
Abstract
Background: Agents targeting HSP90 and GRP94 are seldom tested in stressed contexts such as heat shock (HS) or the unfolded protein response (UPR). Tumor stress often activates HSPs and the UPR as pro-survival mechanisms. This begs the question of stress effects on chemotherapeutic efficacy, particularly with drugs targeting chaperones such as HSP90 or GRP94. We tested the utility of several HSP90 inhibitors, including PU-H71 (targeting GRP94), on a primary canine lung cancer line under HS/UPR stress compared to control conditions.
Methods: We cultured canine bronchoalveolar adenocarcinoma cells that showed high endogenous HSP90 and GRP94 expression; these levels substantially increased upon HS or UPR induction. We treated cells with HSP90 inhibitors 17-DMAG, 17-AAG or PU-H71 under standard conditions, HS or UPR. Cell viability/survival was assayed. Antibody arrays measured intracellular signalling and apoptosis profiles.
Results: HS and UPR had varying effects on cells treated with different HSP90 inhibitors; in particular, HS and UPR promoted resistance to inhibitors in short-term assays, but combinations of UPR stress and PU-H571 showed potent cytotoxic activity in longer-term assays. Array data indicated altered signalling pathways, with apoptotic and pro-survival implications. UPR induction + dual targeting of HSP90 and GRP94 swayed the balance toward apoptosis.
Conclusion: Cellular stresses, endemic to tumors, or interventionally inducible, can deflect or enhance chemo-efficacy, particularly with chaperone-targeting drugs. Stress is likely not held accountable when testing new pharmacologics or assessing currently-used drugs. A better understanding of stress impacts on drug activities should be critical in improving therapeutic targeting and in discerning mechanisms of drug resistance.
Introduction
Tumor cells are seemingly perpetually stressed because of their rapid cell division in inhospitable environments, often of the tumor's own making [Citation1,Citation2]. The unregulated proliferation is energy intensive and metabolically disruptive, frequently leaving the tumor in a hypoxic state amidst nutrient deprivation [Citation3]. However, tumors may be able to use this constant state of stress to their advantage, as upregulation of chaperones/heat shock (HS) proteins produced under stress accomplishes similar functions in overcoming protein folding diseases [Citation4]. These activities would aid in tumor growth, survival and progression.
The HS response has been long documented in cancer, with awareness of both the utility it provides for tumor progression [Citation1,Citation2,Citation5] as well as the potential for therapeutic intervention [Citation2,Citation6]. An additional cellular stress mechanism, the unfolded protein response (UPR), is an endoplasmic reticulum (ER)-based response to conditions that may impede or detrimentally affect that organelle’s function, generally recognized by the accumulation of unfolded or misfolded proteins within the ER lumen [Citation7,Citation8]. The response includes changes in the transcriptional and translational landscapes of stressed cells that lead to recovery of proteostasis or to apoptotic cell death if the cells cannot overcome the stress. However, the UPR appears as a fixture in many cancers, implying a survival benefit from the tumor’s perspective [Citation9].
The premise that cancers survive or even thrive on stress responses suggests that targeting those responses or elements within them may be valuable anti-cancer strategies, particularly the targeting of chaperone activities [Citation2,Citation10,Citation11]. Notably, heat shock protein 90 (HSP90, HSP90A family) and its ER paralog glucose regulated protein 94 (GRP94, HSP90B1) are considered high-value targets for pharmacologic development [Citation2,Citation12–18]. Both of these proteins are important players in the HS and UPRs; their upregulated expression in tumors begs the question, are these proteins viable targets in stressed tumors? Testing of therapeutic agents on stressed cells is rare [Citation19], but may be an important parameter in assessing anti-cancer effects, especially if induction of stress may be part of the therapeutic mode.
In this study, we tested the effects of three HSP90 inhibitors, 17-DMAG, 17-AAG and PU-H71 (PU-H71 also targets GRP94 [Citation20]), on a novel primary canine lung adenocarcinoma cell line. We conducted testing under unstressed (control), heat-stressed and UPR stress conditions. We analyzed phospho-signaling patterns and cell viability before focusing on PU-H71 for short- and long-term soft agar growth and apoptotic impacts. Our results show that at least for this cell line, heat stress actually protects cells from HSP90 inhibition in both short- and long-term treatments, while UPR induction initially increases proliferation in the presence of drug, but later leads to profound growth inhibition. These data suggest that stressors do matter when assessing drug impacts on cells, particularly if the drugs may be targeting proteins involved in the stress responses. This sets the stage for rational combination therapies that may induce effective stresses or that may utilize existing stresses (i.e. for a tumor in vivo) to improve therapeutic efficacy of available chemotherapies.
Materials and methods
Normal lung and tumor tissue
Primary tumor tissue, and recurrent/metastatic tumor tissues were obtained as described [Citation21]. Normal lung tissue was obtained from Dr. Christine Olver, DVM, Colorado State University.
Cell culture
STAR cells (a canine bronchoalveolar adenocarcinoma cell line) were cultured as described [Citation21] with standard conditions at 37 °C, 5% CO2. For HS conditions, cell culture medium was replaced with pre-warmed (42 °C) medium, and cells were incubated for 2 h at 42 °C, 5% CO2. At the end of the incubation, spent medium was removed and replaced with medium that was 37 °C, and cells were allowed to recover for 24 h. To induce the UPR, cell medium was replaced with sterile-filtered medium containing 1 mM dithiothreitol (DTT, Sigma-Aldrich, St. Louis, MO) for 4 h (37 °C, 5% CO2). At the end of the incubation, cells were washed with PBS (Gibco Life Technologies/ThermoFisher, Waltham, MA) and medium was replaced with standard medium for 24 h recovery. For treatment with HSP90 inhibitors, cells were treated with DMSO (Sigma, St. Louis, MO) carrier or were treated with geldanamycin analogues 17-dimethylamino-geldanamycin (17-DMAG), 17-allylamino-demethoxygeldamycin (17-AAG) (both from Enzo/Biomol, Farmingdale, NY) or a purine-based analogue, 8-[(6-iodo-1,3-benzodioxol-5-yl)sulfanyl]-9-[3-(propan-2-ylamino)propyl]purin-6-amine (PU-H71) (Tocris, Bristol, UK) with doses and times as described in the text and figure legends.
Microscopy
Cells in culture were visualized and imaged by phase-contrast microscopy using an Olympus inverted IX73 microscope with a 20X Plan Achromat lens (Tokyo, Japan).
Western blots
Lung and tumor tissues were ground to a powder in liquid nitrogen, and proteins were extracted by overnight rocking in radioimmune precipitation assay (RIPA) buffer (Sigma, St. Louis, MO) with protease and phosphatase inhibitors included (Roche, Indianapolis, IN). Debris was pelleted (16 000×g, 30 min, 4 °C), and lysates were quantified by BCA assay (Pierce ThermoFisher, Waltham, MA). Cell lysates were prepared as described, and Western blots were run as described [Citation22,Citation23]. Antibodies for HS proteins included anti-HSP110 mAb (cat # 610511, BD Biosciences, Franklin Lakes, NJ), used at 1:500 dilution; anti-HSP90 mAb (cat # 610419, BD Biosciences, San Jose, CA), used at 1:500 dilution; anti-HSP70/HSP72 mAb (clone C92F3A-5, ADI-SPA-810, Enzo, Farmingdale, NY), used at 1:1000 dilution; anti-HSC70/HSP73 (ADI-SPA-757, Enzo, Farmingdale, NY), used at 1:1000 dilution; anti-HSP60 Ab (D307, cat # 4870, Cell Signaling, Danvers, MA), used 1 1:500 dilution; anti-HspBP1 mAb (clone MAB-10201, Orbigen, San Diego, CA), used at 1:500 dilution; anti-aB crystallin mAb (clone 1B6.1-3G4, ADI-SPA-222, Enzo, Farmingdale, NY), used at 1:500 dilution; anti-HSF1 Ab (H-311, cat # sc-9144, Santa Cruz Biotechnology, Santa Cruz, CA), used at 1:200 dilution; anti-GRP75/Mortalin mAb (clone 30A5, ADI-SPA-225, Enzo, Farmingdale, NY), used at 1:500 dilution; anti-TRAPd mAb (clone C-6, cat # sc-376706), used at 1:500 dilution. Antibodies for ER chaperones and the UPR, including AKT, pAKT, SAPK/JNK, pSAPK/JNK, were used as described previously [Citation22]. Antibody to PDI, PERK and ERO1L were from Cell Signaling (ER Stress Antibody Sampler Kit #9956), as was a rabbit mAb to ATF4 (D4B8, #11815), all used at 1:500 dilutions. Anti-EGFR mAb was from Novus (clone 29-1; Littleton, CO) used at 1:500 dilution. Anti-actin and anti-tubulin antibodies used for control staining were from Sigma, St. Louis, MO [Citation22,Citation23].
ER stress transcription factor profiling assay
Canine lung, and primary and metastatic tumor samples underwent nuclear extractions with a nuclear extraction kit (ThermoFisher, NE-PER kit, Waltham, MA). UPR/ER stress transcription factors were profiled using the ER Stress (UPR) TF Activation Profiling Plate Array (Signosis, Santa Clara, CA) following kit instructions. Wells were developed using SuperSignal West Femto substrate (ThermoFisher, Waltham, MA). Readouts were obtained using a FluorChem Q Image III device (Protein Simple, Santa Clara, CA). Fold change was determined for the tumor readouts compared to lung readouts, the latter set = 0.
MTS assay
MTS assays of drug-treated cells were performed using the Cell Titer 96 AQueous One Solution MTS assay (Promega, Madison, WI).
Intracellular signaling array
Changes in phosphorylation and cleavage status of 18 signaling molecules were assessed using a PathScan Intracellular Signaling Array (Cell Signaling, Danvers, MA). Cells were treated as described in the text and figure legends. After treatment, cells were harvested by centrifugation (1100×g, 5 min); supernatant was aspirated and cell pellets rinsed twice in PBS. Cells were lysed in buffers provided in the kit, and arrays were probed according to the manufacturer instructions. Arrays were developed as described [Citation22,Citation24].
Clonogenic/soft agar assay
STAR cells were grown in NBA to a logarithmic growth phase. For clonogenic assays, we used the Cyto-select 96-well Tumor Sensitivity Assay (cat# CBA-150, Cell Biolabs, San Diego, CA), according to the recommended guidelines. Prior to soft agar plating, STAR cells were at first left untreated, heat shocked (42 °C, 2 h), or treated with 1 mM DTT for 4 h (UPR induction). Cells were then washed in fresh media and recovered for 24 h. Cells were then treated (or not) with 5 nM PU-H71 for 24 h. Drug was removed and cells were replated in soft agar in a 24 well plate. For short-term exposure, no additional drug was added, but for long-term exposure, 5 nM PU-H71 was added by media replenishment every day for eight days. The agar matrix was solubilized, and cell viability was determined by an MTT assay according to manufacturer’s instructions.
Apoptosis protein array
STAR cells were left untreated, UPR induced and/or treated with PU-H71 as described above. After 24 h, cells were lysed and lysates were used to probe an R&D Apoptosis Antibody Array Kit (Catalog # ARY009) (Minneapolis, MN). The array was developed as described [Citation22,Citation24].
Ingenuity pathway analysis
Data from the apoptosis array were converted to log2 ratios (treated/controls) and entered into Ingenuity Pathway Analysis (IPA, http://www.ingenuity.com/). Data were analyzed by Comparison Analysis, with Networks (interactomes) identified as significant. Data were also analyzed in Comparison Analysis by Diseases and Biofunctions, quantified by z-activation scores (50 categories) or listed by the top 15 Canonical Pathways derived from the dynamics of the data.
Additional statistics
Other than IPA-developed statistical analyses, in other cases comparisons were made by Student t-test using Excel.
Results
STAR cell line
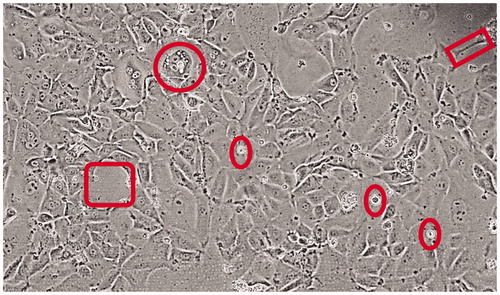
The STAR cell line was generated from a recurrent (metastatic) tumor, arising from a primary tumor originally described as a grade III bronchioalveolar adenocarcinoma [Citation21]. The line grew in serum-free Neurobasal A medium as a tenaciously adherent line, with floating/suspension elements. Those suspension cells, if passaged separately, can recapitulate the same adherent/suspension properties of the original cell line. The micrograph in displays traits of the cell line showing overall lung epithelial features, but with larger gaps between cells (box), cells with dendritic-like connections (rectangle) and the aforementioned floating cells (ovals). The circle shows a larger, cuboidal cell with numerous vacuolar intracellular structures. The diversity of cell types indicates a complex, non-clonal nature likely reminiscent of the original tumor.
Figure 1. STAR cells in culture – morphology. Micrograph shows the diversity of STAR cells, with a somewhat cuboidal appearance of lung epithelium but with frequent large gaps between cells and clusters (square to the left) and lengthy dendritic-like connections to neighboring cells (rectangle to the upper right). The circle denotes a large cuboidal cell with numerous vacuolar intracellular structures, and ovals point out (viable) floating cells.
Tumor expression of stress proteins
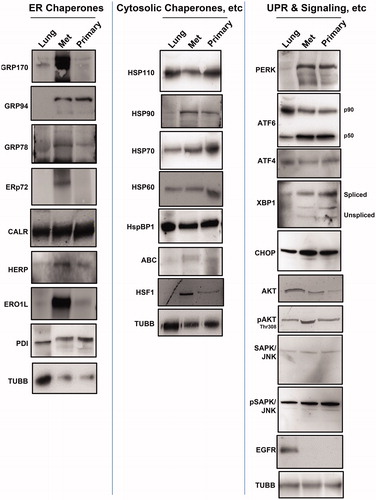
The Western blots in show normal (canine) lung ("Lung") lysate, recurrent/metastatic (“Met”) tumor lysate and lysates prepared from the primary (“Primary”) tumor, probed with various antibodies against stress-related proteins. We see a general, but not universal, increased chaperone expression of the ER chaperones (, left panel) in the recurrent and primary tumors compared to normal lung. This is especially notable for GRP170/HYOU1, GRP94/HSP90B1, ERp72/PDIA4, HERP/HERPUD1 and ERO1L. Western blots probing for cytosolic chaperones, co-chaperones and transcription factors (, middle panel) show relatively high expression levels in the tumors of HSP90/HSP90A, HSP70/HSPA1A/B, alpha B crystallin/CRYAB/HSPB5 and heat shock factor 1 (HSF1), one of the major stress transcription factors. Certain components of the UPR transduction and downstream transcriptional drivers (, right panel) show higher expression in tumors compared to normal lung, with the transducer PERK clearly elevated, as are the processed active forms of ATF6 (i.e. the p50 cleavage product) and XBP1 (the higher molecular weight spliced form). CHOP/GADD153, downstream of PERK activation and often associated with eventual apoptosis [Citation12], shows some change in the tumors compared to lung, and there are elevated levels of phospho-AKT and phospho-SAPK/JNK. Surprisingly, EGFR, while demonstrated to have none of the mutations associated with lung cancer [Citation21], shows little or no expression compared to lung. These results indicate that the HS and ER stress pathways are activated in both the primary and recurrent/metastatic tumors compared to normal lung tissue.
Figure 2. Chaperone and UPR and related signaling molecule expression in canine lung and tumors. Western blots of normal canine lung lysate (“Lung”) STAR metastatic/recurrent (“Met”) and original primary (“Primary”) tumor lysates were probed for the ER chaperones (left panel) listed, for the (typically) cytosolic chaperones (middle panel) listed, and for proteins involved in the UPR and downstream signaling (right panel). Anti-TUBB (beta tubulin) staining is used as an internal loading control.
We performed an additional stress transcription factor evaluation with a targeted DNA binding array, comparing fold changes from the recurrent and primary tumor tissues to lung (Supplemental Figure 1). All of the factors measured were elevated – in some cases, dramatically so – further suggesting that the UPR is highly upregulated in both the primary and metastatic tumor tissues.
STAR cell line expression of stress proteins
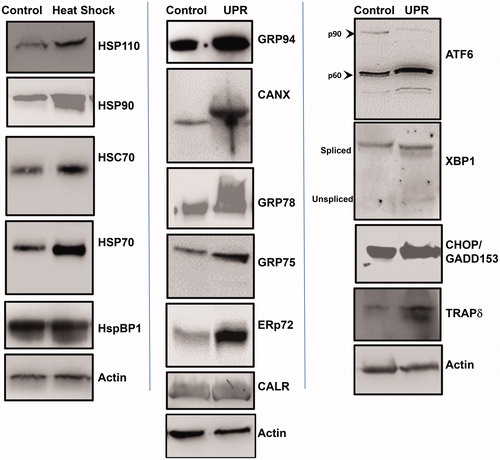
STAR cells were cultured under normal, unstressed conditions (“Control”), under HS conditions (“Heat Shock”; 42 °C for 2 h, with 24 h recovery), or with induction of the UPR (“UPR”; 1 mM DTT for 4 h, with 24 h recovery). Cells were lysed and proteins were separated by SDS-PAGE for Western blotting (). Following HS, STAR cells clearly upregulate HSP110/HSPH1, HSP90, HSC70/HSPA10, HSP70 and show slight upregulation of the HSP70 co-chaperone HspBP1 [Citation25] (left panel). With UPR stress we saw increased expression of GRP94, calnexin/CANX, GRP78/HSPA5, the mitochondrial GRP75/HSPA9, ERp72 and calreticulin/CALR (middle panel). The UPR transducer ATF6 shows some increase in the active (p50) form upon UPR induction compared to control. However, with UPR stress there is an overall increase in both spliced (active) and unspliced forms of the downstream transcription factor XBP1. There is notable upregulation of CHOP/GADD153 with the UPR, as well as the translocon component TRAP delta/TRAPD/SSR4 (right panel).
Figure 3. Heat shock and induction of the unfolded protein response (UPR) in STAR cell line. A cell line was derived from the metastatic lung tumor, which was subjected to heat shock (42 °C, 2 h) or 1 mM DTT, 4 h (to induce the UPR). Cells recovered for 24 h, and were lysed. Western blots were performed to compare protein levels in untreated (“Control”) cell lysates vs. treated (“Heat Shock”) (“UPR”) cell lysates. Left panel shows heat shock protein responses; middle panel shows ER chaperone protein responses; right panel shows UPR components. Actin probe was used as an internal loading control.
The generally upregulated components of the HS and UPR systems suggest a pronounced stress landscape for the solid tumors and the cell line generated from the metastatic tumor. In particular, for the tissue culture cells, the upregulation of HSP90 following HS, and GRP94 following UPR induction, along with their high expression in the tumors themselves, suggested that these might be pharmacologically targetable moieties.
Effects of HSP90 inhibitors on STAR cells
Supplemental Figure 2 shows the viability of STAR cells treated with various HSP90 inhibitors. Cells were treated with DMSO carrier (“Cont”) or were treated with geldanamycin analogues 17-dimethylamino-geldanamycin (“17-DMAG”), 17-allylamino-demethoxygeldamycin (“17-AAG”) or a purine-based analogue, 8-[(6-iodo-1,3-benzodioxol-5-yl)sulfanyl]-9-[3-(propan-2-ylamino)propyl]purin-6-amine ("PU-H71"), for 24 h. Cell viability was determined by MTS assay. All concentrations of 17-DMAG and PU-H71 had significant impacts on cell viability (compared to control cells), while at this time point and drug concentrations, 17-AAG had no discernable effect. We chose to use 5 nM of each of the inhibitors in the ensuing experiments because for two of the three drugs, it had significant, but not total, reduction in viability, which allowed for further assays. We chose to use the same concentration for 17-AAG treatments for the purpose of a functionally inactive control.
Changes in intracellular signaling molecule phosphorylation or cleavage states following stress and treatment with HSP90 inhibitors
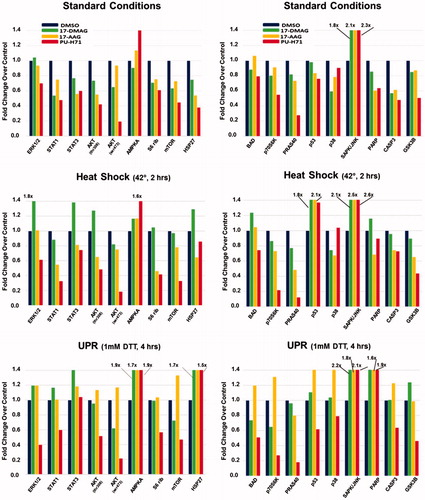
To examine signaling pathway changes (some involving HSP90 client proteins) in STAR cells in the context of stresses and HSP90 inhibition, we treated cells with HSP90 inhibitors following non-stressed (standard) conditions, following HS, or following UPR induction (; an example of the developed array itself with some protein spots annotated is shown in Supplemental Figure 3). Cells were kept in standard conditions, were heat-shocked at 42 °C for 2 h, or were treated with 1 mM DTT for 4 h. After 24 h recovery, cells were treated with 5 nM HSP90 inhibitors for 8 h (>80% of the cells were alive at that dose and time point, data not shown). Cells were then lysed and incubated on an intracellular signaling (antibody) array that measures phospho-protein status (and some cleavage products) to generate a signaling profile. Across all the conditions, PU-H71 in general has the greatest impact on reduced phosphorylation status compared to the effects of the other drugs. We note here that the following proteins in the array are considered HSP90 clients (https://www.picard.ch/downloads/Hsp90interactors.pdf): AKT; AMPKA; mTOR; p70S6K; p53; p38; SAPK/JNK; GSK3B.
Figure 4. Altered signaling profiles in STAR cells pre-treated with stress, followed by HSP90 inhibitors. STAR cells were kept at typical culture conditions (“Standard Conditions”, 37 °C, 5% CO2), or were heat shocked (“Heat Shock”, 42 °C, 2 h), or were subjected to UPR induction (“UPR”, 1 mM DTT, 4 h). Cells recovered for 24 h, and were then treated with 5 nM HSP90 inhibitors for 8 h. Cells were then lysed, and lysates exposed to an intracellular signaling array (antibodies that recognize phosphorylation of specific proteins, or cleavage products). Spot intensities were quantified and compared to controls for each condition (control values set = 1).
Under standard conditions
Breaking the results down by pre-treatment conditions, we see that three major signaling pathways (MAPK/ERK, JAK/STAT and PI3K/AKT) show reduced phosphorylated components with PU-H71 treatment, with similar, but not identical, patterns with 17-DMAG (where ERK1/2 phosphorylation is not reduced) and 17-AAG treatments. Curiously, these three pathways are often downstream of receptor tyrosine kinases such as EGFR [Citation26], which presumably does not play a role in this tumor (). S6 ribosomal protein (RPS6) phosphorylation is also reduced, possibly due to reduced activities of ERK and AKT pathways. The latter also involves the mTOR complex mTORC1 – and here, we also see reductions in phospho-mTOR and phospho-PRAS40/AKT1S1 (especially with PU-H71 treatment), which signal to RPS6 through p70S6K, whose phosphorylation is also reduced. It is not entirely clear if inhibited phosphorylation/activities of these signaling components drive reduced pRPS6, or if they shift the equilibrium in favor of the phosphatase PP1, which de-phosphorylates RPS6 [Citation27]. Also, AKT, mTOR and p70S6K are all clients of HSP90 and may themselves be degraded upon HSP90 inhibitor treatment. HSP27/HSPB1 phosphorylation is critical to its function (and oncogenic activity in cancer cells) [Citation28]. Numerous kinases have known direct or downstream interactions with HSP27, including p38 MAPK (with 17-DMAG producing the most de-phosphorylation and PU-H71 the least), AKT and p70S6K [Citation29], so their reduced phosphorylation status may lead to reduced phospho-HSP27 levels. Also, the reduced kinase activity could result in a shift in equilibrium towards phosphatase activity via the protein phosphatase PP2A [Citation29]. Bcl-2 associated agonist of cell death (BAD) is BCL2 family member whose phosphorylation state results in pro- or anti-apoptotic activities (reviewed in [Citation30]). Numerous signaling pathways phosphorylate BAD, such as PI3K/AKT, RAS/RAF (and therefore, p38 MAPK) mTOR/S6K, etc. Phospho-BAD is sequestered in the cytosol by 14-3-3 family members, and is functionally anti-apoptotic; the hypophosphorylated form (perhaps generated by protein phosphatase PP1) binds anti-apoptotic BCL2 members Bcl-xl/BCL2L1 and BCL2, allowing apoptosis to progress [Citation31]. Curiously, 17-AAG treatment slightly increased pBAD status, while other inhibitors reduced it. Interestingly, non-phosphorylated BAD binds p53 in the cytosol, and that complex engages BAK at the mitochondrial surface, promoting apoptosis [Citation32]. The impact phosphorylation status of p53 is complex, but serine 15 phosphorylation likely recruits transcriptional co-activators [Citation33]. There are only slight reductions of p53 phosphorylation with HSP90 inhibitor treatment (very minimal with 17-DMAG), suggesting that in the unstressed cells, p53 roles remain unchanged with drug treatment. All three inhibitor treatments promote CASP3 cleavage, which could presumably lead to PARP cleavage en route to an apoptotic cascade [Citation34], but 17-DMAG appears to affect this the least. Among other kinases, AKT and p70S6K may phosphorylate GSK3B, which is inhibitory to the latter [Citation35]; thus, reduced activity of those kinases may reduce pGSK3B (or, as a client of HSP90, GSK3B itself may be subject to degradation). With over 100 potential substrates, activation of GSK3B may critically alter any number of pathways [Citation36]. Only PU-H71 seemed to substantially decrease pGSK3B.
Of proteins whose phosphorylation status conspicuously increased following HSP90 inhibitor treatment, AMPKA (alpha subunit of AMPK) can be activated during a variety of stresses, including HS [Citation37]. Under conditions of stasis, HSF1 is in a multi-chaperone complex (including HSP90), and treatment with HSP90 inhibitors cause its release from the complex [Citation38], essentially mimicking the HS response, which could lead to AMPKA activation. 17-AAG somewhat increased pAMPKA, while PU-H71 potently increased pAMPKA. SAPK/JNK belongs to a family of stress kinases with potentially both pro- and anti-apoptotic functions [Citation39]. It may be recruited to lipid rafts under various circumstances [Citation40,Citation41], where HSP90 may chaperone JNK-containing complexes. All three inhibitor compounds profoundly increased pSAPK/JNK, suggesting that these may be pro-apoptotic events.
Under HS conditions
Cells were heat shocked and allowed to recover before drug treatment; compared to "standard" conditions, the general trends of reduced phosphorylation and increased cleavage of the assayed proteins continue for 17-AAG and PU-H71 treatments (with certain exceptions), but the pattern is quite different for 17-DMAG treatment. With that drug, phospho-states of ERK1/2, STAT3, AKTS308, AMPKA, RPS6, mTOR, HSP27 and p53 increased, along with increased PARP and CASP3 cleavage. For the other two drug treatments, pAMPKA remains relatively high, as does pSAPK/JNK (both stress-responsive kinases [Citation37,Citation39]), but all three inhibitors promote a spike in phospho-p53. Curiously, previous studies have reported that heat stress promotes HSP90 interactions with increased phosphorylation of p53, but those can be abrogated with HSP90 inhibitors [Citation42].
Under UPR conditions
Cells were treated with the reducing agent DTT to generate misfolded proteins in the ER, and then allowed to recover, followed by drug treatment. For 17-DMAG treatment, compared to “standard” conditions, we saw increased phosphorylation of ERK1/2, STATs, AKTS308, AMPKA, RPS6, HSP27, PRAS40, p53, p38 and GSK3B, along with enhanced cleavage of PARP and CASP3. Phospho-BAD and phospho-p70S6K were reduced. Compared to “heat shock” conditions, pSTAT1, pAMPKA, pHSP27, pPRSA40, phospho-p38 and pGSK3B are increased, as is PARP cleavage, while expression of pAKT, p-mTOR, pBAD and phospho-p70S6K are decreased. For 17-AAG treatments, compared to “standard” conditions, all the phospho-protein statuses assayed were increased except for PRAS40 and SAPK/JNK, which remained approximately the same, and pGSK3B may have decreased slightly. PARP and CASP3 cleavage were also both higher. The same trends hold true when compared to the “heat shock” condition, although phospho-p53 may not be as highly elevated as with HS. For PU-H71, compared to “standard” conditions, pSTAT3, pAMPKA and pHSP27 are elevated, and PARP cleavage is enhanced, while other phosphorylation states are the same or reduced even further. Compared to “heat shock” conditions, pSTAT, pAMPKA and pHSP27 are increased, as is PARP cleavage, while phospho-p53 and p38 levels are down.
Overall, the general reduction in potentially critical phosphorylation states of numerous signaling molecules, along with the increases in PARP cleavage with treatment following UPR induction, led us to focus on the HSP90/GRP94 inhibitor PU-H71 for the rest of our studies.
Pre-treatment of cells alters short- and long-term responses to PU-H71
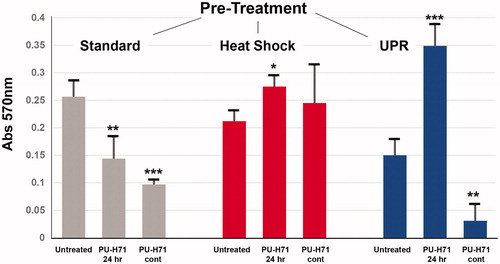
Given the interesting signaling pathway changes induced in STAR cells upon treatment with PU-H71 following stresses, we measured soft agar survival/growth outcomes of the cells after stress treatments followed by short- or long-term exposure to drug. Cells were pre-treated under standard conditions (“Standard”), or were heat shocked (“Heat Shock”), or were UPR-induced (“UPR”) as described above. After 24 h recovery, cells were treated with 5 nM PU-H71 for an additional 24 h in culture. Cells were then plated in soft agar with no further drug exposure (“PU-H71 24 h”), or received continuous (daily treatment) with drug (“PU-H71 cont”). Cells were kept in soft agar for eight days, after which they were extracted from the matrix, and growth/survival was measured by MTT assay. shows those results. Under standard, unstressed conditions, the HSP90 inhibitor significantly reduces cell growth and survival after both short- and long-term exposures. However, following HS, cell growth is actually increased in the short term, with no significant die-off in the long term. With UPR pre-treatment, cell growth is dramatically increased after a short exposure to the drug, but with an almost equally dramatic reduction in viability after continuous exposure. Given the make-or-break effects of the UPR in combination with PU-H71 on cell viability, we measured pro- and anti-apoptotic components with stress and drug treatment.
Figure 5. Soft-agar growth of STAR cells pre-treated with stresses, and then treated with an HSP90 inhibitor. STAR cells were first either left under typical culture conditions, heat shocked or had the UPR induced (described above). After 24 h recovery, cells were treated in culture for 24 h with the HSP90 inhibitor PU-H71 (5 nM), and were then plated in a soft-agar growth assay. Some cells received no drug (“Untreated”), some received no further drug treatment (“PU-H71 24 h”), and some cells received continuous daily dosing with 5 nM PU-H71 (“PU-H71 cont”) for the remainder of the culture growth. After eight days in soft agar, the matrix was solubilized and growth was measured by MTT assay.
Measurements of pro- and anti-apoptotic molecules from UPR-induced cells treated with PU-H71
STAR cells were left untreated, were stressed with DTT to induce the UPR (with 24 h recovery), were treated with 5 nM PU-H71 for 24 h, or were stressed and then drugged with the HSP90/GRP94 inhibitor. Cells were lysed, and lysates were subjected to an antibody array that probes for 35 proteins with differential apoptotic functions (; developed arrays themselves are shown in Supplemental Figure 4). “Control” cell lysate values were used to normalize the rest (the only protein whose level decreased during these studies was clusterin in the combination setting of stress + drug, where levels were only 10% below control levels, data not shown). In this array, protein quantities, rather than phosphorylation or cleavage events, are measured (except for the different phospho-states of p53 and RAD17, and distinctions between pro-caspase 3 and its cleaved version). One sees that the UPR induces clear changes in outputs, with drug treatment leading to the highest value changes overall, while the combination of stress and drug treatment providing intermediate values. Analysis of those changes in the context of the proteins involved reveals a variety of pro- and anti-apoptotic changes for the various array constituents. BAD protein levels are increased with drug treatment, and that increase is maintained following UPR stress. As mentioned above, reduced BAD phosphorylation (as seen in the UPR and drug treatment conditions in ) is associated with apoptosis [Citation31]; thus, these changes are presumed to be pro-apoptotic. BAX is another BCL2 family member with pro-apoptotic activity; increasing its capacity or expression (as seen here) is considered therapeutically relevant [Citation43]. Increases in proCASP3 and cleaved CASP3 would presumably be pro-apoptotic, but this is somewhat in disagreement with , where cleaved CASP3 was reduced (but cleaved PARP, downstream of CASP3 and also pro-apoptotic [Citation34], went up). The timing drug dosing and downstream analyses in the experiments for and are different (and for , the timing coincides with cell proliferation, ), and could explain this discrepancy. Upstream of CASP3 are the Death Receptors DR4 (TNFRSF10A), DR5 (TNFRSF10B), FAS/CD95 and the adapter molecule FADD [Citation44], all increasingly upregulated from UPR conditions to drug treatment to the combination of stress and drug treatment. However, a related receptor, TNFRSF1A, shows a slight increase with UPR induction, and is considerably higher with drug treatment, but again shows reduced levels in the combination of stress and drug. Triggering of the intrinsic death pathway alters mitochondrial outer membrane potential and releases such components as CYTC, HTRA2 and SMAC/DIABLO into the cytosol [Citation45], promoting apoptosome assembly and cell death [Citation46]. Curiously, CYTC and HTRA2 both show patterns similar to TNSFRSF1A with ultimately low levels in the combination scenario, while SMAC is increased in both the drug alone and drug combination. As mentioned above, p53 phosphorylation is complicated, with serine 15 phosphorylation being a major step in recruitment of transcription factors [Citation33]. It is also required for phosphorylation of serine 46, which is related to p53’s apoptotic function [Citation47] (and phospho-p53-Ser15 may not be that important in apoptosis), whereas serine 392 phosphorylation is anti-proliferative [Citation48]. The result for phospho-Ser15 here differs from , but as mentioned, time and dose may bear responsibility for that.
Figure 6. STAR cell outputs of proteins involved in apoptosis after stress and/or HSP90 inhibitor treatment. STAR cells were kept in standard culture conditions (“Control”), were subjected to the UPR (“UPR”), were left unstressed but treated with 5 nM PU-H71 for 24 h (“PU-H71”, or had the UPR induced, and after 24 h recovery, were treated with 5 nM PU-H71 for 24 h (“UPR + PU-H71”). Cells were lysed, and then applied to an antibody array measuring quantities of proteins listed. Spots were quantified and normalized to control values as in . Shown is a heat map representation of those data with the scoring scale at the bottom.
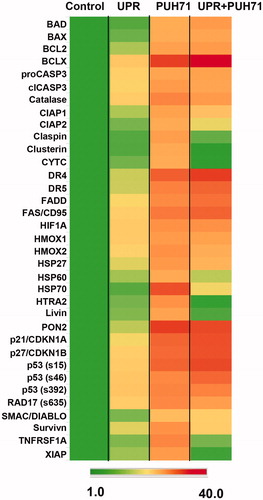
Numerous proteins that are considered anti-apoptotic also demonstrated altered expression during this experiment. Anti-apoptotic BCL2 family members BCL2 and BCLX [Citation49] were both upregulated by stress, PU-H71 treatment, and the combination of stress and drug. These results suggest that there are conflicting actors in the pro- vs. anti-apoptotic scenarios created during UPR stress and HSP90 inhibition. This is further demonstrated in the increased levels of the anti-oxidants catalase, which plays a role in tumor cell apoptosis avoidance [Citation50], and PON2, which is also protective against ER stress [Citation51]. Other negative regulators of apoptosis include CIAP family members, survivin, livin/BIRC7 and XIAP [Citation52,Citation53], which are differentially regulated in these settings. CIAP1/2 are slightly induced with the UPR, considerably upregulated with drug treatment, and are slightly reduced (but still above control levels) in the stress/drug combination. Survivin follows a similar pattern, while XIAP is quite reduced in the combination setting. Claspin, a cell-cycle checkpoint regulator, also follows the previous patterns resulting ultimately in low levels, which may be due to its degradation during apoptosis [Citation54], but another checkpoint protein, RAD17 [Citation55], increases in its phospho-state, which may be related to the stress response [Citation56]. Of the chaperones in this panel with anti-apoptotic properties [Citation57,Citation58], the clusterin expression pattern again resembles XIAP and claspin, while other chaperones (HSP32/HMOX1 and related HMOX2 [Citation59], HSP27 and HSP70) show increased expression during UPR stress, greater expression with drug treatment, and maintain relatively high levels with the combination of stress and drug treatment. HSP60 differs, with a reduced expression in the combination setting.
Some proteins in the array have complicated relationships with apoptosis, such as HIF1A [Citation60], which also has a complicated relationship with EIF2A in the UPR [Citation61]. In this experiment, stress and drug treatment increased its expression (and despite it being an HSP90 client). The cell cycle inhibitors p21/CDNK1A/CIP1/WAF1 and p27/CDKN1B/KIP1 also have pro- and anti-apoptotic functions depending on subcellular localization and mutation status [Citation62–64]; here, their expression increases with stress and drug applications.
These results reveal complicated patterns for pro- and anti-apoptotic effects of UPR stress and HSP90 + GRP94 inhibition; the overall outcome () suggests that the pro-apoptotic forces eventually prevail to prevent tumor cell proliferation.
Using Ingenuity Pathway Analysis we performed a Comparison Analysis to profile the expression changes in the apoptosis assay (from ). The top 2 networks/interactomes are shown in , where A reveals an expected outcome of extensive interactions between numerous players in the apoptotic cascade, including particularly dense nodes around apoptotic drivers such as SMAC/DIABLO, the caspases, cytochrome C and the protease HTRA2. The second network, in , emphasizes connections between the mostly anti-apoptotic array members with the signaling pathways deeply embedded in the UPR (e.g. ), such as the GSK3, Ras, MAPK, PI3K, JNK and NFKB. Supplemental Figure 5A lists the top Diseases and Biofunction categories and the heat map of their z-activation scores, along with the list of the top 15 Canonical Pathways (Supplemental Figure 5C) derived from the data. Supplemental Figure 5B again shows the conflicting impacts of the various pro- and anti-apoptotic outcomes on Cell Survival, which is emblematic of the results presented here.
Figure 7. Ingenuity Pathway Analysis (IPA) comparisons of the quantified readouts from . Values were converted to log2 ratios of experimental/control and entered into a Comparison Analysis in IPA. The top 2 networks (interactomes) are shown. Network names are also shown. Proteins with elevated values from the array are depicted in large bold font with dark background. For B, signaling proteins with known involvement in the UPR are highlighted in lighter background. The “scores” listed are − log [p values] and are based on the probabilities of random associations of these genes/proteins. The significance threshold is set at by default as 1.25. “Focus molecules” refer to nodes from which networks initiate. Solid dark lines show direct connections between proteins found in the array; these are well-known or literature-documented interactions. Broken lines represent indirect connections arising from speculation, or via known intermediaries. Light colored lines connect identified proteins within the network with other proteins which were not part of the array. Line lengths (“edges”) between the protein “nodes” correlate to the amount of supportive literature documenting those interactions. Please note, we have shortened numerous edges to fit the interactomes into the figure.
![Figure 7. Ingenuity Pathway Analysis (IPA) comparisons of the quantified readouts from Figure 6. Values were converted to log2 ratios of experimental/control and entered into a Comparison Analysis in IPA. The top 2 networks (interactomes) are shown. Network names are also shown. Proteins with elevated values from the array are depicted in large bold font with dark background. For B, signaling proteins with known involvement in the UPR are highlighted in lighter background. The “scores” listed are − log [p values] and are based on the probabilities of random associations of these genes/proteins. The significance threshold is set at by default as 1.25. “Focus molecules” refer to nodes from which networks initiate. Solid dark lines show direct connections between proteins found in the array; these are well-known or literature-documented interactions. Broken lines represent indirect connections arising from speculation, or via known intermediaries. Light colored lines connect identified proteins within the network with other proteins which were not part of the array. Line lengths (“edges”) between the protein “nodes” correlate to the amount of supportive literature documenting those interactions. Please note, we have shortened numerous edges to fit the interactomes into the figure.](/cms/asset/1751cbf5-07c6-4a36-aa94-3f3778190895/ihyt_a_1256503_f0007_c.jpg)
Discussion
HSP90 plays a critical role in the proteostasis of numerous signaling and transcription factor complexes, and thus, is important to cellular homeostasis [Citation65]. GRP94, the ER paralog of HSP90, is vital to protein quality control of proteins targeted for membrane localization and secretion [Citation66], as well as in optimized ER stress responses [Citation67]. HSP90 has a significant history as a target of cancer therapy [Citation68] that has become modified to include the potential of HSP90 therapeutics as sensitizers in combination therapies [Citation69]. Moreover, other organellar-specific HSP90 family members are topics of discussion for therapeutic targeting, as well [Citation70], implying that GRP94 could be a valuable focus for treatment.
In this study, we have tested the effects of several HSP90 inhibitors on a unique primary canine lung cancer cell line under typical culture conditions, and under the stresses of HS, or the UPR. One of those inhibitors, the purine-based PU-H71 [Citation71], also targets GRP94 [Citation20], which we thought may have bearing on drug treatment in the context of the UPR. Our results suggest that this particular cell line was most sensitive to PU-H71 (of the three inhibitors) under standard conditions (Supplemental Figure 2), but that stress pre-treatment and timing mattered. Comparisons of signaling pathway activation () following HS (42 °C, 2 h) or UPR induction (1 mM DTT, 4 h) before drug treatment further suggested that PU-H71 was the most effective at reducing potentially important signaling activities, particularly in the stressed states. We focused on PU-H71 treatment of cells in soft agar assays of cells grown initially under standard conditions, or following HS or UPR induction (). Both short-term (24 h treatment followed by eight days of soft agar culture) and long-term drug exposure (supplied daily for eight days) reduced cell survival under standard conditions, but had no beneficial effect for heat-stressed cells. However, following UPR conditions, cells in the short term grew extensively, but suffered a deep decline in cell survival with continuous treatment. This would suggest that the combination of UPR stress – but not heat stress – and drug might be a worthy approach. Finally, we evaluated an apoptosis profile of cells with the UPR and drug combination to see that both pro- and anti-apoptotic factors were promoted ( and ), where apoptosis presumably prevails.
The effect of heat stress leading to lack of PU-H71 efficacy may be of some concern, but it is not clear how widespread this phenomenon is across various cell lines/types. In one report, HS of HUVECs before LPS treatment prevented apoptosis [Citation72], but direct inhibition of HSP90 was not attempted. Most studies involving hyperthermia in combination with HSP90 inhibition have involved inhibitors encased in thermoresponsive nanopolymers [Citation73–75], with few published studies of heat stress separate from drug treatment. The concurrent effects of hyperthermia and HSP90 inhibition on cell lines with mutated p53 did increase apoptosis of those cancer cells [Citation76].
The metastatic tumor and STAR cell line also have a mutated p53 (a missense mutation in the DNA-binding domain) [Citation21], but we do not know what effect this protein has on the cells (i.e. tumor suppressive or oncogenic). In general, similar to humans, p53 mutations appear deleterious in canines with cancer [Citation77–79], suggesting that the tumor suppressor functions are affected. Tumors lacking p53 function were susceptible to HSP90 inhibition by 17-DMAG [Citation80], but again, we do not know the impact of the STAR p53 mutation. However, the effects of mutations in the DNA-binding domain can impact recruitment of other transcription factors/co-factors and influence phosphorylation [Citation81]. Heat shock was demonstrated to increase phosphorylation of p53 [Citation82], and gain-of-function mutants of p53 can promote HSF1 activity, effectively mimicking HS [Citation83] (as do HSP90 inhibitors resulting in the release of HSP90-sequestered HSF1 [Citation38,Citation84]). Glucose starvation, which can induce the UPR, also increases phospho-p53 [Citation85], and there is a p53 isoform (p53/47) that may be generated during UPR/ER stress that has translational effects as well [Citation86,Citation87], which further complicates matters. Clearly, this is an area ripe for further study.
Like p53, p38 MAPK is another HSP90 client, and is widely known as a stress-activated kinase [Citation88], whether via heat stress [Citation89] or the URP [Citation90]. UPR inducement via arsenic leads to activated p38 signaling and GRP94 upregulation [Citation91]. Palmitate has similar UPR-inducing effects, but activation of AMPK (seen in our studies with HSP90 inhibition) attenuates that stress [Citation92]. ER stress can promote apoptosis via the apoptosis signal-regulating kinase 1 (ASK1/MAP3K5) and MAPK cascades (with involvement of p38 [Citation93]). Inhibition of HSP90 can release the protective HSP90/AKT/ASK1 complex to promote apoptosis [Citation94], suggesting that stress signaling via activated p38 may be overcome by drugging HSP90 and/or GRP94. This, too, is a targetable signaling cascade with inhibitors in clinical trials [Citation95].
Curiously, generation of “enhanced proteotoxic stress” by a combination of hyperthermia and the proteasome inhibitor bortezomib was effective in vitro against several cancer cell lines, and also in vivo against a murine lymphoma, when puromycin was used to promote protein misfolding. p53 status had no noticeable effect in those settings [Citation96]. As bortezomib acts as an inducer of the UPR [Citation97], the combination of heat stress (or a form of biologic mimicry, such as HSP90 inhibition) and UPR induction may show promise in anti-cancer therapies [Citation98].
We have recently described signaling effects of the UPR in brain tumor cell lines [Citation22] and highlighted the complicated and often contradictory results from various works. UPR stress signaling is frequently dysregulated in cancer [Citation99], often leading to “inappropriate” survival, but perhaps displaying exploitable weaknesses. For instance, cross-talk between UPR signaling and MAPK cascades, with differential outcomes of cell survival vs. cell death, could have numerous points of intervention [Citation90]. In the context of HSP90 inhibitors, there are conflicting outcomes for the UPR from different inhibitors and different cell types. In multiple myeloma cells – which already have a partially activated UPR through IRE1 and ATF6, due to the production and secretion of high quantities of monoclonal antibody fragments – the geldanamycin derivative IPI-504 blocked the UPR by inactivating XBP1 and ATF6. The loss of transcription factor activity induces apoptosis [Citation100]. In a similar case, a structurally novel HSP90 inhibitor, SNX-2112, downregulated expression of various UPR components to drive apoptosis in hepatocellular carcinoma cells, and this cell death was exacerbated with the application of additional UPR stress [Citation101]. In a unique scenario of UPR induction, low energy focused ultrasound (LOFU) drove the UPR in prostate cancer cells, increasing their apoptotic sensitivity to 17-AAG [Citation102]. Using a variety of different cancer cell lines, PU-H71 itself induced the UPR and triggered apoptosis via the intrinsic mitochondrial pathway (activation of BAX, release of CYTC, caspase activation) along with reductions in the resistance-promoting BCL2 [Citation103]. We saw similar upregulations of the apoptotic effectors upon treatment of STAR cells with PU-H71, but also saw upregulation of the apoptotic resistors BCL2 and BCLX (). Clearly, the effects of UPR stress amidst HSP90 inhibition vary considerably, and will require further analyses to discern possible trends.
Our data reveal complex interplays between the extrinsic (e.g., death-receptor initiated) and intrinsic (e.g., mitochondrial-based) forms of apoptosis [Citation104] ( and ). Often the intrinsic pathways received the most attention in UPR-mediated apoptosis, driven via IRE1/TRAF2/JNK signaling [Citation105], but CHOP/GADD153 can also mediate TRAIL-induced apoptosis, particularly when faced with TRAIL resistant cancer cells [Citation106]. This suggests that there may be heightened sensitivity to HSP90/GRP94 inhibition if the UPR is engaged, potentially enhancing extrinsic pathway apoptosis alongside mitochondrial-based apoptosis. We note that PARP cleavage, frequently used as an indicator of intrinsic-mitochondrial driven apoptosis, was generally reduced following treatment with HSP90 inhibitors in control or heat-shock settings (). Other studies have shown differential PARP cleavage in tumor cells, using 20–200 fold higher concentrations of drugs than we used [Citation107], but induction of the UPR with upregulation of GRP94 may promote PARP cleavage [Citation108]. Given the upregulation of numerous anti-apoptotic members that prevent apoptosis at the level of the intrinsic mitochondrial pathway (e.g., BCL2, BCLX, ), it may be that UPR stress combined with HSP90/GRP94 inhibition overcomes the regulators and drives apoptosis, as indicated by PARP cleavage.
Based on this work, we propose that exploiting the UPR in the context of HSP90/GRP94 inhibition has value in a cancer therapeutic setting. We need a clearer understanding of tumor stress biology to achieve this, as tumors themselves often have active engagement of the UPR, which is generally protective. We would need to distinguish which tumors/cells would be susceptible to promotion or inhibition of the UPR, and if pharmacologic agents are available for these interventions (e.g., UPR inhibitory compounds, chemical chaperones, xenobiotic inducers, proteasome inhibitors, etc. [Citation109,Citation110]). Combinations with HSP90 inhibitors could focus on outcomes such as ratios of apoptotic drivers to anti-apoptotic components as potential determinants for cell survival, as it is not clear from our work that definitive on/off pathways are obvious. At the very least, we need to acknowledge that most of our cell culture-based therapies probably lack the influence of in vivo stresses such as upregulated HSP expression or UPR induction which may profoundly sway the results of drug therapy studies.
We also note that we used a novel canine cancer cell line in this study, derived from a metastatic recurrent tumor in a patient that had been treated with immunotherapy [Citation21]. While perhaps somewhat esoteric compared to human and murine cell studies, extension of tumor biology investigations to true spontaneous forms as found in companion animals has long been seen as a boon to cancer research [Citation111,Citation112]. The use of HSP90 inhibitors has reached clinical stages in canines [Citation113], and we hope our contributions add to rational designs for further utility with applications to both veterinary and human clinical situations.
Conclusions
Stress conditions are prevalent in tumors and may provide resilience to therapeutic interventions; on the other hand, such stress responses may also display exploitable weaknesses for appropriate treatments. Here we have shown that promoting a stress response (the UPR) enhanced responses to an HSP90 inhibitor that also targets one of the players in the UPR. These results suggest that combination strategies to “push tumors over the edge” with stress along with suitable targeted treatments may overcome tumors’ stress-driven resistance and improve anti-cancer responses.
Supplemental_File.zip
Download Zip (6.6 MB)Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
Related Research Data
References
- Graner MW, Bigner DD. (2005). Chaperone proteins and brain tumors: potential targets and possible therapeutics. Neuro Oncol 7:260–78.
- Graner MW, Bigner DD. (2006). Therapeutic aspects of chaperones/heat-shock proteins in neuro-oncology. Expert Rev Anticancer Ther 6:679–95.
- Ackerman D, Simon MC. (2014). Hypoxia, lipids, and cancer: surviving the harsh tumor microenvironment. Trends Cell Biol 24:472–8.
- Cattaneo M, Dominici R, Cardano M, et al. (2012). Molecular chaperones as therapeutic targets to counteract proteostasis defects. J Cell Physiol 227:1226–34.
- Shah SP, Lonial S, Boise LH. (2015). When cancer fights back: multiple myeloma, proteasome inhibition, and the heat-shock response. Mol Cancer Res 13:1163–73.
- Sanghera SS, Skitzki JJ. (2013). Targeting the heat shock response in cancer: tipping the balance in transformed cells. Surg Oncol Clin N Am 22:665–84.
- Walter P, Ron D. (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334:1081–6.
- Graner MW. (2013). The unfolded protein response in glioblastomas: passing the stress test. CNS Oncol 2:1–4. Epub November, 2013.
- Dicks N, Gutierrez K, Michalak M, et al. (2015). Endoplasmic reticulum stress, genome damage, and cancer. Front Oncol 5:11.
- van Ommeren R, Staudt MD, Xu H, Hebb MO. (2016). Advances in HSP27 and HSP90-targeting strategies for glioblastoma. J Neurooncol 127:209–19.
- Taldone T, Ochiana SO, Patel PD, Chiosis G. (2014). Selective targeting of the stress chaperome as a therapeutic strategy. Trends Pharmacol Sci 35:592–603.
- Graner MW. (2015). The unfolded protein response in glioblastomas: targetable or trouble? Future Sci OA 1(2). doi: 10.4155/fso.15.45.
- Tatokoro M, Koga F, Yoshida S, Kihara K. (2015). Heat shock protein 90 targeting therapy: state of the art and future perspective. EXCLI J 14:48–58.
- Butler LM, Ferraldeschi R, Armstrong HK, et al. (2015). Maximizing the therapeutic potential of HSP90 inhibitors. Mol Cancer Res 13:1445–51.
- Whitesell L, Santagata S, Lin NU. (2012). Inhibiting HSP90 to treat cancer: a strategy in evolution. Curr Mol Med 12:1108–24.
- Wu BX, Hong F, Zhang Y, et al. (2016). GRP94/gp96 in cancer: biology, structure, immunology, and drug development. Adv Cancer Res 129:165–90.
- Ansa-Addo EA, Thaxton J, Hong F, et al. (2016). Clients and oncogenic roles of molecular chaperone gp96/grp94. Curr Top Med Chem 16:2765–78.
- Crowley VM, Khandelwal A, Mishra S, et al. (2016). Development of glucose regulated protein 94-selective inhibitors based on the BnIm and radamide scaffold. J Med Chem 59:3471–88.
- Bourseau-Guilmain E, Menard JA, Lindqvist E, et al. (2016). Hypoxia regulates global membrane protein endocytosis through caveolin-1 in cancer cells. Nat Commun 7:11371.
- Usmani SZ, Bona RD, Chiosis G, Li Z. (2010). The anti-myeloma activity of a novel purine scaffold HSP90 inhibitor PU-H71 is via inhibition of both HSP90A and HSP90B1. J Hematol Oncol 3:40.
- Epple LM, Bemis LT, Cavanaugh RP, et al. (2013). Prolonged remission of advanced bronchoalveolar adenocarcinoma in a dog treated with autologous, tumour-derived chaperone-rich cell lysate (CRCL) vaccine. Int J Hyperther 29:390–8.
- Redzic JS, Gomez JD, Hellwinkel JE, et al. (2016). Proteomic analyses of brain tumor cell lines amidst the unfolded protein response. Oncotarget. [Epub ahead of print]. doi: 10.18632/oncotarget.10032.
- Epple LM, Dodd RD, Merz AL, et al. (2013). Induction of the unfolded protein response drives enhanced metabolism and chemoresistance in glioma cells. PLoS One 8:e73267.
- Hellwinkel JE, Redzic JS, Harland TA, et al. (2016). Glioma-derived extracellular vesicles selectively suppress immune responses. Neuro Oncol 18:497–506.
- Raynes DA, Guerriero V. Jr. (1998). Inhibition of Hsp70 ATPase activity and protein renaturation by a novel Hsp70-binding protein. J Biol Chem 273:32883–8.
- Karachaliou N, Rosell R, Molina MA, Viteri S. (2014). Predicting resistance by selection of signaling pathways. Transl Lung Cancer Res 3:107–15.
- Meyuhas O. (2015). Ribosomal protein S6 phosphorylation: four decades of research. Int Rev Cell Mol Biol 320:41–73.
- Katsogiannou M, Andrieu C, Rocchi P. (2014). Heat shock protein 27 phosphorylation state is associated with cancer progression. Front Genet 5:346.
- Kostenko S, Moens U. (2009). Heat shock protein 27 phosphorylation: kinases, phosphatases, functions and pathology. Cell Mol Life Sci 66:3289–307.
- Danial NN. (2008). BAD: undertaker by night, candyman by day. Oncogene 27:S53–S70.
- Garcia A, Cayla X, Guergnon J, et al. (2003). Serine/threonine protein phosphatases PP1 and PP2A are key players in apoptosis. Biochimie 85:721–6.
- Jiang P, Du W, Heese K, Wu M. (2006). The Bad guy cooperates with good cop p53: bad is transcriptionally up-regulated by p53 and forms a Bad/p53 complex at the mitochondria to induce apoptosis. Mol Cell Biol 26:9071–82.
- Dai C, Gu W. (2010). p53 post-translational modification: deregulated in tumorigenesis. Trends Mol Med 16:528–36.
- Decker P, Muller S. (2002). Modulating poly (ADP-ribose) polymerase activity: potential for the prevention and therapy of pathogenic situations involving DNA damage and oxidative stress. Curr Pharm Biotechnol 3:275–83.
- Beurel E, Grieco SF, Jope RS. (2015). Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther 148:114–31.
- An WF, Germain AR, Bishop JA, et al. (2010). Discovery of potent and highly selective inhibitors of GSK3b. Probe reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010?2012 Apr 16 [updated 2014 May 13].
- Hardie DG. (1999). Roles of the AMP-activated/SNF1 protein kinase family in the response to cellular stress. Biochem Soc Symp 64:13–27.
- Zou J, Guo Y, Guettouche T, et al. (1998). Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 94:471–80.
- Chen F. (2012). JNK-induced apoptosis, compensatory growth, and cancer stem cells. Cancer Res 72:379–86.
- Lotocki G, Alonso OF, Dietrich WD, Keane RW. (2004). Tumor necrosis factor receptor 1 and its signaling intermediates are recruited to lipid rafts in the traumatized brain. J Neurosci 24:11010–16.
- Nieto-Miguel T, Gajate C, Gonzalez-Camacho F, Mollinedo F. (2008). Proapoptotic role of Hsp90 by its interaction with c-Jun N-terminal kinase in lipid rafts in edelfosine-mediated antileukemic therapy. Oncogene 27:1779–87.
- Wang C, Chen J. (2003). Phosphorylation and hsp90 binding mediate heat shock stabilization of p53. J Biol Chem 278:2066–71.
- Liu Z, Ding Y, Ye N, et al. (2016). Direct activation of Bax protein for cancer therapy. Med Res Rev 36:313–41.
- Thorburn A. (2004). Death receptor-induced cell killing. Cell Signal 16:139–44.
- van Loo G, Saelens X, van Gurp M, et al. (2002). The role of mitochondrial factors in apoptosis: a Russian roulette with more than one bullet. Cell Death Differ 9:1031–42.
- Zou H, Li Y, Liu X, Wang X. (1999). An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem 274:11549–56.
- Smeenk L, van Heeringen SJ, Koeppel M, et al. (2011). Role of p53 serine 46 in p53 target gene regulation. PLoS One 6:e17574.
- Meek DW. (2015). Regulation of the p53 response and its relationship to cancer. Biochem J 469:325–46.
- Weyhenmeyer B, Murphy AC, Prehn JH, Murphy BM. (2012). Targeting the anti-apoptotic Bcl-2 family members for the treatment of cancer. Exp Oncol 34:192–9.
- Bechtel W, Bauer G. (2009). Catalase protects tumor cells from apoptosis induction by intercellular ROS signaling. Anticancer Res 29:4541–57.
- Devarajan A, Shih D, Reddy ST. (2014). Inflammation, infection, cancer and all that…the role of paraoxonases. Adv Exp Med Biol 824:33–41.
- Yan B. (2011). Research progress on Livin protein: an inhibitor of apoptosis. Mol Cell Biochem 357:39–45.
- Aggarwal BB, Bhardwaj U, Takada Y. (2004). Regulation of TRAIL-induced apoptosis by ectopic expression of antiapoptotic factors. Vitam Horm 67:453–83.
- Semple JI, Smits VA, Fernaud JR, et al. (2007). Cleavage and degradation of Claspin during apoptosis by caspases and the proteasome. Cell Death Differ 14:1433–42.
- Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. (2004). Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 73:39–85.
- Xu B, Wang W, Guo H, et al. (2014). Oxidative stress preferentially induces a subtype of micronuclei and mediates the genomic instability caused by p53 dysfunction. Mutat Res 770:1–8.
- Albany C, Hahn NM. (2014). Heat shock and other apoptosis-related proteins as therapeutic targets in prostate cancer. Asian J Androl 16:359–63.
- Wang C, Zhang Y, Guo K, et al. (2016). Heat shock proteins in hepatocellular carcinoma: molecular mechanism and therapeutic potential. Int J Cancer 138:1824–34.
- Schipper HM. (2004). Heme oxygenase expression in human central nervous system disorders. Free Radic Biol Med 37:1995–2011.
- Piret JP, Mottet D, Raes M, Michiels C. (2002). Is HIF-1alpha a pro- or an anti-apoptotic protein? Biochem Pharmacol 64:889–92.
- Zhu K, Chan W, Heymach J, et al. (2009). Control of HIF-1alpha expression by eIF2 alpha phosphorylation-mediated translational repression. Cancer Res 69:1836–43.
- Ohkoshi S, Yano M, Matsuda Y. (2015). Oncogenic role of p21 in hepatocarcinogenesis suggests a new treatment strategy. World J Gastroenterol 21:12150–6.
- Dutto I, Tillhon M, Cazzalini O, et al. (2015). Biology of the cell cycle inhibitor p21 (CDKN1A): molecular mechanisms and relevance in chemical toxicology. Arch Toxicol 89:155–78.
- Maddika S, Ande SR, Panigrahi S, et al. (2007). Cell survival, cell death and cell cycle pathways are interconnected: implications for cancer therapy. Drug Resist Updat 10:13–29.
- Taipale M, Jarosz DF, Lindquist S. (2010). HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol 11:515–28.
- Marzec M, Eletto D, Argon Y. (2012). GRP94: an HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim Biophys Acta 1823:774–87.
- Eletto D, Dersh D, Argon Y. (2010). GRP94 in ER quality control and stress responses. Semin Cell Dev Biol 21:479–85.
- Miyata Y, Nakamoto H, Neckers L. (2013). The therapeutic target Hsp90 and cancer hallmarks. Curr Pharm Des 19:347–65.
- Solarova Z, Mojzis J, Solar P. (2015). Hsp90 inhibitor as a sensitizer of cancer cells to different therapies (review). Int J Oncol 46:907–26.
- Seo YH. (2015). Organelle-specific Hsp90 inhibitors. Arch Pharm Res 38:1582–90.
- Trendowski M. (2015). PU-H71: an improvement on nature's solutions to oncogenic Hsp90 addiction. Pharmacol Res 99:202–16.
- Liu Z, Zhong T, Zheng D, et al. (2016). Heat stress pretreatment decreases lipopolysaccharide-induced apoptosis via the p38 signaling pathway in human umbilical vein endothelial cells. Mol Med Rep 14:1007–13.
- Rochani AK, Balasubramanian S, Ravindran Girija A, et al. (2016). Dual mode of cancer cell destruction for pancreatic cancer therapy using Hsp90 inhibitor loaded polymeric nano magnetic formulation. Int J Pharm 511:648–58.
- Yang R, Tang Q, Miao F, et al. (2015). Inhibition of heat-shock protein 90 sensitizes liver cancer stem-like cells to magnetic hyperthermia and enhances anti-tumor effect on hepatocellular carcinoma-burdened nude mice. Int J Nanomed 10:7345–58.
- Chen Y, Youn P, Pysher TJ, et al. (2014) Tumour eradication using synchronous thermal ablation and Hsp90 chemotherapy with protein engineered triblock biopolymer-geldanamycin conjugates. Int J Hyperthermia 30:550–64.
- Ohnishi K, Takahashi A, Yokota S, Ohnishi T. (2004). Effects of a heat shock protein inhibitor KNK437 on heat sensitivity and heat tolerance in human squamous cell carcinoma cell lines differing in p53 status. Int J Radiat Biol 80:607–14.
- Kirpensteijn J, Kik M, Teske E, Rutteman GR. (2008). TP53 gene mutations in canine osteosarcoma. Vet Surg 37:454–60.
- Queiroga FL, Raposo T, Carvalho MI, et al. (2011). Canine mammary tumours as a model to study human breast cancer: most recent findings. In Vivo 25:455–65.
- Koshino A, Goto-Koshino Y, Setoguchi A, et al. (2016). Mutation of p53 gene and its correlation with the clinical outcome in dogs with lymphoma. J Vet Intern Med 30:223–9.
- Ayrault O, Godeny MD, Dillon C, et al. (2009). Inhibition of Hsp90 via 17-DMAG induces apoptosis in a p53-dependent manner to prevent medulloblastoma. Proc Natl Acad Sci USA 106:17037–42.
- Lambrughi MD, Gioia L, Gervasio FL, et al. (2016). DNA-binding protects p53 from interactions with cofactors involved in transcription-independent functions. Nucleic Acids Res 44:9096–109.
- Miyakoda M, Suzuki K, Kodama S, Watanabe M. (2002). Activation of ATM and phosphorylation of p53 by heat shock. Oncogene 21:1090–6.
- Li D, Yallowitz A, Ozog L, Marchenko N. (2014). A gain-of-function mutant p53-HSF1 feed forward circuit governs adaptation of cancer cells to proteotoxic stress. Cell Death Dis 5:e1194.
- Song S, Kole S, Precht P, et al. (2010). Activation of heat shock factor 1 plays a role in pyrrolidine dithiocarbamate-mediated expression of the co-chaperone BAG3. Int J Biochem Cell Biol 42:1856–63.
- Isono T, Chano T, Kitamura A, Yuasa T. (2014). Glucose deprivation induces G2/M transition-arrest and cell death in N-GlcNAc2-modified protein-producing renal carcinoma cells. PLoS One 9:e96168.
- Lopez I, Tournillon AS, Nylander K, Fahraeus R. (2015). p53-mediated control of gene expression via mRNA translation during endoplasmic reticulum stress. Cell Cycle 14:3373–8.
- Bourougaa K, Naski N, Boularan C, et al. (2010). Endoplasmic reticulum stress induces G2 cell-cycle arrest via mRNA translation of the p53 isoform p53/47. Mol Cell 38:78–88.
- Obata T, Brown GE, Yaffe MB. (2000). MAP kinase pathways activated by stress: the p38 MAPK pathway. Crit Care Med 28:N67–77.
- Dorion S, Landry J. (2002). Activation of the mitogen-activated protein kinase pathways by heat shock. Cell Stress Chaperones 7:200–6.
- Darling NJ, Cook SJ. (2014). The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim Biophys Acta 1843:2150–63.
- Li C, Xu J, Li F, et al. (2011). Unfolded protein response signaling and MAP kinase pathways underlie pathogenesis of arsenic-induced cutaneous inflammation. Cancer Prev Res (Phila) 4:2101–9.
- Lu J, Wang Q, Huang L, et al. (2012). Palmitate causes endoplasmic reticulum stress and apoptosis in human mesenchymal stem cells: prevention by AMPK activator. Endocrinology 153:5275–84.
- Sekine Y, Takeda K, Ichijo H. (2006). The ASK1-MAP kinase signaling in ER stress and neurodegenerative diseases. Curr Mol Med 6:87–97.
- Zhang R, Luo D, Miao R, et al. (2005). Hsp90-Akt phosphorylates ASK1 and inhibits ASK1-mediated apoptosis. Oncogene 24:3954–63.
- Yong HY, Koh MS, Moon A. (2009). The p38 MAPK inhibitors for the treatment of inflammatory diseases and cancer. Expert Opin Investig Drugs 18:1893–905.
- Neznanov N, Komarov AP, Neznanova L, et al. (2011). Proteotoxic stress targeted therapy (PSTT): induction of protein misfolding enhances the antitumor effect of the proteasome inhibitor bortezomib. Oncotarget 2:209–21.
- Ri M. (2016). Endoplasmic-reticulum stress pathway-associated mechanisms of action of proteasome inhibitors in multiple myeloma. Int J Hematol 104:273–80.
- Zismanov V, Drucker L, Gottfried M. (2014). Combined inhibition of Hsp90 and the proteasome affects NSCLC proteostasis and attenuates cell migration. Anticancer Drugs 25:998–1006.
- Kato H, Nishitoh H. (2015). Stress responses from the endoplasmic reticulum in cancer. Front Oncol 5:93.
- Patterson J, Palombella VJ, Fritz C, Normant E. (2008). IPI-504, a novel and soluble HSP-90 inhibitor, blocks the unfolded protein response in multiple myeloma cells. Cancer Chemother Pharmacol 61:923–32.
- Wang X, Wang S, Liu Y, et al. (2014). The Hsp90 inhibitor SNX-2112 induces apoptosis of human hepatocellular carcinoma cells: the role of ER stress. Biochem Biophys Res Commun 446:160–6.
- Saha S, Bhanja P, Partanen A, et al. (2014). Low intensity focused ultrasound (LOFU) modulates unfolded protein response and sensitizes prostate cancer to 17AAG. Oncoscience 1:434–45.
- Gallerne C, Prola A, Lemaire C. (2013). Hsp90 inhibition by PU-H71 induces apoptosis through endoplasmic reticulum stress and mitochondrial pathway in cancer cells and overcomes the resistance conferred by Bcl-2. Biochim Biophys Acta 1833:1356–66.
- Green DR, Llambi F. (2015). Cell death signaling. Cold Spring Harbor Perspect Biol 7:a006080. doi: 10.1101/cshperspect.a006080.
- Verma G, Datta M. (2012). The critical role of JNK in the ER-mitochondrial crosstalk during apoptotic cell death. J Cell Physiol 227:1791–5.
- Siegelin MD. (2012). Utilization of the cellular stress response to sensitize cancer cells to TRAIL-mediated apoptosis. Expert Opin Ther Targets 16:801–17.
- McCleese JK, Bear MD, Fossey SL, et al. (2009). The novel HSP90 inhibitor STA-1474 exhibits biologic activity against osteosarcoma cell lines. Int J Cancer 125:2792–801.
- Sacco A, Aujay M, Morgan B, et al. (2011). Carfilzomib-dependent selective inhibition of the chymotrypsin-like activity of the proteasome leads to antitumor activity in Waldenstrom's Macroglobulinemia. Clin Cancer Res 17:1753–64.
- Rivas A, Vidal RL, Hetz C. (2015). Targeting the unfolded protein response for disease intervention. Expert Opin Ther Targets 19:1203–18.
- Lafleur MA, Stevens JL, Lawrence JW. (2013). Xenobiotic perturbation of ER stress and the unfolded protein response. Toxicol Pathol 41:235–62.
- MacEwen EG. (1990). Spontaneous tumors in dogs and cats: models for the study of cancer biology and treatment. Cancer Metastasis Rev 9:125–36.
- Knapp DW, Dhawan D, Ostrander E. (2015). “Lassie,” “Toto,” and fellow pet dogs: poised to lead the way for advances in cancer prevention. Am Soc Clin Oncol Educ Book 2015:e667–72. doi: 10.14694/EdBook_AM.2015.35.e667.
- London CA, Bear MD, McCleese J, et al. (2011). Phase I evaluation of STA-1474, a prodrug of the novel HSP90 inhibitor ganetespib, in dogs with spontaneous cancer. PLoS One 6:e27018.