
Abstract
Purpose: Heat shock induces DNA double-strand breaks (DSBs), but the precise mechanism of repairing heat-induced damage is unclear. Here, we investigated the DNA repair pathways involved in cell death induced by heat shock.
Materials and methods: B02, a specific inhibitor of human RAD51 (homologous recombination; HR), and NU7026, a specific inhibitor of DNA-PK (non-homologous end-joining; NHEJ), were used for survival assays of human cancer cell lines with different p53-gene status. Mouse embryonic fibroblasts (MEFs) lacking Lig4 (NHEJ) and/or Rad54 (HR) were used for survival assays and a phosphorylated histone H2AX at Ser139 (γH2AX) assay. MEFs lacking Rad51d (HR) were used for survival assays. SPD8 cells were used to measure HR frequency after heat shock.
Results: Human cancer cells were more sensitive to heat shock in the presence of B02 despite their p53-gene status, and the effect of B02 on heat sensitivity was specific to the G2 phase. Rad54-deficient MEFs were sensitive to heat shock and showed prolonged γH2AX signals following heat shock. Rad51d-deficient MEFs were also sensitive to heat shock. Moreover, heat shock-stimulated cells had increased HR.
Conclusions: The HR pathway plays an important role in the survival of mammalian cells against death induced by heat shock via the repair of heat-induced DNA DSBs.
Introduction
DNA double-strand breaks (DSBs) are the most lethal DNA damage and, if left unrepaired, can cause cell death or carcinogenesis [Citation1]. DSBs are repaired by two distinct pathways: non-homologous end-joining (NHEJ) and homologous recombination (HR) [Citation2]. NHEJ is active throughout all cell cycle phases and is mediated by DNA-dependent protein kinase (DNA-PK; consisting of Ku70/80 and its catalytic subunit [DNA-PKcs]), X-ray repair complementing defective repair in Chinese hamster cells 4 (XRCC4), DNA Ligase IV (Lig4) and XRCC4-like factor (XLF, also known as Cernunnos) [Citation3,Citation4]. In contrast, HR is only active when cells are in the late S and G2 phases of the cell cycle where homologous sequences in the sister chromatid are available as a repair template.
The HR process includes several distinct steps, starting with resection of DNA ends, mediated by exonuclease 1 (Exo1), meiotic recombination 11 (MRE11), C-terminal-binding protein (CtBP) interacting protein (CtIP, also known as retinoblastoma-binding protein 8 [RBBP8]) and replication protein A (RPA) [Citation5]. Homologous DNA pairing is mediated by RAD51 and RAD54 in an ATP-dependent manner [Citation6,Citation7]. The five RAD51 paralogs (RAD51B, RAD51C, RAD51D, XRCC2 and XRCC3) promote the binding of RAD51 to single-stranded DNA (ssDNA) and stabilisation of the nucleoprotein filament [Citation8,Citation9].
DSBs are generated by ionising radiation (IR) as well as a variety of DNA modifying reagents [Citation10]. Heat shock also induces DSBs, as shown by neutral comet assays and phosphorylated histone H2AX at Ser139 (γH2AX) foci formation [Citation11]. It has been proposed that heat shock leads to cell death through the induction of DNA DSBs [Citation12], but the pathway involved in repairing heat-induced damage is still to be elucidated [Citation13]. In this study, we investigated which DNA repair pathways preferentially repair heat-induced DNA DSBs. Here, we show that the HR pathway plays an important role in protecting cells by repairing heat-induced DNA DSBs.
Materials and methods
Cell culture
Cells were cultured in Dulbecco’s modified Eagle’s medium (D-MEM) with high glucose and L-glutamine and supplemented with 10% heat-inactivated foetal bovine serum, penicillin (100 U/ml), streptomycin (100 μg/ml) and HEPES (10 mM) at 37 °C in a humidified atmosphere of 5% CO2. Two human glioblastoma cell lines, A172 (wild-type (wt)p53) and U251MG (mutated (m)p53) were purchased from ATCC (Manassas, VA). H1299 (p53-deficient lung cancer cells) cells were provided by Dr Moshe Oren (Weizmann Institute of Science, Rehovot, Israel), and synchronisation of H1299 was performed as previously described [Citation11]. Mouse embryonic fibroblasts (MEFs) lacking Lig4 and/or Rad54 [Citation14] were provided by Dr Frederick W. Alt (Harvard Medical School, Boston, MA), and all of these cells were on a p53-deficient background. MEFs lacking Rad51d in a p53-deficient background were generated as described previously [Citation15]. SPD8 cells, derived from V79 Chinese hamster cells, were provided by Dr Thomas Helleday (Karolinska Institute, Stockholm, Sweden). SPD8 cells that have a mutation in hypoxanthine guanine phosphoribosyl transferase (hprt), which reverts to a functional hprt gene with HR, were used for the recombination assay as described previously [Citation16].
Inhibitors
Two inhibitors were used for colony formation assays: B02 [3-(Phenylmethyl)-2- [(1E)-2–(3-pyridinyl)ethenyl]-4(3H)-quinazolinone] (a specific inhibitor of human RAD51; EMD Millipore, Darmstadt, Germany) [Citation17] and NU7026 [2-(Morpholin-4-yl)-benzo[h]chromen-4-one] (a specific inhibitor of DNA-PK; EMD Millipore) [Citation18]. Dimethyl sulfoxide (DMSO) was used at a final concentration of 0.5% as a vehicle control. Both of these two inhibitors were used at 10 μM, unless otherwise mentioned. Cells were treated with NU7026, B02 or DMSO beginning at 6 h prior to heat shock. All inhibitors were removed 18 h after the beginning of heat shock, and the cells were cultured with fresh medium after heat shock.
Heat treatment
Culture flasks were immersed in a water bath (Thermominder EX; Taitec Co., Ltd., Saitama, Japan) maintained at a constant temperature. Under the present experimental conditions, no marked change in pH values was detected in the medium during this treatment.
Colony formation assay
Cell survival was measured using a standard colony-forming assay as previously described [Citation11]. Briefly, three T-25 flasks were used, and three independent experiments were repeated for each survival point. Human cancer cells and MEFs were trypsinized and seeded into flasks at defined densities several hours before and immediately after treatments, respectively. Colonies obtained after 7–10 days were fixed with methanol and stained with 2% Giemsa solution. Microscopic colonies composed of more than approximately 50 cells were counted as surviving cells. A sensitisation enhancement ratio (SER) for the inhibitors was calculated to quantify differences between survival curves. The SER was defined as the heating period resulting in a 10% survival rate (D10) divided by that for inhibitor-treated cells.
Cell synchronisation
After serum starvation for 28 h, G1-synchronised H1299 cells were cultured in fresh medium for 28 h. Then, the mitotic cells in the supernatant medium were collected by centrifugation. The mitotic cell pellet was dispersed in fresh medium. The cell-cycle phase of the synchronised mitotic cells at 2.5 h and 17 h after plating were G1 and G2 phase, respectively, as previously reported [Citation11].
H2AX phosphorylation assay by flow cytometry
The amounts of phosphorylated histone H2AX at Ser139 signal were measured by a flow cytometer as previously described [Citation11]. Briefly, cells were incubated with the anti-phospho-H2AX monoclonal antibody (JBW301; EMD Millipore; 1:300) and AlexaFluor 488-conjugated anti-mouse IgG secondary antibody (Thermo Fisher Scientific Inc., Waltham, MA; 1:400).
Recombination assay in SPD8 cells
Recombination assays were performed as previously described [Citation16]. Briefly, SPD8 cells were grown in the presence of 6-thioguanine to suppress spontaneous recombination. Cells were exposed to heat at 42 °C for the indicated times and recovered for 48 h. Revertant cells which were positive for hprt were selected by plating cells in the presence of HAsT (50 μM hypoxanthine, 10 μM L-azaserine, and 5 μM thymidine). Colonies were fixed and stained following 7 (for cloning efficiency) or 10 (for reversion) days of incubation.
Statistics
Data represent the mean ± standard deviation of three independent experiments. Student’s t-test was performed to calculate the statistical significance. *p < .05, **p < .01, ***p < .001.
Results
To determine whether heat sensitivity was enhanced by blocking the HR or NHEJ pathways, a specific inhibitor against the HR pathway of human RAD51 (B02) or the NHEJ pathway of DNA-PK (NU7026) was applied to human glioblastoma cell lines with different p53 status. Based on the dose responses of these two inhibitors when incubated with A172 (wtp53) or U251MG (mp53) cells (), 10 μM of inhibitors was used for all experiments. B02 at 10 μM resulted in a survival fraction of 0.64 ± 0.10 in A172 cells and 0.72 ± 0.15 in U251MG cells, and NU7026 at 10 μM resulted in a survival fraction of 0.92 ± 0.03 in A172 cells and 0.817 ± 0.653 in U251MG cells.
Figure 1. Effect of B02 or NU7026 on cell death induced by heat shock or X-rays in human cancer cell lines. Colony-forming survival assay. A172 (wtp53) and U251MG (mp53) cells were heat-treated at 44 °C for 10 min or irradiated with X-rays (2 Gy) in the presence of B02 or NU7026 at the indicated concentrations. Filled circles, inhibitor alone; grey triangles, heat shock; open triangles, X-rays. *, ** and *** represent p < .05, p < .01 and p < .001, respectively.
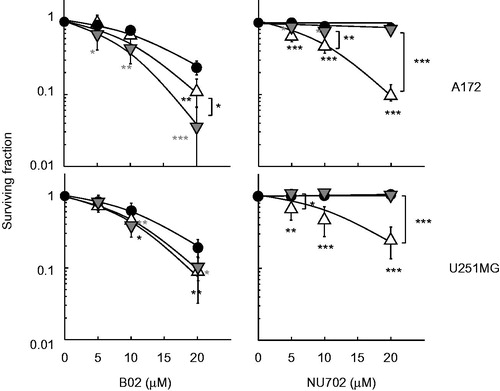
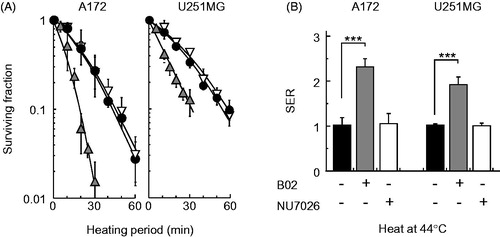
B02 enhanced heat-induced cell death in both the A172 and U251MG cell lines, but NU7026 had no effect on heat sensitivity (). The SERs for B02 were 2.30 ± 0.16 in A172 cells and 1.90 ± 0.15 in U251MG cells, and for NU7026, they were 1.03 ± 0.22 in A172 cells and 0.99 ± 0.05 in U251MG cells ().
Figure 2. Effect of B02 or NU7026 on heat-induced cell death in human cancer cell lines. (A) Colony-forming survival assay. A172 (wtp53) and U251MG (mp53) cells were heat-treated at 44 °C for the indicated times in the presence of B02 (10 μM) or NU7026 (10 μM). Filled circles, no inhibitor; grey triangles, B02; open triangles, NU7026. (B) SER of inhibitors at 10% survival rate. Filled columns, no inhibitor; grey columns, B02, open columns, NU7026. *** represents p < .001.
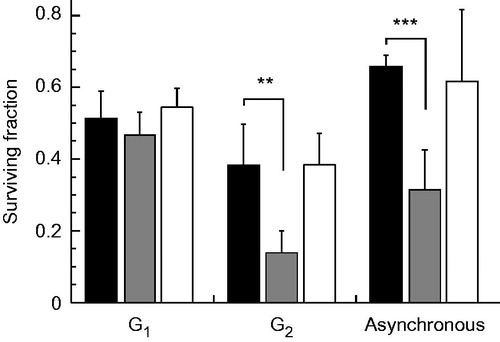
To clarify the effect of B02 or NU7026 on heat sensitivity for cells at different cell cycle phases, H1299 cells were synchronised at the G1 and G2 phases, exposed to heat at 44 °C for 20 min in the presence or absence of B02 (10 μM) or NU7026 (10 μM) and grown for colony formation. H1299 cells in the G2 phase were more sensitive to heat shock in the presence of B02, but the cells in the G1 phase showed no sensitivity (). Heat sensitivities of H1299 cells at the G2 phase without inhibitors and with NU7026 or B02 were 0.38 ± 0.11, 0.38 ± 0.09 and 0.11 ± 0.03, respectively, and those at the G1 phase were 0.51 ± 0.07, 0.54 ± 0.05 and 0.46 ± 0.06, respectively.
Figure 3. Effect of B02 or NU7026 on heat-induced cell death at different cell cycle phases. H1299 cells were synchronised at G1 and G2 phases, exposed to heat at 44 °C for 20 min in the presence of B02 (10 μM) or NU7026 (10 μM), and grown until the formation of colonies. Filled columns, no inhibitors; grey columns, B02; open columns, NU7026. ** and *** represent p < .01 and p < .001, respectively.
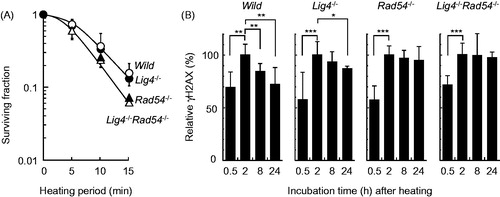
Next, using MEFs lacking either Lig4 (a NHEJ factor) and/or Rad54 (an HR factor), heat sensitivity was measured using a colony forming assay. Rad54−/− cells were more sensitive to heat shock, but Lig4−/− cells showed similar sensitivity to heat as the wild-type cells (). In addition, there was no additional sensitisation in Rad54−/−Lig4−/− cells compared with Rad54−/− cells. The enhancement ratio of Rad54-deficient cells compared with the wild-type cells calculated from the heating period corresponding to a D10 was 1.24. To test whether heat-induced damage was repaired in cells lacking the NHEJ (Lig4) and/or HR (Rad54) repair pathways, heat-induced phosphorylation of H2AX was monitored by flow cytometry. The wild-type and Lig4−/− cells showed a peak intensity at 2 h after heat-treatment, and Rad54−/− and Lig4−/− Rad54−/− cells showed prolonged γH2AX signals up to 24 h ().
Figure 4. Heat sensitivity and H2AX phosphorylation after heat shock in MEFs. (A) survival assay. The cells were exposed to heat at 45.5 °C for the indicated times and grown until the formation of colonies. Open circles, wild-type; filled circles, Lig4−/−; filled triangles, Rad54−/−; open triangles, Lig4−/−Rad54−/−. (B) H2AX phosphorylation assay. The cells were exposed to heat at 45.5 °C for 20 min and allowed to recover for the indicated times. *, ** and *** represent p < .05, p < .01 and p < .001, respectively.
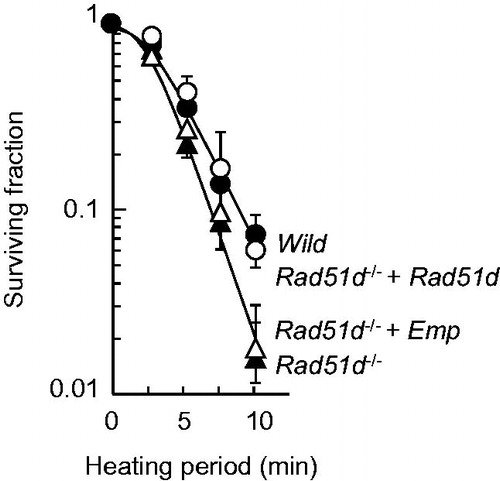
To confirm that HR deficiency leads to heat sensitivity, MEFs lacking Rad51d, one of the Rad51 paralogs, were used. Rad51d−/− and Rad51d−/− complemented with an empty vector were heat sensitive compared with Rad51d+/+ and Rad51d−/− complemented by a wild-type Rad51d expressing plasmid (). The enhancement ratio of Rad51d-deficient cells compared with the wild-type cells calculated from the heating period corresponding to a D10 was 1.31.
Figure 5. Heat sensitivity of Rad51d-proficient and Rad51d-deficient MEFs. The cells were exposed to heat at 45.5 °C for the indicated times and grown until the formation of colonies. Open circles, wild-type; filled circles, Rad51d−/−+ Rad51d; filled triangles, Rad51d−/−; open triangles, Rad51d−/− + empty vector.
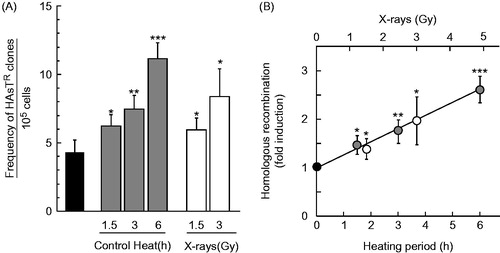
To detect HR events caused by heat shock, SPD8 cells were exposed to a temperature of 42 °C, and the frequency of HAsT-resistant clones was calculated. The frequency of spontaneous HAsT-resistant clones in untreated controls was 4.24 ± 0.93 (× 10−5). As a result, SPD8 cells with heat shock had more HAsT-resistant clones compared with untreated control cells (). The frequency of HAsT-resistant clones induced by heat shock at 42 °C for 1.5, 3 and 6 h was 6.22 ± 0.76, 7.42 ± 0.98 and 11.1 ± 1.12 (× 10−5), respectively. The fold induction of HR/heating period (min−1) at 42 °C was 4.45 × 10−3 (r = .997) (). X-rays were used as a positive control, and the frequency of HAsT-resistant clones induced by X-rays at 1.5 and 3 Gy were 5.90 ± 0.85 and 8.34 ± 2.03 (× 10−5), respectively. The fold induction of HR/irradiation dose (Gy−1) was 3.11 × 10−1 (r = .992) ().
Figure 6. Homologous recombination frequencies induced by heat shock or X-rays in SPD8 cells. The cells were exposed to heat at 42 °C for the indicated times or irradiated with X-rays at the indicated doses and used for recombination assays. (A) frequency of HAsTR clones per 105 cells. (B) fold induction of HR. Grey circles, heat shock; open circles, X-rays. *, ** and *** represent p < .05, p < .01 and p < .001, respectively.
Discussion
To study the role of HR factors during cellular response to heat shock, we have used several approaches. We used B02 in this study, which specifically disrupts DNA binding of RAD51 [Citation19]. The enhancement ratio from blocking RAD51 with B02 in response to heat shock was approximately twofold (), while the contribution of both RAD54 and RAD51D in response to heat shock was approximately 1.3-fold ( and ). Additionally, the enhancing effect of B02 on heat sensitivity was obvious in cells in the G2 phase but not the G1 phase of the cell cycle (). It is important to note that these results were obtained using different cell lines from different species (human glioblastoma cell lines and mouse embryonic fibroblasts) and that cell cycle properties, DNA repair efficacy, and the ratio of NHEJ and HR likely differ between these cell lines.
Based on the current study () and our previous work [Citation20], the contribution of RAD54 in response to heat shock and X-rays was approximately 1.3 and 1.8, respectively, while those of Lig4 were approximately 1.0 and 8.9, respectively. Lig4 (NHEJ) plays major roles in repairing DSBs and in the cellular resistance in response to IR regardless of the cell cycle phase; however, it seems to have no role in the response to heat shock throughout the cell cycle. It is also important to note that the Rad54-deficient chicken B-lymphocyte cell line DT40 was more sensitive to heat shock compared with the wild-type cell line [Citation21]. Their results and our current study suggest that the function of RAD54 in protecting cells against heat shock is conserved among vertebrates, at least between the mouse- and chicken-derived cell lines.
Following the detection of heat-induced DSBs using a neutral comet assay and γH2AX focus formation [Citation11], additional reports showed that heat shock induces H2AX phosphorylation [Citation22–24] and DSBs [Citation23]. An explanation for the difference between heat-induced damage and IR-induced damage might be that heat-induced damage causes DNA damage, alterations in higher-order chromatin structures [Citation25], and/or the masking of DNA damage [Citation26] by heat-labile proteins, whereas IR mainly induces DNA damage such as DSBs. Wild-type MEFs showed that heat-induced γH2AX reached a peak at approximately 2 h (). For IR, H2AX phosphorylation reached a peak approximately 15–30 min after DSB induction [Citation11]. It may be possible that heat-induced γH2AX originates in part from base damage and/or nicks at replication forks, which results in arrest or replication fork collapse. Furthermore, one-ended DBSs arising from collapsed replication forks are preferentially repaired by the HR pathway, but not by NHEJ [Citation27,Citation28]. The results showing that Rad54-deficient cells showed prolonged γH2AX signals up to 24 h () suggest that heat shock induces DNA damage (either DSBs, collapsed replication forks, or other types) resulting in the phosphorylation of H2AX and that heat-induced DNA DSBs are repaired by the RAD54-mediated pathway leading to a reduced amount of γH2AX. Together with the survival data, this study suggests that RAD54 protects cells from heat shock, likely through its role in HR.
One remaining question is in regards to what represents the heat-induced γH2AX foci of cells during S phase, stalled or collapsed replication forks? The characteristic of stalled replication forks is its readiness for quick recovery after the removal of the stress, but on the other hand, collapsed replication forks persist longer [Citation27]. Compared with other DSB-inducing stressors (i.e. IR), heat-induced γH2AX foci remain for a long period [Citation11]. A recent study showed that persistent DSBs induced by heat shock were generated by encountering DNA replication forks with single-stranded DNA breaks generated by DNA topoisomerase I [Citation29]. Together, these data indicate that heat-induced γH2AX foci of cells in the S phase represent collapsed replication forks, rather than stalled replication forks.
Heat shock causes cell cycle arrest at the S phase, possibly due to stalled replication forks [Citation23,Citation30]. Heat shock activates checkpoint kinase 1, ataxia telangiectasia and the Rad3-related protein, the kinases responsible for the arrest and collapse of replication forks [Citation31,Citation32]. RAD51 has two distinct roles: (1) restart of stalled replication forks and (2) HR repair of collapsed replication forks [Citation27]. In this study, we showed that RAD51/RAD54/RAD51D, factors involved in the HR pathway, protect cells from heat shock (). In addition, the recombination assay with SPD8 cells showed that heat shock generates DNA damage, which is required to be repaired by the HR pathway (). These results strongly suggest that the HR pathway repairs heat-induced DSBs rather than the NHEJ pathway, most likely at collapsed replication forks (one-ended DSB), which is distinct from the role of HR factors in restarting stalled replication forks.
The majority of DNA DSBs generated by IR are quickly repaired, predominantly by NHEJ; however, very small numbers of DSBs are complex and more difficult to repair, likely leading to cell death. Several lines of evidence show that factors involved in DSB repair are likely to be affected by heat shock, for example, Ku heterodimer (Ku70/Ku80) [Citation33–36], MRN complex [Citation37–39], breast cancer susceptibility gene 1 (BRCA1) [Citation40], BRCA2 [Citation41] and RAD51 [Citation41,Citation42]. Heat shock affects these proteins by causing denaturation, cytoplasmic translocation from the nucleus, and protein degradation by the proteasome pathway [Citation43]. These biological observations are currently proposed as possible reasons why IR combined with heat shock results in increased cell death with excessive unrepaired DSBs compared with IR alone [Citation44].
DSBs induced by heat shock are now widely accepted to be generated through biological processes such as DNA replication. Therefore, unlike the combination of IR and heat shock, heat shock alone does not generate large amounts of DSBs. This is probably why heat shocked cells can still perform critical biological functions, such as proliferation by restarting replication forks and DNA repair by protein synthesis or by the refolding of denatured protein. We propose that after recovery for a certain time following heat shock, HR factors, but not NHEJ factors, translocate to the nucleus and localise to DSBs to facilitate accurate repair, resulting in cell survival.
Conclusions
The current study shows that HR factors play an important role in protecting cells and that DNA DSBs induced by heat shock are repaired by the HR pathway. We report that heat shock has unique modes of action on DNA compared with other DNA damaging conditions and conclude that HR is the predominant pathway that repairs heat-induced DSBs.
Acknowledgements
The authors thank Dr Moshe Oren, Dr Frederick W. Alt and Dr Thomas Helleday for kindly providing cell lines used in this work and Dr Thomas Helleday for valuable advice and helpful criticisms of the manuscript.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the article.
Funding
This work was supported by grants from the Program to Disseminate the Tenure Tracking System of MEXT granted to Gunma University (to A.T.), JSPS KAKENHI [Grant Number: JP18390335, JP20390330, JP22659224, JP23390303, JP16K10341 to A.T., JP26463052 to A.K.], and the American Cancer Society, 10.13039/100000048 [RSG-030158–01-GMC to D.L.P.].
References
- Ohnishi T, Mori E, Takahashi A. (2009). DNA double-strand breaks: their production, recognition, and repair in eukaryotes. Mutat Res 669:8–12.
- Chapman JR, Taylor MR, Boulton SJ. (2012). Playing the end game: DNA double-strand break repair pathway choice. Mol Cell 47:497–510.
- Davis AJ, Chen DJ. (2013). DNA double strand break repair via non-homologous end-joining. Transl Cancer Res 2:130–43.
- Davis AJ, Chen BP, Chen DJ. (2014). DNA-PK: a dynamic enzyme in a versatile DSB repair pathway. DNA Repair (Amst) 17:21–9.
- Longhese MP, Bonetti D, Manfrini N, Clerici M. (2010). Mechanisms and regulation of DNA end resection. EMBO J 29:2864–74.
- Sigurdsson S, Van Komen S, Petukhova G, Sung P. (2002). Homologous DNA pairing by human recombination factors Rad51 and Rad54. J Biol Chem 277:42790–4.
- Baumann P, Benson FE, West SC. (1996). Human Rad51 protein promotes ATP-dependent homologous pairing and strand transfer reactions in vitro. Cell 87:757–66.
- Sigurdsson S, Van Komen S, Bussen W, et al. (2001). Mediator function of the human Rad51B-Rad51C complex in Rad51/RPA-catalyzed DNA strand exchange. Genes Dev 15:3308–18.
- Masson JY, Tarsounas MC, Stasiak AZ, et al. (2001). Identification and purification of two distinct complexes containing the five RAD51 paralogs. Genes Dev 15:3296–307.
- Takahashi A, Ohnishi T. (2005). Does gammaH2AX foci formation depend on the presence of DNA double strand breaks? Cancer Lett 229:171–9.
- Takahashi A, Matsumoto H, Nagayama K, et al. (2004). Evidence for the involvement of double-strand breaks in heat-induced cell killing. Cancer Res 64:8839–45.
- Mori E, Takahashi A, Ohnishi T. (2008). The biology of heat-induced DNA double-strand breaks. Thermal Med 24:39–50.
- Oei AL, Vriend LE, Crezee J, et al. (2015). Effects of hyperthermia on DNA repair pathways: one treatment to inhibit them all. Radiat Oncol 10:165.
- Mills KD, Ferguson DO, Essers J, et al. (2004). Rad54 and DNA Ligase IV cooperate to maintain mammalian chromatid stability. Genes Dev 18:1283–92.
- Smiraldo PG, Gruver AM, Osborn JC, Pittman DL. (2005). Extensive chromosomal instability in Rad51d-deficient mouse cells. Cancer Res 65:2089–96.
- Helleday T, Arnaudeau C, Jenssen D. (1998). A partial hprt gene duplication generated by non-homologous recombination in V79 Chinese hamster cells is eliminated by homologous recombination. J Mol Biol 279:687–94.
- Huang F, Motlekar NA, Burgwin CM, et al. (2011). Identification of specific inhibitors of human RAD51 recombinase using high-throughput screening. ACS Chem Biol 6:628–35.
- Veuger SJ, Curtin NJ, Richardson CJ, et al. (2003). Radiosensitization and DNA repair inhibition by the combined use of novel inhibitors of DNA-dependent protein kinase and poly(ADP-ribose) polymerase-1. Cancer Res 63:6008–15.
- Huang F, Mazina OM, Zentner IJ, et al. (2012). Inhibition of homologous recombination in human cells by targeting RAD51 recombinase. J Med Chem 55:3011–20.
- Takahashi A, Kubo M, Ma H, et al. (2014). Nonhomologous end-joining repair plays a more important role than homologous recombination repair in defining radiosensitivity after exposure to high-LET radiation. Radiat Res 182:338–44.
- Yin HL, Suzuki Y, Matsumoto Y, et al. (2004). Radiosensitization by hyperthermia in the chicken B-lymphocyte cell line DT40 and its derivatives lacking nonhomologous end joining and/or homologous recombination pathways of DNA double-strand break repair. Radiat Res 162:433–41.
- Kaneko H, Igarashi K, Kataoka K, Miura M. (2005). Heat shock induces phosphorylation of histone H2AX in mammalian cells. Biochem Biophys Res Commun 328:1101–6.
- Velichko AK, Petrova NV, Kantidze OL, Razin SV. (2012). Dual effect of heat shock on DNA replication and genome integrity. Mol Biol Cell 23:3450–60.
- Takahashi A, Mori E, Somakos GI, et al. (2008). Heat induces gammaH2AX foci formation in mammalian cells. Mutat Res 656:88–92.
- Kampinga HH, Dikomey E. (2001). Hyperthermic radiosensitization: mode of action and clinical relevance. Int J Radiat Biol 77:399–408.
- RotiRoti JL. (2008). Cellular responses to hyperthermia (40-46 °C): Cell killing and molecular events. Int J Hyperthermia 24:3–15.
- Petermann E, Orta ML, Issaeva N, et al. (2010). Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell 37:492–502.
- Britton S, Coates J, Jackson SP. (2013). A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. J Cell Biol 202:579–95.
- Velichko AK, Petrova NV, Razin SV, Kantidze OL. (2015). Mechanism of heat stress-induced cellular senescence elucidates the exclusive vulnerability of early S-phase cells to mild genotoxic stress. Nucleic Acids Res 43:6309–20.
- Wong RS, Thompson LL, Dewey WC. (1988). Recovery from effects of heat on DNA synthesis in Chinese hamster ovary cells. Radiat Res 114:125–37.
- Furusawa Y, Iizumi T, Fujiwara Y, et al. (2012). Inhibition of checkpoint kinase 1 abrogates G2/M checkpoint activation and promotes apoptosis under heat stress. Apoptosis 17:102–12.
- Tuul M, Kitao H, Iimori M, et al. (2013). Rad9, Rad17, TopBP1 and claspin play essential roles in heat-induced activation of ATR kinase and heat tolerance. PLoS One 8:e55361.
- Burgman P, Ouyang H, Peterson S, et al. (1997). Heat inactivation of Ku autoantigen: possible role in hyperthermic radiosensitization. Cancer Res 57:2847–50.
- Ihara M, Suwa A, Komatsu K, et al. (1999). Heat sensitivity of double-stranded DNA-dependent protein kinase (DNA-PK) activity. Int J Radiat Biol 75:253–8.
- Beck BD, Dynlacht JR. (2001). Heat-induced aggregation of XRCC5 (Ku80) in nontolerant and thermotolerant cells. Radiat Res 156:767–74.
- Ihara M, Takeshita S, Okaichi K, et al. (2014). Heat exposure enhances radiosensitivity by depressing DNA-PK kinase activity during double strand break repair. Int J Hyperthermia 30:102–9.
- Zhu WG, Seno JD, Beck BD, Dynlacht JR. (2001). Translocation of MRE11 from the nucleus to the cytoplasm as a mechanism of radiosensitization by heat. Radiat Res 156:95–102.
- Seno JD, Dynlacht JR. (2004). Intracellular redistribution and modification of proteins of the Mre11/Rad50/Nbs1 DNA repair complex following irradiation and heat-shock. J Cell Physiol 199:157–70.
- Takahashi A, Mori E, Ohnishi T. (2010). The foci of DNA double strand break-recognition proteins localize with gammaH2AX after heat treatment. J Radiat Res 51:91–5.
- Xian Ma Y, Fan S, Xiong J, et al. (2003). Role of BRCA1 in heat shock response. Oncogene 22:10–27.
- Krawczyk PM, Eppink B, Essers J, et al. (2011). Mild hyperthermia inhibits homologous recombination, induces BRCA2 degradation, and sensitizes cancer cells to poly(ADP-ribose) polymerase-1 inhibition. Proc Natl Acad Sci USA 108:9851–6.
- Genet SC, Fujii Y, Maeda J, et al. (2013). Hyperthermia inhibits homologous recombination repair and sensitizes cells to ionizing radiation in a time- and temperature-dependent manner. J Cell Physiol 228:1473–81.
- van den Tempel N, Horsman MR, Kanaar R. (2016). Improving efficacy of hyperthermia in oncology by exploiting biological mechanisms. Int J Hyperthermia 32:446–54.
- Horsman MR. (2016). Realistic biological approaches for improving thermoradiotherapy. Int J Hyperthermia 32:14–22.