Abstract
Purpose. Recent progress suggests that short peptide motifs can be engineered into biopolymers with specific temperature dependent behavior. This review discusses peptide motifs capable of thermo-responsive behavior, and broadly summarizes design approaches that exploit these peptides as drug carriers. This review focuses on one class of thermally responsive peptide-based biopolymers, elastin-like polypeptides in greater detail.
Analysis. Four peptide motifs are presented based on leucine zippers, human collagen, human elastin, and silkworm silk that are potential building blocks for thermally responsive biopolymers. When these short motifs (<7 amino acids) are repeated many times, they generate biopolymers with higher order structure and complex temperature triggered behaviors. These structures are thermodynamically modulated, making them intrinsically temperature sensitive. These four motifs can be categorized by the directionality and reversibility of association. Elastin-like polypeptides (ELPs) are one promising motif that reversibly associates during heating. ELPs aggregate sharply above an inverse phase transition temperature, which depends on polymer hydrophobicity, molecular weight, and concentration. ELPs can be modified with chemotherapeutics, are biodegradable, are biocompatible, have low immunogenicity, and have terminal pharmacokinetic half-lives >8 h. ELP block copolymers can reversibly form micelles in response to hyperthermia, and this behavior can modulate the binding avidity of peptide ligands. When high molecular weight ELPs are systemically administered to mice they accumulate in tumors; furthermore, hyperthermia can initiate the ELP phase transition and double the concentration of peptide in the tumor.
Conclusions. Temperature sensitive peptides are a powerful engineering platform that will enable new strategies for hyperthermia-directed drug delivery.
Introduction
Ongoing research in clinical hyperthermia suggests that primary tumors can be either directly ablated by heat or indirectly sensitized by mild hyperthermia Citation[1–4]. Ablative therapies directly kill cells using high temperatures (>43°C) of sufficient duration Citation[3]. At the periphery of ablative thermal doses are tissues that experience mild hyperthermia (40° to 43°C), which is insufficient to cause tissue necrosis under short duration. In view of its synergies with these front-line therapies, the application of mild hyperthermia is under clinical development as an adjunct to chemotherapy and external beam irradiation Citation[4]. Regions of tissue exposed to mild hyperthermia may further benefit from combined treatment with therapeutics that can respond to modest increases in temperature Citation[5], Citation[6]. This review discusses categories of temperature sensitive peptides that could potentially be used for the thermally targeted delivery of therapeutics to heated tissues, with a primary focus on recent advances using elastin-like polypeptides (ELPs) in the treatment of cancer.
Under ideal ablative hyperthermia, every tumor cell would be exposed to a sufficient temperature to initiate cell death, while leaving healthy tissues unharmed. In practice, hyperthermia application is limited by the resolution and penetration of the heat deposition modality, the ability to image the tumor boundaries, and the need to avoid causing edema and necrosis of nearby healthy tissues Citation[7]. For example, during trans-urethral thermal ablation of prostate tumors, damage to the nearby rectal wall has significant implications to patient health and quality of life. These implications may prevent ablation of the entire tumor, leaving tumor cells that can recur Citation[8]. Mild hyperthermia also carries a risk of compromising surrounding healthy tissue via edema upon prolonged application Citation[3]. To better utilize established hyperthermia modalities, sophisticated temperature responsive therapeutics are under development that piggyback on mild hyperthermia Citation[5], Citation[6]. In conjunction with regional or local hyperthermia, temperature sensitive delivery strategies are attractive if they enhance the accumulation and penetration of therapeutics in heated regions that contain a malignancy.
Peptide structures and activities are inherently sensitive to temperature, making them excellent platforms for designing hyperthermia responsive therapies. Composed of a linear chain of natural (or unnatural) Citation[9] amino acids, the primary peptide sequence gives rise to complex secondary, tertiary, and quaternary structures that provide the fundamental molecular recognition that underlies most of biology. Thermodynamic considerations drive the assembly of these structures (e.g. hydrogen bonding, electrostatic interactions, van der Waals interactions, and the hydrophobic effect) and most proteins are therefore temperature sensitive. Upon heating, many proteins denature, leading to deactivation or aggregation; in contrast, a sub-set of proteins is known to denature upon cooling Citation[10]. In live cells, protein denaturation can either directly cause cell death or sensitize cells to other treatment modalities such as chemotherapy or radiation therapy, which is currently the primary rationale for the development of mild hyperthermia as a treatment modality for cancer Citation[1].
Here we focus on the application of mild hyperthermia (e.g. with mean temperatures less than 43°C) to spatially enhance the accumulation or activity of drugs that are coupled to or encapsulated within temperature sensitive polypeptide constructs, as opposed to thermal ablation via high temperature fields. A variety of repetitive peptide motifs have been characterized for temperature dependent switching of structure Citation[11], Citation[12] and some of these peptides are attractive candidates for drug delivery using focused, mild hyperthermia Citation[13–15].
Temperature sensitive peptides can be derived from protein sequences and structures Citation[11], Citation[12], Citation[14], Citation[16], and these motifs can be re-engineered with new functions. The simplest of these peptides are capable of forming basic structural themes, such as β sheets and α helices (). The transition between different secondary structures imparts dramatic changes in peptide properties. For instance, a random coil peptide may remain monomeric in solution at low temperatures. Upon heating, the peptide may adopt structure, such as a helix, which enables aggregation and formation of physically larger, immobilized particles. For hyperthermia applications, these transitions must be engineered to occur in physiological solutions between body temperature, 37°C, and 43°C, above which sustained hyperthermia causes cell death. The goal is to develop peptide transitions that occur in a narrow thermal window around 40°C. As the field of biomolecular engineering matures, it will become increasingly feasible to engineer functional peptides that transition between two or more structural states. Recent progress in this area is encouraging, and several peptide motifs are enabling the rational design of clinically relevant thermo-responsive biopolymers.
Table I. Categories of thermo-responsive peptides.
Classes of temperature sensitive peptides
Temperature sensitive peptides must respond to heating with a significant change in properties. From the perspective of hyperthermia triggered delivery of imaging agents or drugs, two obvious properties are directionality and reversibility. Directionality refers to whether the peptide undergoes association or disassociation with increasing temperature. Reversibility refers to whether or not the peptide returns to its original state upon cooling to normal temperature. Of course, there are many other properties that are specific to the secondary structure formed, such as rigidity, radius of hydration, and the formation of nanostructures. Nonetheless, directionality and reversibility are useful properties to categorize temperature sensitive peptides ().
The property of directionality determines whether a therapeutic peptide will selectively associate or disassociate in regions of heated tissue. When peptides associate in an aqueous environment, typical examples of which are aggregation (e.g. microphase separation) and nano-mesoscale self-assembly (e.g. micelle or vesicle formation), they form nano- to microscale structures with reduced mobility, which can alter the distribution of the therapeutic peptide within the tumor (). Association events such as self-assembly can also mediate binding of the agent to the vascular wall by promoting multivalency or transport into extravascular space. For example, we have designed a peptide that self-associates (aggregates) and thereby doubles the concentration of polymer in the tumor during hyperthermia Citation[15]. Many chemotherapeutics have a therapeutic index close to unity; therefore, a two-fold increase in tumor concentration can significantly impact the efficacy of the treatment. In an alternative strategy, if a peptide disassociates in response to hyperthermia, larger diameter particles or micelles could rapidly break up in the tumor, initiating faster diffusion of the monomeric peptides through the tumor tissue (). In unheated tissues, such a peptide could circulate as micelles, which limits vascular permeability and renal clearance. Thus, a disassociating peptide could extend plasma circulation time, prevent accumulation in healthy tissues, and reduce dose-limiting toxicity. The role of directionality should be considered when designing a peptide for a new hyperthermia-directed strategy.
Figure 1. Strategies for designing anti-tumor temperature sensitive peptides. Images depict peptide drug carriers flowing through permeable tumor vasculature during or after local hyperthermia. (A) An associating peptide accumulates into depots within hyperthermic tumor regions. (B) A disassociating peptide micelle disassembles within hyperthermic tumor, and monomeric peptides diffuse rapidly into the tumor. (C) An irreversible-associating peptide is retained in depots after the tissue cools to body temperature, contacting a subset of cells. (D) A reversible-associating peptide floods the tissue with concentrated monomeric peptides after the tissue cools to body temperature, contacting most of the cells.
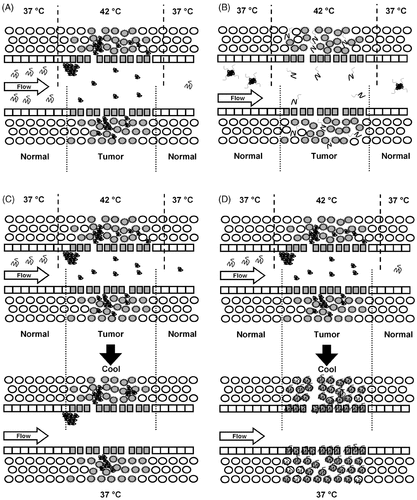
In contrast, reversibility defines whether a peptide returns to its initial state after heating ceases. Peptides in thermodynamic equilibrium may transition reversibly to and from conformations with changes in temperature. During hyperthermia, a reversible peptide may be soluble at body temperature, aggregate upon heating, and return to a soluble state upon cooling; this behavior is typical of a lower critical solution temperature phase transition, which is exhibited by many water soluble macromolecules Citation[14], Citation[17]. In contrast, an irreversible peptide may be soluble at body temperature, aggregate upon hyperthermia, and remain aggregated indefinitely upon cooling. This is because irreversible peptides are kinetically trapped in thermodynamically unstable conformations at low temperatures. An increase in temperature overcomes the activation energy barrier and accelerates the transition to a lower free energy state that is thermodynamically favored (and is therefore stable) and which does not reverse upon cooling. Such an irreversible peptide would persist in its insoluble, aggregated state for extended periods after cooling. Depending on the therapeutic cargo and its preferred mode of delivery, this behavior may or may not be desirable (). For example, irreversible aggregation may be a viable approach for delivering radionuclide γ-emitters, as they do not need to be intracellular to initiate cell death. Alternatively, a reversibly associating peptide may aggregate in regions of heating, forming concentrated localized deposits that disassociate upon cooling (). Such an approach could aid the delivery of chemotherapeutic agents, which must move freely across cell membranes and intracellular barriers to access their molecular targets inside cells. Clearly, the effect of reversibility on the therapeutic strategy and drug cargo should be evaluated during selection of peptide motif.
Based on directionality and reversibility, a variety of therapeutic schemes utilizing temperature sensitive peptides can be envisioned (). Each cargo varies in size, the tissue and cellular location of the target, and their mechanism of action. The choice of scheme depends on the therapeutic cargo, and optimal strategies may differ between chemotherapeutics, radionuclides, or enzymes. Ideally, the cargo should guide the selection of peptide motif, and multiple schemes for every cargo should be compared to determine the optimal scheme for delivery.
Representative temperature sensitive peptides
Temperature sensitive peptides can be classified by directionality and reversibility into four categories, and an example of each category is shown in . Both leucine zippers and collagen peptide motifs disassociate during heating, while heating initiates association of elastin and silk motifs. When classified by reversibility, the leucine zipper and elastin motifs can return to their original states after cooling; however, collagen and silk do not cool into their pre-heated states. These four peptide motifs thus fall into distinct categories, and discussion of their properties is useful for designing hyperthermia-directed therapeutics that use these peptide motifs Citation[11].
A category I peptide, the leucine zipper motif disassociates reversibly (). Originally the leucine zipper was observed in DNA binding domains, including that of C/EBP Citation[18], Citation[19]. The C/EBP leucine zipper contains 5 repeats of a 7 amino acid sequence (abcdefg)n, where the d residue is hydrophobic, frequently leucine. The a residue is also hydrophobic, while the e and g residues are charged. Proline residues are not typically found in the motif. In biological solutions, two to four zippers associate and adopt an α-helical structure. The function of the zipper is to join two or more peptides into a bundle, termed a coiled-coil. The coiled-coil is stabilized along its axis by hydrophobic interactions between the a and d residues and also by electrostatic interactions between oppositely charged e and g residues at the surface. Leucine zippers disassociate above a melting temperature that is a strong function of the specific peptide sequence. Their melting temperature is concentration dependent and occurs over a range of 10°–20°C, above which the peptide monomers adopt a random coil conformation Citation[20]. Shortly after discovery, synthetic proteins containing multiple zippers were described Citation[21]. More recently, tri-block copolymers with leucine zipper end caps were found to assemble into temperature dependent hydrogels Citation[22]. A simplified leucine (bold) zipper motif was identified, (VSSLESK)6, with a melting temperature around 95°C, and into which disruptive lysines residues (underlined) successfully shifted the melting temperature downward to around 40°C, (VSSLESK)2(VSKLESK)1(KSKLESK)1(VSKLESK)1(VSSLESK)1Citation[23]. The melting of these coiled-coils is reversible, making the leucine zipper an attractive candidate for engineering protein biopolymers that destabilize under hyperthermia.
A category II temperature sensitive peptide, the collagen motif, disassociates with increasing temperature; however, upon cooling collagen does not typically return to its original structure (). Instead, re-cooling of collagen generates intermittent peptide crosslinks between neighboring collagen molecules, leading to the formation of hydrogels. Heating in combination with limited hydrolysis transforms collagens into gelatins. Thus, collagens are distinct from leucine zippers, in that the leucine zipper cools to the same conformation as before heating while collagen does not. Collagen is formed by a peptide triple helix, directed by proline rich repeats of the tri-peptide (Gab)n where a is frequently proline and b is frequently hydroxyproline Citation[24]. Natural collagens have significant variability at the a and b position, with different compositions for each type. Typically, collagens have a melting temperature between 38° and 42°C Citation[25], within the therapeutic hyperthermia window. Collagens can be very long with molecular weights >100 kD, such as human pro collagen type I (α1), which contains n = 361 repeats of the motif (Gab)n. Within this sequence, the complete canonical motif (GPPOH)n is repeated a maximum of five times. Natural collagen contains extensive proline hydroxylation, a post-translational modification. Over-expression of prolyl-4-hydroxylase has enabled efficient expression of collagen in yeast Citation[25], and collagen-like-peptides (CLPs) have also been produced in bacteria. To produce CLPs in Escherichia coli, the bacteria are supplemented with hydroxyproline, thus modifying proline residues randomly and not exclusively at the b position Citation[26]. Nonetheless, bacterial CLPs form a triple helix that can be denatured at high temperatures, and does not return to the native conformation as determined by circular dichroism Citation[26]. Collagens and their denatured derivatives, gelatins, are widely used additives in pharmaceuticals, and their biocompatibility and melting profiles make them suitable for engineering thermoresponsive peptides, under applications where reversibility is undesirable.
A category III peptide, the elastin-like-polypeptide (ELP) is a pentapeptide identified from human tropoelastin (). The pioneering work characterizing ELPs was done by Urry and coworkers, initially using synthetic peptides and later using recombinant protein expression from E. coli Citation[27]. In contrast to collagens or leucine zippers, ELPs reversibly self-associate and phase separate in response to heating. ELPs consist of repeating (aPGbG)n where the guest residue, b, can be any amino acid except proline Citation[14]. Residue a is typically valine or isoleucine. ELPs undergo an endothermic phase transition above an inverse phase transition temperature. The transition temperature is a function of the guest residues (a and b), the molecular weight, the concentration, and the presence of co-solutes. The transition temperature decreases if guest residue hydrophobicity, polymer molecular weight, concentration, or NaCl concentration increases. Heated ELPs form type II β-turns, as detected using circular dichroism Citation[27], which can result in phase separation over a narrow temperature window of less than 2°C for many ELP. The ELP phase transition is a useful biotechnology tool, enabling purification of recombinant ELPs, and more importantly of ELP fusion proteins, from bacterial lysate by multiple rounds of temperature cycling Citation[28]. In addition, the ELP phase transition enables the temperature dependent formation of nanoscale self-assembled structures from ELP diblocks Citation[29], Citation[30]. The ELP motif is an extremely useful peptide for designing hyperthermia-directed peptides, and its behavior and applications for drug delivery will be discussed in detail through the remainder of the article.
A representative category IV temperature sensitive peptide () is derived from the heavy fibroin protein produced by the silkworm Bombyx mori, which undergoes an irreversible association in response to heating Citation[31]. While several insect silks contain distinct peptide motifs Citation[11], the silkworm fibroin motif is the most widely studied, (GAGAGS)n, where n have been reported between 2 and 170 repeats Citation[11]. Within natural fibroin, there is much less variability in amino acid usage than in previously mentioned leucine zippers, elastins, and collagens. Typically, alterations to silk-like polypeptides (SLPs) have been studied by combining this motif with other peptides, such as hydrophilic spacers or ELP. Purified SLP motifs form highly crystalline β-sheets, in an exothermic reaction that proceeds faster at higher temperatures and concentrations Citation[31], Citation[32]. The crystalline product is very stable, does not dissolve in water, and is difficult to process. Despite an absence of human homologs, silk peptides are relatively biocompatible Citation[31]. For hyperthermia applications, the silk motif is useful for initiating non-reversible association of peptide subunits; however, the rate of association is not as rapid as the association of elastin motifs.
Properties of elastin-like polypeptides
ELPs have a number of interesting properties, which are useful for engineering hyperthermia-responsive therapeutics: (1) ELPs undergo a reversible phase transition upon heating, similar to the LCST transition observed in some synthetic, water soluble polymers Citation[27–29], Citation[31], Citation[32]; raising the solution temperature above a critical transition temperature, Tt, triggers an aqueous demixing phase transition in which ELPs go from being solvated and hence highly water soluble to a desolvated aggregated state (also called coacervate). (2) The ELP phase transition occurs in very narrow 1–2°C range (at µM concentration or above; the width of the transition is a function of ELP concentration). (3) The phase transition temperature is sensitive to numerous variables, including molecular weight, concentration, co-solutes, and guest residue identity. Each of these factors is an engineering variable that is useful to control the phase transition behavior of ELPs. In particular, the ELP composition (i.e. type and fraction of the different residues at the fourth guest residue position) and MW are two orthogonal ‘Design’ variables in that they allow one to encode a specified transition temperature (at a specified concentration of ELPs and cosolute type and concentration) into the design of the ELPs. (4) ELPs have good pharmacokinetics and are retained for significant periods in the central blood compartment Citation[33]. (5) ELPs are tolerated by the immune system, and show very low ability to induce neutralizing antibodies Citation[34]. (6) ELPs are biocompatible and do not cause inflammation near site of injection Citation[34]. (7) A concern with many high molecular weight polymers, ELPs are degradable by proteolysis, reducing issues of long-term accumulationCitation[35]. (8) ELPs are amenable to biosynthesis using recombinant DNA techniques and often the phase transition can efficiently purify polymer from bacterial cell lysates Citation[28]. (9) ELPs can be designed for facile attachment of radionuclides or chemotherapeutics via precisely positioned conjugation sites (e.g. unique lysine or cysteine residues); the ability to precisely position a desired number of conjugation sites along the ELP chain (and potentially other repetitive polypeptides) provides the important ability to design the architecture of the ELP-drug conjugate. An equally important attribute of ELPs for delivery of biological protein and peptide based therapeutics stems from the fact that they are genetically encoded, so that it is trivial to synthesize these biological conjugates as genetically encoded fusions with ELPs; to date all ELP fusions that we have successfully expressed display phase transition behavior similar to the parent ELP. These factors make ELPs a flexible platform for the synthesis of thermo-responsive therapeutics.
In this regard, the fact that one can synthesize ELPs using genetic engineering confers a number of advantages over synthetic polymers for drug delivery. First, genetically engineered polymers are monodisperse Citation[13]. Polydispersity is an important variable in the development of high molecular weight therapeutics, as different molecular weight fractions experience different fates in the body. While chemical polymerization methods exist that generate narrow polydispersity, cellular expression enables near perfect monodispersity. Second, genetically encoded synthesis provides precise control over the specific peptide sequence; the placement and identity of each amino acid is particularly important to control the transition temperature by specifying the guest residue composition, in the design of block copolymers by imparting amphiphilicity to ELPs as a function of temperature or pH, and in the placement of conjugation sites for small molecule therapeutics that are covalently attached or chelated to the ELP. Third, the fact that ELPs undergo a soluble-insoluble phase transition is advantageous, as it enables efficient purification of mg-g scale quantities of ELPs from the cells in which they are expressed. Using mild heating or the addition of salt, the phase transition causes ELPs and covalently linked peptides or proteins to precipitate under centrifugation. Since ELP phase transition is reversible, the polymer can usually be resuspended in cold buffer and centrifuged again to remove denatured cellular contaminants. By repeating this step three to five times, it is possible to purify >500 mg protein/l of bacterial broth to >99% purity Citation[36], and this purification can proceed without time-consuming and costly chromatography.
The ELP inverse phase transition is observed as the abrupt formation of two liquid phases, a process called coacervation, which occurs over less than a 2°C change in temperature. Recently a mechanism was proposed that describes the ELP transition as the product of multiple temperature dependent steps Citation[37]. The first step is the formation of the β-turn spiral; however, there is additional evidence that ELPs can also adopt a distorted β-strand Citation[38]. Observed by circular dichroism, the spirals form even in dilute solutions that do not coacervate. At low temperatures ELPs are in a random coil conformation Citation[37], and hydrophobic guest residues (valine, alanine, isoleucine, etc.) are surrounded by structured shells of water Citation[27]. As temperature increases, the entropic cost of the structured water increases. To compensate, the peptide backbone adopts an ordered β-turn spiral, which reduces the exposed hydrophobic surface area and extent of water ordering. The second step is the coacervation of spiral folded peptides, which occurs at the inverse phase transition temperature. This step is highly concentration dependent and easily observed by the formation of turbid aggregates Citation[37]. This multi-step process generates abrupt peptide associations, which can respond dramatically to the moderate temperature changes prevalent in clinical hyperthermia.
The rapid and reversible ELP phase transition also lends itself to developing temperature-responsive switches Citation[39], Citation[40]. Our group recently described such an approach called temperature triggered multivalency () Citation[30], in which ELPs are synthesized with a peptide ligand at one end of the polymer chain that is specific for a cell-surface receptor. Upon systemic injection, the ELPs are designed to circulate as ‘unimers’ (i.e. soluble ELP molecules) in the body until it reaches areas of locally applied, mild hyperthermia (∼40°C). The higher local temperature initiates self-assembly of the ELPs into micelles that present multiple copies of the ligand on their corona, thus increasing the binding avidity of the micelle for the cell-surface receptor compared to unimers that only present a single copy of the ligand (). These micelles are therefore expected to bind to target cells in the heated region and to internalize to a greater degree in heated tissue (e.g. tumor) than in the rest of the body. To implement this strategy of temperature triggered multivalency, ELP diblock copolymers were synthesized with a hydrophobic block [V5-60] with a transition temperature ∼40°C attached to a hydrophilic block [V1A8G7-96] with a transition temperature above 50°C. Using our ELP nomenclature, a is always valine and b () is an alternating mixture of the indicated guest residues at the ratio denoted in subscript, followed by the total number of pentapeptides, n. For example, [V1A8G7-96]/[V1-60] represents a hydrophilic block consisting of 96 pentamers (480 amino acids), [VG (VPGAG VPGGG)7 VGGAG VPG]6, followed by a hydrophobic block consisting of 60 pentamers (300 amino acids) of [VG VPG]60. Dynamic light scattering as a function of temperature indicated that as the hydrophobic block transitions at its Tt, it triggers the formation of micelles with a hydrodynamic radius of ∼30 nm (). Upon further heating the micelle is stable until the transition temperature of the other, hydrophilic block is reached (∼50°C), and at higher temperature the micelles aggregate into larger microscopic aggregates. The critical micelle temperature was a function of the hydrophobic block (∼41°C), and the critical micelle concentration for this block copolymer was ∼8 µM Citation[30]. An RGD and NGR tripeptide functionalized ELP diblock exhibited temperature dependent micelle behavior, showing that the system is fairly tolerant to the inclusion of non-ELP sequences at the corona without disrupting self-assembly (). Uptake of the NGR-presenting ELPs (with a fluorophore label) was studied by confocal microscopy on cells positive for the NGR receptor, HT-1080; furthermore, the NGR block copolymer demonstrated 19-fold greater colocalization with the cytoplasm after a 42°C incubation as compared to a 37°C incubation Citation[30]. These results highlight the usefulness of ELPs in the development of hyperthermia-directed drug carriers.
Figure 2. Elastin-like polypeptide block copolymers: affinity modulated switches. (A) An ELP block copolymer with a hydrophilic [V1A8G7-96] amino terminus and a hydrophobic [V5-60] carboxy terminus. Also indicated are an amino terminal NGR peptide ligand (half-ring) and a carboxy terminal therapeutic agent (lightning bolt). At body temperature the ELPs are monomeric, but at 42°C the ELPs assemble into spherical micelles decorated with multiple ligands. (B) Hydrodynamic radius and optical density as a function of temperature for ELP block copolymers with and without the NGR peptide ligand. For 25 µM peptide, Both ELPs are monomeric at 37°C and multimeric at 42°C. Error bars indicate polydispersity. (Reproduced with permission Citation[30]).
![Figure 2. Elastin-like polypeptide block copolymers: affinity modulated switches. (A) An ELP block copolymer with a hydrophilic [V1A8G7-96] amino terminus and a hydrophobic [V5-60] carboxy terminus. Also indicated are an amino terminal NGR peptide ligand (half-ring) and a carboxy terminal therapeutic agent (lightning bolt). At body temperature the ELPs are monomeric, but at 42°C the ELPs assemble into spherical micelles decorated with multiple ligands. (B) Hydrodynamic radius and optical density as a function of temperature for ELP block copolymers with and without the NGR peptide ligand. For 25 µM peptide, Both ELPs are monomeric at 37°C and multimeric at 42°C. Error bars indicate polydispersity. (Reproduced with permission Citation[30]).](/cms/asset/b69f4fca-06b9-479f-b7d7-68fe38562171/ihyt_a_315123_f0002_b.gif)
As a peptide therapeutic, ELP biopolymers have reasonably good pharmacokinetics with terminal circulation half-lives of 8–11 h in nude mice Citation[33]. In designing macromolecular drug carriers, it is beneficial to develop polymers that are close to or slightly larger than the renal filtration cut-off, thus enabling circulation in the plasma for the entire period of applied hyperthermia (typically a few hours). The pharmacokinetics of a 59 kD ELP [V5A2G3-150] with a transition temperature >37°C was evaluated in mice (). A 14C label was incorporated into ELPs during biosynthesis and used to track the fate of ELPs in vivo Citation[41]. Following intravenous administration, ELPs displayed two-compartment pharmacokinetics with a distribution half-life of 7.3 min, a terminal half-life of 8.4 h, an initial volume of distribution of 1.4 mL, and a clearance of 0.32 mL/h Citation[33]. After intravenous administration, ELPs degraded at a rate of 2.5 weight%/day into short fragments (MW < 10 kD), as determined by trichloroacetic acid precipitation Citation[33]. Thus, ELPs are biodegradable, which is desirable from the perspective of their ultimate clearance from the body, but not so quickly as to limit blood pharmacokinetics.
Figure 3. Pharmacokinetics and tumor accumulation of elastin-like polypeptides. 14C labeled ELPs were administered via tail vein to nude mice for an initial plasma concentration of 25 µM. (A) Whole blood pharmacokinetics were determined over 24 hours. The ELP1[V5A2G3-150] concentration was determined and fit with a bi-exponential solution to a two compartment model to obtain the indicated half-lives. (B) A temperature sensitive ELP1[V5A2G3-150] (Tt = 39°C) and a temperature insensitive ELP2[V1A8G7-160] (Tt = 53°C) were administered to mice with subcutaneous FADU tumors in the right hind leg. Immersed in a water bath, the leg and tumor were heated to 41.5°C for 1 h before isolation. * indicates that ELP1(heat) was significantly (p < 0.05, Fischer's PLSD) greater than either control. Error bars indicate the standard error of the mean (n = 5). (Reproduced with permission Citation[33]).
![Figure 3. Pharmacokinetics and tumor accumulation of elastin-like polypeptides. 14C labeled ELPs were administered via tail vein to nude mice for an initial plasma concentration of 25 µM. (A) Whole blood pharmacokinetics were determined over 24 hours. The ELP1[V5A2G3-150] concentration was determined and fit with a bi-exponential solution to a two compartment model to obtain the indicated half-lives. (B) A temperature sensitive ELP1[V5A2G3-150] (Tt = 39°C) and a temperature insensitive ELP2[V1A8G7-160] (Tt = 53°C) were administered to mice with subcutaneous FADU tumors in the right hind leg. Immersed in a water bath, the leg and tumor were heated to 41.5°C for 1 h before isolation. * indicates that ELP1(heat) was significantly (p < 0.05, Fischer's PLSD) greater than either control. Error bars indicate the standard error of the mean (n = 5). (Reproduced with permission Citation[33]).](/cms/asset/63bb9440-c0f2-43ed-9a64-bb8b1ef35cad/ihyt_a_315123_f0003_b.gif)
In addition to possessing favorable pharmacokinetics, ELPs are biocompatible and have low immunogenicity Citation[31], Citation[34], Citation[42]. Urry and coworkers evaluated a γ-irradiated cross-linked poly(VPGVG)∼120, and found it to be extremely well tolerated. ELPs were found non-cytotoxic (mouse fibroblasts), without acute systemic toxicity (mice: i.p. and i.v.), without dermal irritation (rabbit: i.c.), and without systemic antigenicity (guinea pigs: i.v.). In addition, ELPs did not alter whole blood clotting time (dog) or induce red blood cell hemolysis (rabbit) Citation[42]. Urry and coworkers reported that complete Freund's adjuvant was needed to generate a low titer antibody response to subcutaneous injected ELPs Citation[34]. Cappello and coworkers found similar results when investigating the immunogenicity of a peptide containing silk and ELP sequences [(GVGVP)8(GAGAGS)2]18 in rabbits Citation[34]. Without adjuvant, they injected polymer (10 mg) at time zero and gave boosters of (0.5 mg polymer) at 6 and 8 weeks. After 9 weeks, samples of sera were quantified by ELISA for binding to the pure ELP peptide (VPGVG)8 sequence. Under these injection conditions, no reactivity was identified (titer < 2); however, the administration of (VPGVG)8 with complete Freund's adjuvant at a dose of 10 mg of polymer at week 0, 2, 3, 5, and 7 did generate a serum titer of 480-fold Citation[34]. Combined with good pharmacokinetics, the fact that ELPs are biocompatible, biodegradable, and do not stimulate an antibody response without the administration of adjuvant suggests that ELPs are reasonable platforms for building therapeutic temperature sensitive peptides.
Anti-tumor applications of ELP
ELPs are being developed as carriers for chemo- and radiotherapeutics; current efforts are directed at various aspect of delivery including ligand optimization, cargo encapsulation or conjugation, and evaluation of the in vivo behavior of ELPs laden with their drug cargo. To optimize triggered tumor binding/uptake, peptide ligands are being examined as already discussed above (). Additionally, both small molecules Citation[15], Citation[43–47] and cell cycle arrest peptides are being formulated as cargo Citation[48], Citation[49]. As discussed, the evidence for favorable pharmacokinetics and biocompatibility suggests that ELPs may carry cargo similarly to synthetic macromolecular drug carriers Citation[50]; furthermore, ELPs have added-potential to generate useful hyperthermia-directed therapeutics.
ELPs have been covalently modified with small molecule drugs Citation[43–47] and with peptide cell cycle inhibitors Citation[48], Citation[49]. The potent anthracycline doxorubicin was fused to ELPs both via pH reversible hydrazone linkages Citation[43], Citation[44] and also via a cathepsin mediated peptide GFLG spacer Citation[46], Citation[47]. Using other high molecular weight polymers, both linkage strategies have successfully reduced tumor growth in animals Citation[50]. For both strategies, the mechanism of release depends upon cellular uptake and endo/lysosomal processing at the site of the tumor. After cellular internalization, trafficking to the low pH vesicles initiates either acid mediated cleavage of the hydrazone linkage or cathepsin mediated proteolysis of GFLG. For the hydrazone-mediated approach, release regenerates doxorubicin; however, the cathepsin-mediated release generates a doxorubicin adduct with a short modification at the drug's primary amine. Both of these mechanisms of drug release depend on cellular uptake, and there are two reasons that ELPs are expected to efficiently traffic to the lysosome. First, the heat-induced phase transition has been shown to increase cellular uptake in cell culture Citation[51]. Second, ELPs have been shown to initiate trafficking of ELP-doxorubicin to low pH cellular compartments, even in the absence of hyperthermia Citation[43]. Interestingly, it appears that the attachment of hydrophobic molecules, such as doxorubicin, can have a significant influence on the transition temperature Citation[43]. These shifts in transition temperature can exceed 10°C and vary by drug and chemistry of attachment Citation[43], Citation[44]. To produce ELP-drug conjugates suitable for hyperthermia, it may be necessary to optimize the ELP guest residues and molecular weight on a case-by-case basis in order to generate a therapeutic system with a plasma transition temperature of 40°C.
Initial studies of hyperthermia-directed targeting have characterized the behavior of monoblock ELPs in heated tumor (). When animals are given ELPs systemically, the biopolymer circulates in the blood for reasonably long periods (), and if heating is applied during this time, then significantly greater quantities of ELPs accumulate in the tumor () Citation[5], Citation[15], Citation[33]. illustrates the tumor concentration after one hour of local heating (41.5°C) of either ELP1 (39°C transition temperature, 59 kD) or ELP 2 (53°C transition temperature, 62 kD). At normal temperatures, ELP1 accumulates to 11% injected dose (ID) per gram tissue; however, upon heating ELP1 accumulation increased significantly to 19% ID/g (p < 0.05, Fischer's PSLD). For the temperature insensitive control ELP2, the heating had little effect.
To determine the mechanism of accumulation, in vivo confocal fluorescence microscopy was employed to observe the growth of ELP microaggregates and determine the how the phase transition affects the extravascular appearance of peptide Citation[5]. A FaDu tumor was implanted under a dorsal skin fold window and imaged using confocal scanning laser fluorescence microscopy. After systemic administration, tumors were heated for 45 minutes to 41.5°C and then allowed to cool back to 37°C. ELP1 and ELP2 conjugated to two different color fluorophores were co-administered; in vivo fluorescence microscopy revealed that only ELP1 responded to hyperthermia in vivo. ELP1 formed numerous micron sized aggregates that were predominantly inside the vascular compartment Citation[5]. The most dramatic effect occurred after the removal of heat. As the temperature returned to 37°C, the vascular ELPs aggregates quickly redissolved and the fluorescence in the extravascular compartment increased dramatically Citation[5]. The quantified extravascular signal, demonstrated that that ELP1 concentrations were about twice that of ELP2. It was suggested that the rapid increase in the extravascular concentration of soluble ELPs immediately after cooling drives a greater diffusive flux, yielding tissue penetration superior to that of unheated controls.
These results suggest that the induction of the ELP phase transition roughly doubles the concentration of ELPs in the tumor. Importantly, this concentration boost can be achieved without increasing the concentration at other sites in the body that exhibit dose-limiting toxicity. Chemotherapeutics are frequently dosed at the maximum tolerated level, as determined by a dose-limiting tissue. For efficacious therapeutics, such as doxorubicin, the dose of free drug cannot be significantly increased, thus preventing further increases in the tumor concentration. The data suggest that an ELP-drug will reach twice the tumor concentration with hyperthermia compared to without hyperthermia, and this should substantially increase the efficacy of many drugs that are conjugated to a heat sensitive ELP.
Future directions and barriers to clinical translation
The clinical translation of thermo-responsive peptides requires further development in three areas: optimization of thermal control during pharmacokinetic clearance; improved models describing how cargo, molecular weight, and concentration affect thermal behavior of thermally responsive drug carriers; as well as optimization of standard pharmaceutical considerations. The first two areas concern properties specific to thermo-responsive peptides, while pharmaceutical considerations (purification, quality control, and immunogenicity) are analogous to those encountered for other protein therapeutics and will not be discussed here.
To bring thermo-responsive peptides into the clinic, they must be optimized to meet the duration (∼2 h) and magnitude (38–43°C) of clinically relevant mild hyperthermia. Here, the major outstanding issue that must be addressed is that the thermo-pharmacokinetics of the carrier must be optimized so that the driving force for thermal targeting is maximized over the duration of applied heat. This is a convoluted problem because the same variables that determine pharmacokinetics also influence the thermodynamic and kinetic transition between the soluble and insoluble state of thermo-responsive peptides. After systemic administration, long-circulation of polymers is limited by various parameters, including their net-charge, molecular weight, enzymatic degradation, and off-target binding. For new pharmaceutical agents, the influence of these parameters on pharmacokinetics requires careful study; furthermore, for thermo-responsive peptides these parameters are coupled with their thermal behavior. For example, the (1) MW, (2) systemic concentration and (3) drug stoichiometry are three variables that affect both the pharmacokinetics and thermal behavior of ELP-drug conjugates so that altering one parameter to optimize pharmacokinetics deleteriously impact their thermal behavior or vice versa. Increasing the molecular weight of ELPs–a prototypical thermo-responsive polypeptide–to sustain high plasma concentrations can decrease the transition temperature by more than 10°C Citation[52]. This is undesirable because decreasing the transition temperature below that of the body eliminates thermal targeting and may precipitously alter biodistribution. An additional factor to be taken into account during dose-escalation studies, the transition temperatures of ELPs exhibit an inverse log concentration dependence, so that the transition temperature may decrease significantly with incresing dose. Attachment of a therapeutic agent to a polymeric carrier can also alter the transition temperature, so that the effect of drug loading of the carrier on the thermal behavior of the carrier must be taken into account in addition to its obvious impact on therapeutic efficacy and toxicity. Fortunately, the effect of these variables on both the pharmacokinetics and thermal behavior of ELPs are now reasonably well understood though close to a decade of studies in our group, and can be controlled by virtue of the absolute control one can exercise in the recombinant synthesis of these polypeptides over two orthogonal, molecular design variables: the identity and composition of the residues a and b in the pentameric repeat sequence and MW ().
More generally, in order to engineer a peptide with optimal thermal behavior and pharmacokinetics at the clinically optimal dosage demands a quantitative model. Such a model would accurately predict how control of a small subset of amino acids in a motif could provide a specific thermal response over a range of concentrations and molecular weight. Such a dataset is available for ELPs Citation[27], but further research is required to develop equivalent datasets for other peptides (). An additional influence to be incorporated into such modeling is the significant effect that the pharmacological cargo can exert on peptide behavior. Peptides can carry cargo, including radionuclides, drugs, biofunctional peptides, and fused proteins. Due to their molecular weight, hydrophobicity, mechanism of action, and dosage regimes, each of these cargoes may necessitate individualized approaches to designing thermo-responsive carriers. For instance, radionuclides are so potent that only a small fraction (<1%) of the peptides require modification to reach a maximum tolerated dose. Conversely, chemotherapeutics require relatively high dosages, whereby 5–10% of the polymer mass consists of drug. Under this approximation, each polymer may have between 1–10 drugs per peptide. The modification of a small fraction of peptides with a low molecular weight radionuclide will be expected to have a relatively small impact on the thermal properties of the peptide. However, the inclusion of multiple hydrophobic drugs per peptide significantly influences thermal properties Citation[43]. Presumably, even larger molecular weight peptides and fusion proteins may further distort the thermal properties Citation[53]. To translate optimized approaches to the clinic will require additional research and development into accurate models that explain and predict how critical variables (molecular weight, concentration, and cargo) influence the properties of these thermo-responsive peptides.
Conclusions
Multiple peptide motifs are capable of different temperature dependent transitions, and can be harnessed for drug delivery by focused, mild hyperthermia. Using recombinant DNA methodology, such peptides can be ‘polymerized’ into thermo-responsive biopolymers that target therapeutic cargo to tumors using externally applied mild hyperthermia. This review has discussed a toolbox of peptide motifs that confer the ability to associate or disassociate both reversibly or irreversibly in response to a thermal stimulus. This toolbox of thermo-responsive peptides can generate a range of therapeutic approaches, as best suited for the therapeutic cargo. For example, the elastin-like-polypeptides (ELPs) show promise in their ability to deposit and switch conformations in heated tissues and tumors. ELPs are biodegradable, circulate for reasonable periods in the blood, display good biocompatibility, and appear to be reasonable drug delivery platforms. Recent progress suggests that temperature sensitive peptides, including ELPs, will be successfully engineered with complex behaviors that enable hyperthermia-directed drug delivery.
Acknowledgements
This work was supported with NIH grant 5F32-CA-123889 to J.A.M. and NIH grant R01-EB-00188 to A.C.
Declaration of interest: Ashutosh Chilkoti is a cofounder of a start-up company, Phase Bioscience, in Durham, NC, USA that is commercializing the ELP phase transition technology for application in biotechnology and medicine, including purification of recombinant proteins.
References
- Kampinga HH. Cell biological effects of hyperthermia alone or combined with radiation or drugs: A short introduction to newcomers in the field. Int J Hyperthermia 2006; 22: 191–196
- Falk MH, Issels RD. Hyperthermia in oncology. Int J Hyperthermia 2001; 17: 1–18
- Dewhirst MW, Viglianti BL, Lora-Michiels M, Hanson M, Hoopes PJ. Basic principles of thermal dosimetry and thermal thresholds for tissue damage from hyperthermia. Int J Hyperthermia 2003; 19: 267–294
- Wust P, Hildebrandt B, Sreenivasa G, Rau B, Gellermann J, Riess H, Felix R, Schlag PM. Hyperthermia in combined treatment of cancer. Lancet Oncol 2002; 3: 487–497
- Dreher MR, Liu W, Michelich CR, Dewhirst MW, Chilkoti A. Thermal cycling enhances the accumulation of a temperature-sensitive biopolymer in solid tumors. Cancer Res 2007; 67: 4418–4424
- Ponce AM, Vujaskovic Z, Yuan F, Needham D, Dewhirst MW. Hyperthermia mediated liposomal drug delivery. Int J Hyperthermia 2006; 22: 205–213
- Pisani LJ, Ross AB, Diederich CJ, Nau WH, Sommer FG, Glover GH, Butts K. Effects of spatial and temporal resolution for MR image-guided thermal ablation of prostate with transurethral ultrasound. J Magn Res Imag 2005; 22: 109–118
- Colombel M, Gelet A. Principles and results of high-intensity focused ultrasound for localized prostate cancer. Prostate Cancer Prostatic Diseases 2004; 7: 289–294
- Mendel D, Cornish VW, Schultz PG. Site-directed mutagenesis with an expanded genetic-code. Ann Rev Biophys Biomol Structure 1995; 24: 435–462
- Franks F. Protein destabilization at low temperatures. Adv Protein Chem 1995; 46: 105–139
- Hart DS, Gehrke SH. Thermally associating polypeptides designed for drug delivery produced by genetically engineered cells. J Pharm Sciences 2007; 96: 484–516
- Mart RJ, Osborne RD, Stevens MM, Ulijn RV. Peptide-based stimuli-responsive biomaterials. Soft Matter 2006; 2: 822–835
- Chilkoti A, Dreher MR, Meyer DE. Design of thermally responsive, recombinant polypeptide carriers for targeted drug delivery. Adv Drug Deliv Rev 2002; 54: 1093–1111
- Chilkoti A, Dreher MR, Meyer DE, Raucher D. Targeted drug delivery by thermally responsive polymers. Adv Drug Deliv Rev 2002; 54: 613–630
- Meyer DE, Shin BC, Kong GA, Dewhirst MW, Chilkoti A. Drug targeting using thermally responsive polymers and local hyperthermia. J Controlled Release 2001; 74: 213–224
- Meyer DE, Kong GA, Dewhirst MW, Zalutsky MR, Chilkoti A. Targeting a genetically engineered elastin-like polypeptide to solid tumors by local hyperthermia. Cancer Res 2001; 61: 1548–1554
- Galaev IY, Mattiasson B. Thermoreactive water-soluble polymers, nonionic surfactants, and hydrogels as reagents in biotechnology. Enzyme Microb Tech 1993; 15: 354–366
- Landschulz WH, Johnson PF, Mcknight SL. The leucine zipper - A hypothetical structure common to a new class of DNA-binding proteins. Science 1988; 240: 1759–1764
- Struhl K. Helix-turn-helix, zinc-finger, and leucine-zipper motifs for eukaryotic transcriptional regulatory proteins. Trends Biochem Sci 1989; 14: 137–140
- Thompson KS, Vinson CR, Freire E. Thermodynamic characterization of the structural stability of the coiled-coil region of the bzip transcription factor gcn4. Biochem 1993; 32: 5491–5496
- Regan L, Degrado WF. Characterization of a helical protein designed from 1st principles. Science 1988; 241: 976–978
- Petka WA, Harden JL, McGrath KP, Wirtz D, Tirrell DA. Reversible hydrogels from self-assembling artificial proteins. Science 1998; 281: 389–392
- Xu CY, Breedveld V, Kopecek J. Reversible hydrogels from self-assembling genetically engineered protein block copolymers. Biomacromolecules 2005; 6: 1739–1749
- Creighton TE. Proteins: Structure and molecular properties. W.H. Freeman, New York 1993
- Olsen D, Yang CL, Bodo M, Chang R, Leigh S, Baez J, Carmichael D, Perala M, Hamalainen ER, Jarvinen M, et al. Recombinant collagen and gelatin for drug delivery. Adv Drug Delivery Rev 2003; 55: 1547–1567
- Buechter DD, Paolella DN, Leslie BS, Brown MS, Mehos KA, Gruskin EA. Co-translational incorporation of trans-4-hydroxyproline into recombinant proteins in bacteria. J Biol Chem 2003; 278: 645–650
- Urry DW. Physical chemistry of biological free energy transduction as demonstrated by elastic protein-based polymers. J Phys Chem B 1997; 101: 11007–11028
- Meyer DE, Chilkoti A. Purification of recombinant proteins by fusion with thermally-responsive polypeptides. Nat Biotech 1999; 17: 1112–1115
- Wright ER, Conticello VP. Self-assembly of block copolymers derived from elastin-mimetic polypeptide sequences. Adv Drug Delivery Rev 2002; 54: 1057–1073
- Dreher MR, Simnick AJ, Fischer K, Smith RJ, Patel A, Schmidt M, Chilkoti A. Temperature triggered self-assembly of polypeptides into multivalent spherical micelles. J Am Chem Soc 2008; 130: 687–694
- Megeed Z, Cappello J, Ghandehari H. Genetically engineered silk-elastinlike protein polymers for controlled drug delivery. Adv Drug Delivery Rev 2002; 54: 1075–1091
- Cappello J, Crissman JW, Crissman M, Ferrari FA, Textor G, Wallis O, Whitledge JR, Zhou X, Burman D, Aukerman L, et al. In-situ self-assembling protein polymer gel systems for administration, delivery, and release of drugs. J Cont Rel 1998; 53: 105–117
- Liu W, Dreher MR, Furgeson DY, Peixoto KV, Yuan H, Zalutsky MR, Chilkoti A. Tumor accumulation, degradation and pharmacokinetics of elastin-like polypeptides in nude mice. J Control Release 2006; 116: 170–178
- Domb AJ, Kost J, Wiseman DM. Handbook of biodegradable polymers. Harwood, Amsterdam 1997
- Shamji MF, Betre H, Kraus VB, Chen J, Chilkoti A, Pichika R, Masuda K, Setton LA. Development and characterization of a fusion protein between thermally responsive elastin-like polypeptide and interleukin-1 receptor antagonist - sustained release of a local antiinflammatory therapeutic. Arthritis Rheum 2007; 56: 3650–3661
- Chow DC, Dreher MR, Trabbic-Carlson K, Chilkoti A. Ultra-high expression of a thermally responsive recombinant fusion protein in E-coli. Biotech Prog 2006; 22: 638–646
- Yamaoka T, Tamura T, Seto Y, Tada T, Kunugi S, Tirrell DA. Mechanism for the phase transition of a genetically engineered elastin model peptide (vpgig)(40) in aqueous solution. Biomacromol 2003; 4: 1680–1685
- Yao XL, Hong M. Structure distribution in an elastin-mimetic peptide (vpgvg)(3) investigated by solid-state NMR. J Amer Chem Soc 2004; 126: 4199–4210
- Megeed Z, Winters RM, Yarmush ML. Modulation of single-chain antibody affinity with temperature-responsive elastin-like polypeptide linkers. Biomacromol 2006; 7: 999–1004
- Sallach RE, Wei M, Biswas N, Conticello VP, Lecommandoux S, Dluhy RA, Chaikof EL. Micelle density regulated by a reversible switch of protein secondary structure. J Amer Chem Soc 2006; 128: 12014–12019
- Liu W, Dreher MR, Chow DC, Zalutsky MR, Chilkoti A. Tracking the in vivo fate of recombinant polypeptides by isotopic labeling. J Cont Rel 2006; 114: 184–192
- Urry DW, Parker TM, Reid MC, Gowda DC. Biocompatibility of the bioelastic materials, poly(gvgvp) and its gamma-irradiation cross-linked matrix - Summary of generic biological test-results. J Bioactive Compat Polymers 1991; 6: 263–282
- Dreher MR, Raucher D, Balu N, Michael Colvin O, Ludeman SM, Chilkoti A. Evaluation of an elastin-like polypeptide-doxorubicin conjugate for cancer therapy. J Control Rel 2003; 91: 31–43
- Furgeson DY, Dreher MR, Chilkoti A. Structural optimization of a 'Smart' Doxorubicin-polypeptide conjugate for thermally targeted delivery to solid tumors. J Control Rel 2006; 110: 362–369
- Bae Y, Buresh RA, Williamson TP, Chen THH, Furgeson DY. Intelligent biosynthetic nanobiomaterials for hyperthermic combination chemotherapy and thermal drug targeting of hsp90 inhibitor geldanamycin. J Control Rel 2007; 122: 16–23
- Bidwell GL, Davis AN, Fokt I, Priebe W, Raucher D. A thermally targeted elastin-like polypeptide-doxorubicin conjugate overcomes drug resistance. Investig New Drugs 2007; 25: 313–326
- Bidwell GL, Fokt I, Priebe W, Raucher D. Development of elastin-like polypeptide for thermally targeted delivery of doxorubicin. Biochem Pharmacol 2007; 73: 620–631
- Bidwell GL, Raucher D. Application of thermally responsive polypeptides directed against c-myc transcriptional function for cancer therapy. Mol Cancer Therap 2005; 4: 1076–1085
- Bidwell GL, Raucher D. Enhancing the antiproliferative effect of topoisomerase ii inhibitors using a polypeptide inhibitor of c-myc. Biochem Pharmacol 2006; 71: 248–256
- Duncan R. Polymer conjugates as anticancer nanomedicines. Nature Rev Cancer 2006; 6: 688–701
- Raucher D, Chilkoti A. Enhanced uptake of a thermally responsive polypeptide by tumor cells in response to its hyperthermia-mediated phase transition. Cancer Res 2001; 61: 7163–7170
- Meyer DE, Chilkoti A. Quantification of the effects of chain length and concentration on the thermal behavior of elastin-like polypeptides. Biomacromol 2004; 5: 846–851
- Trabbic-Carlson K, Meyer DE, Liu L, Piervincenzi R, Nath N, LaBean T, Chilkoti A. Effect of protein fusion on the transition temperature of an environmentally responsive elastin-like polypeptide: A role for surface hydrophobicity?. Protein Eng Design Selection 2004; 17: 57–66