were examined (). In a group of cell lines having wtp53, which are HT1080 cells, RKO cells, and MCF-7 cells, both Hsp72 and p53 were present constitutively in the non-heat-treated control. The Hsp72 contents in these cell lines increased significantly after heat shock and were suppressed to the control level by heat shock plus QCT treatment. The exception was RKO cells; heat shock alone did not increase the Hsp72 level, and heat shock plus QCT decreased the level only slightly. In the case of p53, heat shock significantly increased p53 in all of the cell lines in this group, and heat shock plus QCT treatment suppressed the accumulation of p53 almost to the control level in MCF-7 cells. The suppression was even stronger in HT1080 and RKO cells.
Figure 2. Effects of heat shock and heat shock plus QCT on the levels of Hsp72 and p53 in human cancer cell lines. Exponentially growing cells were heat-shocked at 43°C alone for 2 h or concomitantly with QCT (150 µM). Control cells were neither heat-shocked nor treated with QCT. Whole-cell proteins were extracted immediately after heat shock and analysed with Western blotting. (a) Hsp72 and p53 were visualized using anti-Hsp72/73 antibody and anti-p53 antibody, respectively. C, control (without any treatment); HS, heat shock; H/Q, heat shock plus QCT. (b) The intensity of the band indicated in (a) was measured by densitometry and the relative level of Hsp72 protein was calculated. The protein level detected in the control cells was determined as 1.0. Filled column, control; dotted column, heat shock; open column, heat shock plus QCT.
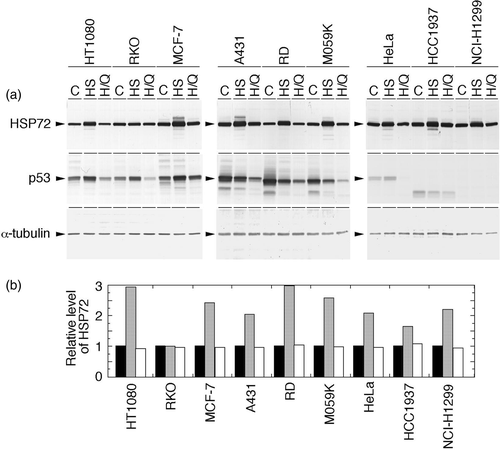
In a group of cell lines having mutant p53, including A431 cells, RD cells, and M059K cells, Hsp72 was present constitutively in the non-heat-treated control, increased significantly after heat shock, and was suppressed to the control level by heat shock plus QCT treatment. The content of constitutively expressed p53 in the non-heat-treated control of this group was higher than that of the group of cell lines with wtp53, but heat shock somewhat decreased p53, and heat shock plus QCT treatment further suppressed p53 in all of the cell lines of this group.
In the case of a group of cell lines having deleted p53 or inactivated wtp53, including HeLa, HCC1937, and NCI-H1299 cells, Hsp72 was present constitutively in the non-heat-treated control, then was significantly increased after heat shock and was suppressed to the control level by heat shock plus QCT treatment. In HeLa cells, p53 was marginally observed; heat shock increased the level slightly, and heat shock plus QCT treatment completely suppressed it. In the other two cell lines of this group, no p53 was observed in the non-heat-treated control, in heat-shocked cells, or in cells treated with heat shock plus QCT.
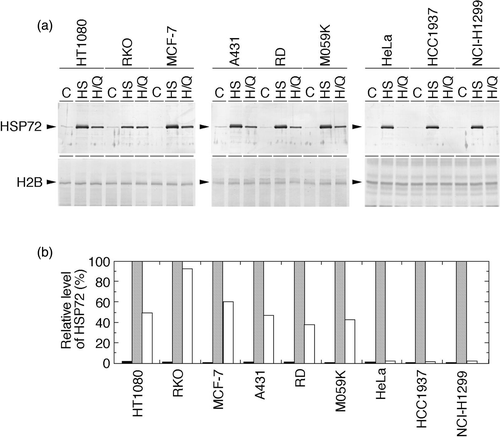
QCT's effects on nuclear Hsp72 levels in heat-shocked human cancer cell lines
Nuclear Hsp72 levels in heat-shocked human cancer cell lines with or without QCT treatment were examined (). In contrast to the total cellular levels, constitutively expressed nuclear Hsp72 was observed only marginally in all the cell lines used. Heat shock significantly increased it, but the effect of heat shock plus QCT treatment differed according to the p53 status. In the group of cell lines with deleted or inactivated p53, nuclear Hsp72 was suppressed dramatically to the control level. In the group of cell lines with p53 function, nuclear Hsp72 was suppressed, but suppressed level is lower than that in cells without p53 function.
Figure 3. Effects of heat shock and heat shock plus QCT on nuclear Hsp72 levels in human cancer cell lines. Exponentially growing cells were heat-shocked at 43°C alone for 2 h or concomitantly with QCT (150 µM). Control cells were neither heat-shocked nor treated with QCT. Nuclear proteins were extracted immediately after heat shock and analysed with Western blotting. (a) Hsp72 and p53 were visualized using anti-Hsp72/73 antibody and anti-p53 antibody, respectively. C, control (without any treatment); HS, heat shock; H/Q, heat shock plus QCT. (b) The intensity of the band indicated in (a) was measured by densitometry and the relative level of Hsp72 protein was calculated. The protein level detected in the heat-shocked cells was determined as 100%. Filled column, control; dotted column, heat shock; open column, heat shock plus QCT.
QCT's effects on the heat sensitivities of human cancer cell lines
To compare QCT's effects on the heat sensitivities of human cancer cell lines having different p53 statuses, cell survival after treatment with QCT (150 µM, 2 h) alone, heat shock (43°C, 2 h) alone, or heat shock plus QCT was determined with a colony formation assay. The results are summarized in . The effects of QCT are evaluated with enhancement ratios (ER).
Table II. Effects of quercetin treatment on lethal effects of heat shock.
QCT treatment alone differently influenced the plating efficiencies of these cancer cell lines regardless of p53 status. It had almost no effect on the survival of MCF-7 cells, slightly suppressed that of HeLa and HCC1937 cells, moderately suppressed that of HT1080, RKO, and RD cells, and suppressed that of the other cell lines to almost half of the respective control levels. Heat shock alone suppressed the survival of each cancer cell line more than did QCT treatment alone; the most affected lines were RKO, NCI-H1299, and RD cells, although these cell lines belong to different p53 status groups. Heat shock plus QCT treatment suppressed the survival of each of these lines far more than did heat shock alone; these effects were statistically synergistic (p < 0.01) in all the cell lines but HCC1937 cells and HeLa cells. Suppression was strongest in HeLa, HCC1937, and NCI-H1299 cells, all of which belong to the group of cell lines with deleted or inactivated p53.
We calculated ER in order to compare QCT's effects on heat sensitivity among the cell lines. ER was obtained as follows. First, survival after heat shock plus QCT treatment was divided by that after QCT treatment alone to delete the lethal effect of QCT treatment alone. Survival after heat shock alone divided by this result gave ER. As shown in , the group of cell lines with deleted or inactivated p53 revealed significantly higher ER (225.0–247.6) than the wtp53 group (2.4–52.3) or the mp53 group (1.6–30.1) (p > 0.01).
Cellular p53 expression and QCT's effects on nuclear Hsp72 levels in p53-inducible cells and their parent cells
99v9 cells are derived from NCI-H1299 cells having deleted p53. The established cell line 99v9–p53 harbors the Ecdysone-inducible expression vectors for human wild-type p53 gene. 99v9–p53 cells, as well as their parent 99v9 cells, expressed cellular p53 only marginally. Administration of ponasterone A (PA) to 99v9–p53 cells for 24 h increased the p53 level significantly, whereas there was no effect on it in 99v9 cells by the same treatment ().
Figure 4. Cellular p53 expression and effects of heat shock and heat shock plus QCT on nuclear Hsp72 levels in human p53-inducible cells. 99v9 cells having deleted p53 and their derivative 99v9–p53 cells having ponasterone A (PA)-inducible vectors for human wild-type p53 gene were used. (a) After treatment with or without PA for 24 h, whole-cell proteins were extracted and analysed with Western blotting. p53 was visualized using anti-p53 antibody. (b) After treatment with or without PA for 24 h, exponentially growing 99v9–p53 cells were heat-shocked at 43°C alone for 2 h or concomitantly with QCT (150 µM). Control cells were neither heat-shocked nor treated with QCT. Nuclear proteins were extracted immediately after heat shock and analysed with Western blotting. Hsp72 was visualized using anti-Hsp72/73 antibody. C, control (without any treatment); HS, heat shock; H/Q, heat shock plus QCT. (c) The intensity of the band indicated in (b) was measured by densitometry and the relative level of Hsp72 protein was calculated. The protein level detected in the heat-shocked cells was determined as 100%. Filled column, control; dotted column, heat shock; open column, heat shock plus QCT.
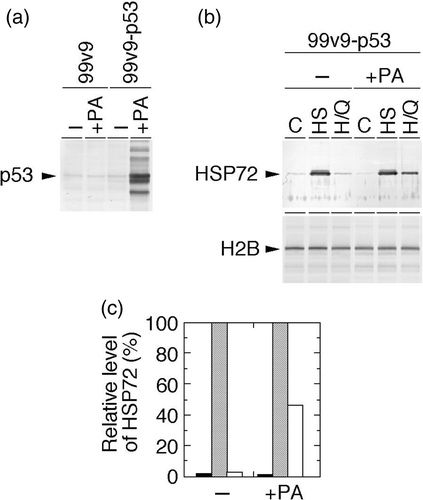
Nuclear Hsp72 was expressed marginally in the non-heat-treated control, and then was significantly increased after heat shock in 99v9–p53 cells irrespective of PA administration. After heat shock plus QCT treatment, it was suppressed dramatically to the control level in the absence of PA, while it was suppressed to a lesser extent in the presence of PA (c).
Effects of p53 induction on QCT enhancement of heat-sensitivity
In 99v9 cells and 99v9–p53 cells, regardless of PA administration, QCT treatment alone suppressed the survival of each of the cell lines to less than half of the respective control level, and heat shock alone suppressed it to almost one fifth of the results of QCT treatment alone. Although heat shock plus QCT treatment synergistically suppressed the survival of each of these lines far more than did heat shock alone (p < 0.01), survival of 99v9–p53 (+PA) cells was higher than that of the other cell lines, in which almost the same suppression was observed (). As a result of these observations, 99v9–p53 cells with PA administration showed lower ER (58.9) than did 99v9–p53 cells without PA administration (222.4) and other cell lines (181.3 and 184.8).
Table III. Effects of p53 induction on QCT enhancement of heat-sensitivity.
Discussion
Cellular Hsp72 was constitutively expressed in the cancer cell lines used in this study, and the amount of Hsp72 was augmented by heat shock in every cell line except RKO. Nuclear Hsp72 was also expressed constitutively, although its amount was marginal. Heat shock augmented the amount of nuclear Hsp72 in every cell line, a result that coincides with those of other reports Citation[9], Citation[10]. We demonstrated that at the cellular level QCT suppressed Hsp72 induction after heat shock, regardless of p53 status. It should be noted that, in every case, QCT suppressed the induction of cellular Hsp72 to the control level, indicating that QCT treatment is only effective until the transactivation mediated by HSF1. We further showed that at nuclear levels QCT completely suppressed the accumulation of Hsp72 after heat shock to the control level in cell lines having no p53 or wtp53 degraded by E6, whereas it did so to a lesser extent in cell lines having wtp53 or mp53. In RKO cells, heat shock did not increase the cellular level of Hsp72, but increased its nuclear level. Furthermore, QCT treatment with heat shock did not affect either the cellular or nuclear levels of Hsp72. Because QCT has been shown to inhibit phosphorylation of HSF1 after heat shock Citation[37], Citation[38], these results suggest that the pathway involved in HSF1 phosphorylation was absent. The absence of Hsp72 induction in RKO cells might contribute to the highest heat sensitivity and the lowest ER in the wtp53 group (). Further experiments will be needed to elucidate the defect in RKO cells.
The p53 response to heat shock differs depending on the p53 status. Augmented p53 levels were observed in cell lines having wtp53, including HeLa cells, whereas the level was rather decreased in cell lines having mp53. We found that QCT suppressed the accumulation of p53 after heat shock both in human tumour cells with wtp53 and in those with mp53. As shown in , QCT completely suppressed the accumulation of p53 to the control level or to a lesser extent in cells with wtp53. In contrast, the level of p53 was somewhat decreased upon heat shock, and QCT treatment additionally lowered the level (). Although the mechanism of QCT effects remains to be determined, a previous study showed that QCT (100 µM) stimulated phosphorylation of p53 Citation[42]. Therefore, it is possible that suppression of p53 level was not due to abrogation of p53 stabilization; rather, degradation of p53 may be promoted because protective Hsp72 accumulation in the nuclei was compromised in cells treated with heat shock plus QCT.
QCT has been shown not only to inhibit the proliferation of human cancer cells, but also to potentiate the lethal effect of hyperthermia on human tumour cell lines, including HeLa cells Citation[32], melanoma cells Citation[33], and prostatic carcinoma cells in vitro and in vivo Citation[34]. This study also indicated that QCT treatment concomitant with heat shock exceedingly sensitized all human cancer cell lines to heat shock. Interestingly, we demonstrated for the first time that the enhancing effects of QCT are strongest in cell lines having no p53 or wtp53 degraded by E6. Heat shock has been reported to induce nuclear accumulation of p53 Citation[13–15], which induces transactivation of downstream target genes. Nuclear accumulation of Hsp72 has also been shown to accelerate the recovery of heat-induced alterations of nuclei and nucleoli Citation[11]. As mentioned above, it is well known that QCT inhibits phosphorylation of HSF1 Citation[37], Citation[38], which disables Hsp72 induction. Because the Hsp72 protein acts as a survival factor by protecting apoptosis induction, the abrogation of Hsp72 induction should decrease cell survival. In addition to these observations, our present study demonstrated that QCT completely suppressed the heat-induced nuclear accumulation of Hsp72 only in cell lines having little or no p53, suggesting that p53 status-dependent suppression of nuclear accumulation of Hsp72 after heat shock by QCT may occur by an alternative mechanism. This could be through an association of wtp53 and mp53 with Hsp72/73, which was demonstrated after heat shock Citation[13], Citation[29]. A previous study examined the protein-protein interaction between Hsp72 and p53 in detail Citation[43], and the high-affinity binding sites were mapped to the hydrophobic core of the central DNA-binding domain of the p53 molecule in addition to the low-affinity binding sites mapped to both the N-terminal and C-terminal regions. Thus, oncogenic mutations, as well as changes to the wild-type molecule including heat denaturation, in the p53 protein, whose structural alteration exposes high-affinity binding sites, have higher probability to be recognized by the Hsp72 protein. Based on our findings and these observations, it can be proposed that Hsp72 might be concomitantly transported into the nuclei of heat-shocked cells in association with the translocation of p53 from the cytoplasm to the nuclei after heat shock. However, in cell lines having deleted p53 or inactivated wtp53, such translocation of p53 would be absent or insufficient, so the nuclear accumulation of Hsp72 is severely impaired and, consequently, the heat-induced lethal effects are strongly enhanced by QCT. To confirm this, we used p53-inducible cell lines and obtained the supportive results that restoration of p53 increased nuclear Hsp72 levels which had been completely suppressed by QCT after heat shock, and attenuated QCT enhancement of heat-sensitivity. In addition, this proposed mechanism is also supported by the recent observation that preincubation of heated cells with QCT suppressed Hsp72 translocation from the cytoplasm to the nucleus in HeLa cells Citation[44], which bear inactivated wtp53.
It is generally accepted that temperatures more than 42°C kill cancer cells efficiently. Especially, hyperthermia at 43°C has been extensively studied for its lethal effects on human cancer cells, and hyperthemic cell death has been shown to be exceedingly enhanced at temperatures above 43°C Citation[45]. However, because higher temperature induces thermoresistance and because hyperthermia equipment faced to the difficulties of achieving effective temperatures, mild hyperthermia whose temperature is between 40°C and 42°C has been emerging as an alternative way to treat cancer cells Citation[46]. On the other hand, recent clinical trials of local hyperthermia in combination with radiotherapy have shown that the higher thermal dose group achieved better outcome than the low dose group Citation[46], Citation[47]. Since QCT has been reported to inhibit the acquisition of thermotolerance by inhibiting Hsp72 induction in human tumour cell lines without increasing the thermosensitivity of non-tolerant cells Citation[48], it is expected from the results of this study using a heating temperature of 43°C that QCT makes it possible to use 43°C clinically for the better efficacy. Furthermore, a recent phase I clinical trial of patients with cancer resistant to standard therapies indicated that QCT administered intravenously can be used clinically without significant irreversible toxicity Citation[49]. These observations suggest that QCT can be applied clinically as a hyperthermic sensitizer. On such occasions, the p53 status of a clinical tumour tissue specimen would be a useful marker to predict the extent of QCT's enhancing effects in hyperthermia. This could also help to reduce the dose of QCT in order to avoid dose-limiting nephrotoxicity and uncomfortable side effects, such as local pain on injection, flushing, and emesis, all of which were reported to be encountered at higher dose levels Citation[46].
In conclusion, this study indicates that QCT treatment concomitant with heat shock suppresses the level of p53, as well as that of Hsp72. It also reveals that QCT remarkably enhances the lethal effects of heat shock, which is based on inhibition of heat-induced nuclear translocation of Hsp72 by QCT, and that the enhancing effects of QCT are strongest in cancer cell lines having deleted p53 or inactivated wtp53. This in turn suggests that p53 is a component for nuclear accumulation of Hsp72 and therefore, p53 status is an important determinant of sensitization of human tumour cells to hyperthermia by QCT. Further study using various tumour samples, including fresh tumour tissues, is needed to elucidate the mechanism of those effects and to validate our results.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Pelham HR. Speculations on the functions of the major heat shock and glucose-regulated proteins. Cell 1986; 46: 959–961
- Becker J, Craig EA. Heat-shock proteins as molecular chaperones. Eur J Biochem 1994; 219: 11–23
- Welch WJ. Mammalian stress response: Cell physiology, structure/function of stress proteins, and implications for medicine and disease. Physiol Rev 1992; 72: 1063–1081
- Morimoto RI. Heat shock: The role of transient inducible responses in cell damage, transformation, and differentiation. Cancer Cells 1991; 3: 295–301
- Morimoto RI. Cells in stress: Transcriptional activation of heat shock genes. Science 1993; 259: 1409–1410
- Li GC, Mivechi NF, Weitzel G. Heat shock proteins, thermotolerance, and their relevance to clinical hyperthermia. Int J Hyperthermia 1995; 11: 459–488
- Baler R, Dahl G, Voellmy R. Activation of human heat shock genes is accompanied by oligomerization, modification, and rapid translocation of heat shock transcription factor HSF1. Mol Cell Biol 1993; 13: 2486–2496
- Cotto JJ, Kline M, Morimoto RI. Activation of heat shock factor 1 DNA binding precedes stress-induced serine phosphorylation. J Biol Chem 1996; 271: 3355–3358
- Watanabe M, Suzuki K, Kodama S, Sugahara T. Normal human cells at confluence get heat resistance by efficient accumulation of Hsp72 in nucleus. Carcinogenesis 1995; 16: 2373–2380
- Welch WJ, Feramisco JR. Nuclear and nucleolar localization of the 72,000-dalton heat shock protein in heat-shocked mammalian cells. J Biol Chem 1984; 259: 4501–4513
- Pelham HR. Hsp70 accelerates the recovery of nucleolar morphology after heat shock. EMBO J 1984; 3: 3095–3100
- Tsukahara F, Maru Y. Identification of novel nuclear export and nuclear localization-related signals in human heat shock cognate protein 70. J Biol Chem 2004; 279: 8867–8872
- Matsumoto H, Shimura M, Omatsu T, Okaichi K, Majima H, Ohnishi T. p53 proteins accumulated by heat stress associate with heat shock proteins Hsp72/HSC73 in human glioblastoma cell lines. Cancer Lett 1994; 87: 39–46
- Ohnishi T, Wang X, Ohnishi K, Matsumoto H, Takahashi A. p53-dependent induction of WAF1 by heat treatment in human glioblastoma cells. J Biol Chem 1996; 271: 14510–14513
- Miyakoda M, Nakahata K, Suzuki K, Kodama S, Watanabe M. Heat-induced G1 arrest is dependent on p53 function but not on RB dephosphorylation. Biochem Biophys Res Commun 1999; 266: 377–381
- Miyakoda M, Suzuki K, Kodama S, Watanabe M. Activation of ATM and phosphorylation of p53 by heat shock. Oncogene 2002; 21: 1090–1096
- Wang C, Chen J. Phosphorylation and Hsp90 binding mediate heat shock stabilization of p53. J Biol Chem 2003; 278: 2066–2071
- Sugano T, Nitta M, Ohmori H, Yamaizumi M. Nuclear accumulation of p53 in normal human fibroblasts is induced by various cellular stresses which evoke the heat shock response, independently of the cell cycle. Jpn J Cancer Res 1995; 86: 415–418
- Nitta M, Okamura H, Aizawa S, Yamaizumi M. Heat shock induces transient p53-dependent cell cycle arrest at G1/S. Oncogene 1997; 15: 561–568
- Harris C, Hollstein M. Clinical implications of the p53 tumor-suppressor gene. N Engl J Med 1993; 329: 1318–1327
- Lavine AJ. p53, the cellular gatekeeper for growth and division. Cell 1997; 88: 323–331
- Hofseth LJ, Hussain SP, Harris CC. p53: 25 years after its discovery. Trends Pharmacol Sci 2004; 25: 177–181
- Agarwal ML, Taylor WR, Chernov MV, Chernova OB. Stark GR. The p53 network. J Biol Chem 1998; 273: 1–4
- Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 in the cellular response to DNA damage. Cancer Res 1991; 51: 6304–6311
- Ehrhart JC, Duthu A, Ullrich S, Appella E, May P. Specific interaction between a subset of the p53 protein family and heat shock proteins Hsp72/hsc73 in a human osteosarcoma cell line. Oncogene 1988; 3: 595–603
- Lehman TA, Bennett WP, Metcalf RA, Welsh JA, Ecker J, Modali RV, Ullrich S, Romano JW, Appella E, Testa JR, et al. p53 mutations, ras mutations, and p53-heat shock 70 protein complexes in human lung carcinoma cell lines. Cancer Res 1991; 51: 4090–4096
- Davidoff AM, Iglehart JD, Marks JR. Immune response to p53 is dependent upon p53/Hsp70 complexes in breast cancers. Proc Natl Acad Sci USA 1992; 89: 3439–3442
- Matsumoto H, Wang X, Ohnishi T. Binding between wild-type p53 and Hsp72 accumulated after UV and gamma-ray irradiation. Cancer Lett 1995; 92: 127–133
- Pinhasi-Kimhi O, Michalovitz D, Ben-Zeev A, Oren M. Specific interaction between the p53 cellular tumour antigen and major heat shock proteins. Nature 1986; 320: 182–184
- Manach C, Scalbert A, Morand C, Rémésy C, Jiménez L. Polyphenols: Food sources and bioavailability. Am J Clin Nutr 2004; 79: 727–747
- Birt DF, Hendrich S, Wang W. Dietary agents in cancer prevention: Flavonoids and isoflavonoids. Pharmacol Ther 2001; 90: 157–177
- Kim JH, Kim SH, Alfieri AA, Young CW. Quercetin, an inhibitor of lactate transport and a hyperthermic sensitizer of HeLa cells. Cancer Res 1984; 44: 102–106
- Piantelli M, Tatone D, Castrilli G, Savini F, Maggiano N, Larocca LM, Ranelletti FO, Natali PG. Quercetin and tamoxifen sensitize human melanoma cells to hyperthermia. Melanoma Res 2001; 11: 469–476
- Asea A, Ara G, Teicher BA, Stevenson MA, Calderwood SK. Effects of the flavonoid drug quercetin on the response of human prostate tumours to hyperthermia in vitro and in vivo. Int J Hyperthermia 2001; 17: 347–356
- Hosokawa N, Hirayoshi K, Nakai A, Hosokawa Y, Marui N, Yoshida M, Sakai T, Nishino H, Aoike A, Kawai K, et al. Flavonoids inhibit the expression of heat shock proteins. Cell Struct Funct 1990; 15: 393–401
- Wei YQ, Zhao X, Kariya Y, Fukata H, Teshigawara K. Uchida A. Induction of apoptosis by quercetin: involvement of heat shock protein. Cancer Res 1994; 54: 4952–4957
- Hosokawa N, Hirayoshi K, Kudo H, Takechi H, Aoike A, Kawai K, Nagata K. Inhibition of the activation of heat shock factor in vivo and in vitro by flavonoids. Mol Cell Biol 1992; 12: 3490–3498
- Nagai N, Nakai A, Nagata K. Quercetin suppresses heat shock response by down regulation of HSF1. Biochem Biophys Res Commun 1995; 208: 1099–1105
- Ghosh JC, Suzuki K, Kodama S, Watanabe M. Effects of protein kinase inhibitors on the accumulation kinetics of p53 protein in normal human embryo cells following X-irradiation. J Radiat Res (Tokyo) 1999; 40: 23–37
- Suzuki K, Mori I, Nakayama Y, Miyakoda M, Kodama S, Watanabe M. Radiation-induced senescence-like growth arrest requires TP53 function but not telomere shortening. Radiat Res 2001; 155: 248–253
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970; 227: 680–685
- Ye R, Goodarzi AA, Kurz EU, Saito S, Higashimoto Y, Lavin MF, Appella E, Anderson CW, Lees-Miller SP. The isoflavonoids genistein and quercetin activate different stress signaling pathways as shown by analysis of site-specific phosphorylation of ATM, p53 and histone H2AX. DNA Repair 2004; 3: 235–244
- Fourie AM, Hupp TR, Lane DP, Sang BC, Barbosa MS, Sambrook JF, Gething MJ. Hsp70 binding sites in the tumor suppressor protein p53. J Biol Chem 1997; 272: 19471–19479
- Jakubowicz-Gil J, Pawlikowska-Pawlega B, Piersiak T, Pawelec J, Gawron A. Quercetin suppresses heat shock-induced nuclear translocation of Hsp72. Folia Histochem Cytobiol 2005; 43: 123–128
- Hildebrandt B, Wust P, Ahlers O, Dieing A, Sreenivasa G, Kerner T, Felix R, Riess H. The cellular and molecular basis of hyperthermia. Crit Rev Oncol Hematol 2002; 43: 33–56
- Dewhirst MW, Vujaskovic Z, Jones E, Thrall D. Re-setting the biologic rationale for thermal therapy. Int J Hyperthermia 2005; 21: 779–790
- Jones EL, Oleson JR, Prosnitz LR, Samulski TV, Vujaskovic Z, Yu D, Sanders LL, Dewhirst MW. Randomized trial of hyperthermia and radiation for superficial tumors. J Clin Oncol 2005; 23: 3079–3085
- Koishi M, Hosokawa N, Sato M, Nakai A, Hirayoshi K, Hiraoka M, Abe M, Nagata K. Quercetin, an inhibitor of heat shock protein synthesis, inhibits the acquisition of thermotolerance in a human colon carcinoma cell line. Jpn J Cancer Res 1992; 83: 1216–1222
- Ferry DR, Smith A, Malkhandi J, Fyfe DW, deTakats PG, Anderson D, Baker J, Kerr DJ. Phase I clinical trial of the flavonoid quercetin: Pharmacokinetics and evidence for in vivo tyrosine kinase inhibition. Clin Cancer Res 1996; 2: 659–668