
Abstract
Intraperitoneal (IP) drug delivery, either as an intraoperative chemoperfusion or as an adjuvant, repeated instillation, is an established treatment modality in patients with peritoneal carcinomatosis. The efficacy of IP drugs depends on its ability to penetrate the tumour stroma in order to reach their (sub)cellular target. It is known, that drug penetration after IP delivery is limited to a few millimetres. Here, we review the basic tissue transport mechanisms after IP delivery and discuss the biophysical barriers and obstacles that limit penetration distance. In addition, we review the physical and pharmaceutical interventions that have been studied in order to improve delivery of small molecular and macromolecular drugs after IP instillation. These interventions could inform the design of future clinical trials aiming at an improved efficacy of IP-based drug delivery in carcinomatosis patients.
Introduction
Intraperitoneal (IP) drug delivery, either during surgery or as an adjuvant therapy, has evolved into an established treatment modality in patients with peritoneal carcinomatosis (PC). The rationale of IP drug delivery is based on a pharmacokinetic advantage: since the peritoneal clearance is much slower than systemic clearance, a high IP dose will result in only moderate systemic drug exposure. The peritoneal-plasma “barrier” consists mostly of the submesothelial connective tissue with its associated microvessel network. Recent data suggest that the endothelial glycocalyx is of major importance in regulating solute and fluid transfer towards and from the peritoneal cavity [Citation1]. The pharmacokinetic advantage of IP drug delivery is usually expressed as the ratio of the area under the concentration-time curve (AUC) in the peritoneal over the plasma compartment (AUCper/AUCplasma), and ranges widely from approximately 2–1000 depending on the drug and model used. Preclinical studies have shown an exponential decrease in survival (S) with increasing dose of a non-cell cycle specific anticancer drug according to the log-linear relationship S = exp[− (αD)], where α is a parameter that depends on the level of susceptibility to the drug (α = 0 for completely resistant cells) and D represents drug dose [Citation2]. Moreover, the higher IP dose may overcome platinum resistance in certain patients [Citation3].
Assuming the systemically absorbed vascular drug delivery is negligible, the efficacy of IP chemotherapy critically depends on its ability to cross a number of tissue barriers and interfaces from the peritoneal cavity to its (intra)cellular target. Little is known about the tissue transport mechanisms after IP drug delivery. What is clear, is that the tissue penetration distance of IP drug into peritoneal cancer nodules is limited to less than a millimetre at most [Citation4]. As a consequence, only patients with the microscopic peritoneal disease or implants not larger than 2–3 mm will, in theory, benefit from IP drug delivery. Limited tissue penetration is explained by a number of physical (high interstitial fluid pressure (IFP), dense extracellular matrix (ECM)) and chemical (binding, sequestration, metabolism and degradation) effects that prevent the drug from reaching an adequate number of target cells. Here, we first provide an overview of the transport mechanisms that govern tissue transport after IP drug delivery. Subsequently, we discuss approaches aimed to enhance drug transport by modification of the drug itself and/or the physicochemical properties of the tumour microenvironment (TME).
Basic mechanisms of tissue drug transport
Fluid transport
Tissue transport after IP delivery of chemotherapy is the result of the interplay between convective, diffusive and reactive transport [Citation5]. When the tumour is considered to be a rigid isotropic porous medium, fluid flow can be described by Darcy’s law [Citation6]:
(1) where u represents the interstitial fluid velocity (m/s), ∇Pi the pressure gradient vector (Pa/m) and K the hydraulic conductivity of the tumour tissue for interstitial fluid (m2/Pa·s). Hydraulic conductivity is defined as the ratio of the intrinsic permeability of the tissue k (m2), which results from tissue properties such as stromal stiffness and cellular density, over the dynamic viscosity of the interstitial fluid μ (Pa·s):
(2)
Solute transport
Mass conservation of the drug is given by:
(3) with Cdrug the time-dependent concentration of the drug present in the interstitium (mol/m3), D the diffusion coefficient (m2/s), ∇2 the Laplacian operator, ∇ the divergence operator and S the sink in drug concentration (mol/m3). The first right-hand term describes diffusion along a concentration gradient, while the second (negative) term accounts for convective transport, which is directed outwards due to the fact that the IFP exceeds the pressure exerted by the fluid column (typically 2–20 cm of H2O) during a clinical chemoperfusion procedure with open abdomen. The sink term S is composed of two different terms:
(4) where Sbl represents the sink in the drug concentration related to the vascular uptake (mol/m3/s) and Scell the sink in drug concentration due to tissue/cellular uptake, binding and metabolism (mol/m3/s). The closure term for the loss due to the cellular uptake of the drug is described by a first-order elimination, an approach commonly used in literature with beta; being a first-order elimination constant (1/s):
(5)
The closure term of the resorption by the vascular system is given by the following equation [Citation5]:
(6) with Fv the fluid velocity across the vascular wall, σ the reflection coefficient of vessels for the drug, Cv and Ci the concentration of drug in the vascular system and in the interstitium respectively (mol/m3), Pc the permeability of the vessel wall for the drug (m/s) and Pev the Peclet number that expresses the ratio of the mass transport contributions by convection to that by diffusion across the microvascular walls given by:
(7)
Furthermore, the third term in EquationEquation 3(3) , represents the reactive drug transport. Within this term, an account is made for the drug that will no longer be available for transport in the IF due to a number of different drug-tissue interactions such as the irreversible binding of cisplatin to plasma proteins as albumin, cell uptake and vascular resorption [Citation7,Citation8].
Although the mechanisms for solute transport are treated separately above, in reality, these occur simultaneously, with drug diffusion representing the dominant mechanism for most low molecular weight drugs that are used in clinical practise.
Several mathematical models of varying complexity have been proposed aiming to provide insight into the transport mechanisms that determine tissue concentrations and drug efficacy after IP drug delivery (for a recent review, the reader may consult Steuperaert et al. [Citation9]) Dedrick and Flessner proposed a simple spatially distributed model to describe diffusion-based tissue penetration of IP drug [Citation10]. The model predicts that the concentration of drug in the intercellular space, Ce, approaches the blood concentration Cb exponentially according to the formula:
(8)
where Cp represents the peritoneal drug concentration, k the rate constant of capillary drug removal, D the drug diffusivity and x the penetration distance from the serosal surface.
The characteristic penetration distance by diffusion X can be written as:
(9)
This relationship illustrates that a considerable increase in drug diffusivity or decrease in vascular removal is required in order to affect tissue penetration distance. Experimental studies suggest that, in tumour tissue, the penetration of intraperitoneally administered drugs is limited to 1–2 mm [Citation11].
Stromal transport barriers after IP drug delivery
Malignant tumours are characterised by a pathologically elevated IFP, which represents a barrier to convective drug transport [Citation12]. While normal tissue IFP ranges from −3 to +3 mm Hg, values as high as 60 mm Hg (8 kPa) have been recorded in tumour tissue. Interstitial hypertension has been associated with worse therapy response and impaired survival in both preclinical and patient studies. Elevated interstitial pressure results from rapid tumour cell proliferation, contraction of the interstitial stroma by activated fibroblasts, hyperpermeable microvessels and deficient lymphatic drainage. As a consequence, drug delivery within tumour tissue primarily depends on diffusion. The extent of diffusive transport depends on properties of the drug (size, charge and configuration) and the ECM (cellular composition, density, viscoelasticity, geometrical arrangement and electrostatic properties) [Citation13]. Tumour stroma mainly consists of adipose tissue, smooth muscle and epithelial cells, but also includes pericytes, endothelial cells, leukocytes and activated fibroblasts (cancer-associated fibroblasts or CAFs) [Citation14,Citation15]. Interestingly, due to intense reciprocal interaction with the TME, stromal cells develop a modified phenotype and altered function [Citation14]. Tumour tissue is characterised by increased deposition of collagen I, the most abundant ECM protein. As a result, tumour stroma is characterised by increased stiffness or rigidity compared to normal tissue. Rigidity (or stiffness) is measured using Young’s modulus of elasticity, which is the ratio of stress to strain along an axis. Tissue elasticity can be measured using a mechanical device that directly applies a mechanical load and measures the resulting deformation. Alternatively, non-invasive elastography techniques can be used with ultrasound or magnetic resonance imaging [Citation16,Citation17]. In addition to increased collagen deposition, tumours were shown to further exacerbate matrix stiffness by increased expression of the collagen cross-linking enzyme lysyl oxidase [Citation18]. Paszek et al. found that tissue stiffness of mouse mammary tumours far exceeded that of normal breast tissue (elastic modulus 4049 ± 938 Pa vs. 167 ± 31 Pa) while mechanical disturbances were shown to enhance the malignant phenotype through “mechanoregulatory” circuits [Citation19,Citation20]. In addition to the density of the collagen fibres that are deposited in the ECM, their geometric arrangement may affect drug diffusion. Fibres that are oriented tangentially from the tumour surface may direct drug diffusion away from the tumour, while the opposite may result from fibres that are radially aligned [Citation21]. In addition to stromal components, the tumour cell population in itself represents a barrier to diffusive drug transport. Using advanced imaging techniques, Chauhan et al. demonstrated a significant improvement of macromolecular diffusion when colorectal xenografts were treated with diphtheria toxin, which causes apoptosis of tumour cells but leaves the mouse stroma intact [Citation22]. Other cell types include fibroblasts, myofibroblasts, mesenchymal cells such as pericytes, endothelial cells and immune cells, all of which contribute to a high cellular density and solid stress, compressing the interstitial matrix.
Strategies to improve interstitial drug transport
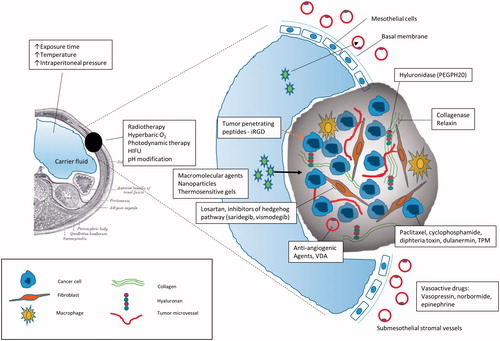
Several physical and pharmacological interventions have been tested in order to improve drug transport and tissue penetration ().
Figure 1. Strategies to improve tissue penetration after intraperitoneal drug delivery. HIFU: high intensity focussed ultrasound; VDA: vascular disrupting agents; TPM: tumour priming microparticles.
Physical methods
Increased intra-abdominal pressure
Higher IP pressures could, in theory, improve convection-driven drug transport. Esquis et al. demonstrated in a rat carcinomatosis model that increasing the IP pressure (22 mm Hg during 1 h) resulted in a significantly higher cisplatin penetration in tumour tissue and improved survival [Citation23]. At the same time, they reported that a 40 mm Hg IP pressure during 2 h was well tolerated in ventilated pigs. Similarly, Jacquet et al. found a significant enhancement of doxorubicin uptake in the abdominal wall and diaphragm of rats when the IP pressure was increased to 20–30 mm Hg [Citation24]. Facy et al. studied the effect of moderately elevated IP pressure (25 cm H2O or 18.3 mm Hg), created by a water column over the abdomen, on tissue distribution of IP oxaliplatin in healthy pigs [Citation25]. They found that elevated IP pressure enhanced diffusion of the drug in both the visceral and parietal peritoneum; the combination with hyperthermia achieved the highest peritoneal tissue concentrations. Interesting results were recently reported by Reymond et al., who comprehensively studied the clinical effects of laparoscopically instilling low dose chemotherapy as an aerosol (pressurised IP aerosol chemotherapy (PIPAC)) in patients with advanced, unresectable carcinomatosis [Citation26–29]. Following administration of a pressurised aerosol of CO2 loaded with doxorubicin (1.5 mg/m2) and cisplatin (7.5 mg/m2) during 30 min at 12 mmHg and 37 °C, low systemic toxicities and excellent tissue penetration were observed.
Increased exposure time
Results from mathematical and experimental models suggest that the exposure time is as important as drug concentration for antitumour efficacy [Citation30,Citation31]. This may be explained by the fact that drug diffusion is a slow process. Toley et al. determined doxorubicin diffusion in colorectal spheroids using a microfluidic device, and found that tissue drug concentration increased with time until 12 h after initiation of the experiment [Citation32]. Over the past few years, novel pharmaceutical carriers have been developed specifically aimed at IP delivery; these include microparticles, nanoparticles, liposomes, micelles, implants and injectable depots [Citation33,Citation34]. The aim of these novel formulations is primarily to achieve improved IP retention of the agent, resulting in a sustained cytotoxic drug exposure [Citation35].
Hyperthermia
In theory, the Einstein–Stokes equation, which states that diffusion is proportional to temperature and inversely proportional to the viscosity of the medium, predicts increased drug diffusion with increasing temperature. Also, the hydraulic conductivity, the primary determinant of convective transport, is increased due to enhanced matrix permeability and a reduced fluid viscosity at a higher temperature. Nevertheless, the potential of hyperthermia to enhance IP drug delivery remains uncertain. Preclinical studies using murine colon, melanoma and mammary tumours showed that whole body hyperthermia (39.5 °C during 6 h) lowers IFP, improves blood flow and enhances the efficacy of radiotherapy [Citation36]. Others, however, were unable to observe IFP lowering upon locoregional heating (41.8 °C during 2 h) in a glioma xenograft model [Citation37]. Along the same line, Klaver et al. was unable to detect a survival benefit by the addition of hyperthermia in a syngeneic rat colorectal carcinomatosis model [Citation38]. Several authors have studied chemotherapy concentrations in tumour tissue after hyperthermic IP delivery. Los et al. found that, compared to normothermic administration, locoregional heating (41.5 °C during 60 min) resulted in a four-fold increase in cisplatin tumour concentration in a rat CC531 colon carcinomatosis model [Citation39]. Of note, plasma cisplatin levels were also significantly elevated after hyperthermic treatment, which raises the question whether the difference in tumour tissue drug concentration resulted from increased peritoneal to tumour transport, or from increased systemic exposure. Later work by Zeamari et al. was unable to confirm the benefit of hyperthermia: mild hyperthermic perfusion with cisplatin (40 °C during 90 min) did not improve drug uptake in small IP tumours in a rat model [Citation40]. Similarly, Facy et al. did not find a significant difference in cisplatin concentration of ovarian peritoneal tumours in a rat model after normothermic or hyperthermic (42 °C during 60 min) chemoperfusion [Citation41]. These authors hypothesised that increased peritoneal drug clearance due to vasodilation and increased blood flow may explain the lack of benefit associated with hyperthermia.
Hyperbaric oxygen
Tumour stroma is characterised by a mismatch between oxygen supply and demand, resulting in hypoxic and even necrotic regions. Administration of oxygen under hyperbaric conditions was shown to potentiate the effect of chemotherapy as a result of enhanced cytotoxicity and neovascularisation. Possibly, hyperbaric oxygen therapy reverses the malignant stroma phenotype allowing improved drug transport. Moen et al. administered hyperbaric oxygen (2 bar) to rats bearing chemically induced mammary tumours, and found a significantly lower IFP compared to control animals [Citation42].
Photodynamic therapy
Photodynamic therapy (PDT) may lower the IFP by impairment of the tumour microcirculation. Leunig et al. treated Syrian gold hamsters bearing a subcutaneously implanted amelanotic melanoma with PDT, and observed a significant decrease in IFP 24 h after PDT therapy, after an initially elevated IFP at 1 and 3 h [Citation43]. Similar findings were reported by Perentes et al. in a subpleurally implanted syngeneic sarcoma model in the rat [Citation44]. They found that low dose PDT not only decreased IFP, but also enhanced tumour tissue distribution of doxorubicin in treated animals.
Radiation therapy
Some patients with peritoneal surface malignancy receive radiation therapy (RT) as part of a multimodal strategy [Citation45]. Radiotherapy has profound and diverse effects on many components of the TME, including the cancer cell population, endothelial cells and stromal cells. Animal experiments and clinical studies in cervical and head and neck cancer patients have shown that fractionated RT results in improved tissue oxygenation and lowered IFP [Citation46]. In theory, therefore, neoadjuvant or concomitant RT should enhance convection-driven delivery of macromolecules. As an example, Davies et al. found a two- to four-fold increase in tumour uptake of doxorubicin after RT in a mouse osteosarcoma xenograft model [Citation47]. There are no in vivo data available on IP drug delivery after RT. In an ex vivo model on porcine peritoneal samples, irradiation of the samples at various doses did not enhance the penetration of doxorubicin after pressurised aerosol delivery (PIPAC) [Citation48].
Ultrasound
High-intensity focussed ultrasound (HIFU) operated in thermal mode allows to concentrate acoustic energy on a focal spot [Citation49]. This form of ultrasound results in increased tissue permeability. The exact causal mechanisms remain unclear but include disruption of matrix protein structure and temporary cavitation effects [Citation50]. HIFU was shown to enhance tissue penetration of small molecule and nanomolecular chemotherapy. Li et al. observed that HIFU treatment of a mouse pancreatic cancer resulted in a 4.5-fold increase in tissue doxorubicine concentration after IV administration [Citation51]. Similarly, pulsed HIFU resulted in significantly improved distribution and penetration of 90Y labelled B3 monoclonal antibody in an A431 epidermoid carcinoma subcutaneous xenograft model [Citation52]. Thus far, studies combining HIFU with IP tumours have not been published.
Pharmacological methods
Agents targeting the tumour vasculature
Tumour microvessels are structurally abnormal, one of their characteristics being increased permeability, leading to loss of macromolecules such as proteins into the interstitium, which contributes to increased IFP. Depending on the dose and timing of administration, agents such as bevacizumab that target new blood vessel formation result in vascular normalisation, i.e. the return to a functionally normal phenotype including a lowered IFP [Citation53]. The effects of pharmacologically induced vascular normalisation on tumour drug uptake are, however, inconsistent in non-peritoneal animal models [Citation54]. We have recently demonstrated that, in a mouse colorectal carcinomatosis model, pre-treatment with either bevacizumab or pazopanib significantly lowers tissue IFP, enhances oxaliplatin penetration and improves tumour control after IP chemoperfusion [Citation55].
Vascular disrupting agents (VDAs), which target the endothelium of existing blood vessels, may improve drug transport through tissue necrosis. Treatment with the VDA, ZD6126 resulted in a lowered IFP in murine and human tumour models [Citation56]. No studies were published that examine cytotoxic drug penetration after VDA administration.
Vasoactive drugs
Theoretically, tissue drug retention could be improved by vasoconstriction of the tumour capillary bed, preventing outward transport of the drug. The proof of principle in a pig model was described by Lindner et al., who noted a significant increase in peritoneal/plasma AUC ratio of IP carboplatin and etoposide following reduction of splanchnic circulation with an IV vasopressin analogue [Citation57]. These observations were later confirmed in a rat model bearing syngeneic carcinomatosis [Citation58]. In this model, IV vasopressin decreased peritoneal blood flow as measured indirectly with the Xe-133-clearance method; the presence of PC did not influence peritoneal blood flow or the effect of vasopressin. Mahteme et al. studied the uptake of radiolabelled 5-FU in a nude rat bearing human colorectal peritoneal nodules [Citation59]. They found that tumour uptake was significantly higher after IP administration of the vasoconstrictive agent norbormide, which acts through modulation of calcium influx. Similar effects were found with epinephrine (adrenaline). Duvillard et al. found that concurrent IP administration of epinephrine lead to a 4 to 12-fold increase of platinum concentration in rat peritoneal tumours [Citation60]. In addition, they noted that rats with nodules 1–2 mm in diameter, insensitive to IP cisplatin alone, were cured when the anticancer drug was combined with epinephrine. Based on these findings, early phase clinical studies were initiated. A phase I study of cytoreductive surgery followed by IP instillation of 30 mg/l of cisplatin at 37 °C and increasing concentrations of IP epinephrine showed that the recommended dose is 2 mg/l, the dose limiting toxicity consisting of cardiac intolerance [Citation61]. As expected, the co-administration of IP epineprine resulted in a 60% decrease in the serum AUC of platinum. A population pharmacokinetic study from the same group based on samples from 55 patients, half of whom treated with IP epinephrine, confirmed that epinephrine halves clearance between peritoneum and serum increases the central volume of distribution of platinum and enhances IP drug exposure [Citation62]. Oman et al. reported a phase I/II study of IP 5-FU in 68 patients with unresectable pancreas cancer, 25% of whom were treated with concurrent IV vasopressin [Citation63]. They found that IV vasopressin did not significantly decrease plasma 5-FU AUC, but reduced Cmax on day 2 of treatment. The overall efficacy of the treatment was low, with only 4% response and a median survival of 8 months.
Vasodilators such as hydralazine were shown to reduce IFP in a subcutaneous murine sarcoma model, yet their effect on drug transport after systemic or IP administration is unknown [Citation64]. One may hypothesise, that vasodilators will enhance systemic exposure after IP delivery through increased peritoneal resorption.
Drugs targeting the stromal components
The tyrosine kinase inhibitor imatinib targets (among others) the platelet derived growth factor beta receptor, which is expressed by a number of mesenchymal cell types including (myo)fibroblasts, smooth muscle cells and pericytes. Inhibition of the receptor reduces tumour IFP and enhances the effect of chemotherapy, possibly through a reprogramming of non-vascular stromal cells to produce a looser ECM with the enhanced diffusion of small molecular drugs [Citation65].
Direct modification of the interstitial stiffness by interfering with the fibrillary or viscoelastic constituents may enhance drug transport. Provenzano et al. treated KPC mice, which spontaneously develop ductal pancreatic cancer, with systemic PEGPH20, a PEGylated recombinant human hyaluronidase enzyme, in order to ablate the tumour hyaluronate density [Citation66]. They demonstrated that this enzymatic therapy effectively ablated stromal hyaluronic acid (HA), reduced IFP, re-expanded blood vessels, dramatically improved tissue distribution of systemically administered doxorubicin and significantly enhanced the antitumour efficacy of gemcitabine. A recently completed phase Ib study of PEGPH20 and gemcitabine suggested a potential benefit of the combination therapy in pancreatic cancer patients with a high pre-treatment HA content [Citation67]. Along with a similar line, Kato et al. showed that treatment of mice bearing subcutaneous lung tumours with intravenous collagenase-I reduced tumour IFP and enhanced tumour accumulation of a liposome/plasmid DNA complex [Citation68]. Similar effects of collagenase on tumour IFP and macromolecule transport were reported by Eikenes et al. [Citation69]. Brown et al. studied the effect of relaxin, a hormone secreted during pregnancy, on collagen fibre length and diffusion of IgG and dextran in mice bearing a dorsal window chamber implanted with a human sarcoma cell line [Citation70]. They found that chronic (12 d) infusion of relaxin significantly shortened average collagen fibre length and enhanced diffusive transport of both IgG and dextran. It was subsequently shown by the same group that relaxin increases the interaction of CAFs with collagen fibres by stimulating beta1-integrin activity [Citation71].
The angiotensin II receptor blocker losartan was shown to inhibit TGF-β activity and other profibrotic cytokines, reduce stromal collagen and hyaluronan levels in orthotopic pancreatic cancer models, and improve delivery of 5-FU and liposomal doxorubicin [Citation72–74]. A phase II clinical trial in locally advanced pancreas cancer combining losartan with chemo- and radiotherapy was recently initiated (NCT01821729).
Alternatively, the desmoplastic stroma in tumours such as pancreas cancer may be altered by targeting the sonic Hedgehog (HH) pathway. In a mouse pancreas cancer model, co-administration of the HH inhibitor IPI-926 (saridegib) increased vascular density and tumour concentrations of gemcitabine [Citation75]. Clinical development of the product was, however, suspended when a phase II trial of IPI-926 with gemcitabine showed detrimental effects [Citation76]. Also, the combination of the HH inhibitor GDC-0449 (vismodegib) with gemcitabine was not superior to gemcitabine alone in metastatic pancreas cancer [Citation77].
Finally, it was recently shown that the vitamin D receptor is abundantly expressed in tumour stroma, and that administration of a vitamin D receptor ligand causes stromal remodelling and increased cancer drug uptake, possibly through interference with the TGF-β/SMAD signalling cascade [Citation78,Citation79].
Drugs targeting tumour cell density
Cellular density may be reduced by sequential cell killing induced by “distribution enhancers” such as the taxanes. Preclinical studies have shown that pre-treatment with paclitaxel reduces tumour IFP, impairs tumour cellularity and enhances subsequent drug delivery [Citation80]. In the clinical setting, Taghian et al. measured tissue IFP and pO2 before and after sequential paclitaxel and doxorubicin in patients with breast cancer [Citation81]. They found that paclitaxel, but not doxorubicin, led to a significantly reduced tumour tissue IFP and increased pO2, irrespective of size. Geretti et al. used cyclophosphamide as a priming agent in a mouse mammary cancer model, and showed that pre-treatment resulted in improved systemic delivery of PEGylated liposomal doxorubicin through reduction of tumour cell density, IFP lowering and increasing vascular perfusion [Citation82]. Similarly, Chauhan et al. showed that treatment of mice bearing window chambers with colorectal xenografts with diphtheria toxin, which leaves murine tissue intact, significantly reduces the hindrance to macromolecular diffusion as measured with in vivo fluorescence recovery after photobleaching [Citation22]. The tumour cell population may also be targeted with agents that cause apoptotic cell death through the tumour necrosis factor related apoptosis inducing ligand (TRAIL) pathway. Hylander et al. studied the effect of recombinant Apo2L/TRAIL (dulanermin) on TME and drug transport in colorectal and pancreatic subcutaneous xenografts [Citation83]. They found that a single dose of Apo2L/TRAIL resulted in a significant IFP decrease and enhanced tissue uptake of gemcitabine and liposomal gemcitabine. Dulanermin was evaluated in a phase II trial in patients with advanced non-small-cell lung cancer [Citation83]. Although there was a trend towards increased caspase-cleaved cytokeratin-18 after dulanermin treatment, the addition of the investigational product did not improve outcome compared to chemotherapy (paclitaxel, carboplatin and bevacizumab) alone.
An interesting approach was reported by Lu et al., who developed paclitaxel-based “tumour priming” microparticles (TPM), consisting of two components with different drug release rates: one component releasing the drug load rapidly to induce tumour priming, and a second component providing sustained drug release [Citation84]. In an ovarian xenograft model, they showed that IP TPM resulted in enhanced tumour tissue concentration of paclitaxel and lower toxicity compared to the commercial paclitaxel/cremophor formulation.
Another interesting approach is the use of tumour-penetrating peptides in order to enhance delivery of small molecule or nano-sized drugs that are co-administered intraperitoneally. Tumour-penetrating peptides such as iRGD require binding to αv integrins, which are abundantly expressed on the surface of cancer and endothelial cells. Upon binding, the peptide is modified through proteolytic cleavage, which exposes a C-terminal amino acid sequence (“CendR”). This CendR motif binds to neuropilin 1 and/or 2, initiating a specific bulk transport mechanism that related to, but distinct from micropinocytosis [Citation85,Citation86]. Importantly, iRGD transports not only compounds that are directly conjugated to its N-terminus, but also compounds that are co-administered with it [Citation87]. Several authors have explored the use of IP iRGD in order to enhance delivery of conjugated or co-administered compounds. Sugahara et al. administered IP doxorubine with or without IP iRGD in mice bearing human gastric (MKN45P) and colorectal (Lovo-6) carcinomatosis [Citation88]. They found that co-administration of iRGD lead to a significant increase in doxorubicine tumour tissue concentration in peritoneal nodules. Simon-Gracia et al. used paclitaxel-loaded iRGD polymersomes in human gastric and colon carcinomatosis mouse models [Citation89]. They found that iRGD functionalization of the polymersomes leads to enhanced tumour penetration and improved antitumour efficacy compared to the untargeted polymersomes or albumin-bound paclitaxel.
Drugs that alter stromal pH
Diffusion of basic chemotherapeutic drugs such as doxorubicin may be impeded by binding to acidic structures and organelles. Therefore, modification of the stromal pH may improve penetration of certain drugs. This concept was recently demonstrated by Patel et al., who showed that the proton pump inhibitor pantoprazole increased endosomal pH, increased nuclear uptake and tissue penetration of doxorubicin in multilayered cell cultures, and led to increased growth delay in a mouse MCF7 xenograft model [Citation90]. The same group reported similar effects on the biodistribution and efficacy of doxorubicin for the proton pump inhibitor lansoprazole [Citation91].
Conclusion and future perspectives
The efficacy of intraperitoneally administered anticancer drugs is limited by poor tissue penetration. The mechanisms that govern interstitial drug transport after IP administration remain incompletely understood. Novel mathematical models, as well as experiments using in vitro setups, are expected to generate important insights.
Investigator has targeted the drug itself, its carrier fluid, the tumour cell population and the stromal components in order to achieve better tissue penetration. While many of these approaches are successful in animal models, very few successful clinical applications are available thus far. Some of the interventions described above, such as administration of antiangiogenic agents, are impossible to implement in the perioperative clinical setting. Most likely, novel pharmaceutical formulations and constructs including thermosensitive gels and tumour penetrating peptides have the greatest potential for future clinical IP therapy.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the article.
References
- Flessner MF. (2008). Endothelial glycocalyx and the peritoneal barrier. Perit Dial Int 28:6–12.
- Gardner SN. (2000). A mechanistic, predictive model of dose-response curves for cell cycle phase-specific and -nonspecific drugs. Cancer Res 60:1417–25.
- Markman M. (2008). Antineoplastic agents in the management of ovarian cancer: current status and emerging therapeutic strategies. Trends Pharmacol Sci 29:515–19.
- El-Kareh AW, Secomb TW. (2004). A theoretical model for intraperitoneal delivery of cisplatin and the effect of hyperthermia on drug penetration distance. Neoplasia 6:117–27.
- Baxter LT, Jain RK. (1989). Transport of fluid and macromolecules in tumors. I. Role of interstitial pressure and convection. Microvasc Res 37:77–104.
- Bird RB, Stewart WE, Lightfoot EN. (2007). Transport phenomena (revised 2nd ed.). New York: John Wiley & Sons.
- Messori L, Merlino A. (2016). Cisplatin binding to proteins: a structural perspective. Coord Chem Rev 315:67–89.
- Ivanov AI, Christodoulou J, Parkinson JA, et al. (1998). Cisplatin binding sites on human albumin. J Biol Chem 273:14721–30.
- Steuperaert M, Debbaut C, Segers P, Ceelen W. (in press). Modelling drug transport during intraperitoneal chemotherapy. Pleura and Peritoneum.
- Dedrick RL, Flessner MF. (1997). Pharmacokinetic problems in peritoneal drug administration: tissue penetration and surface exposure. J Natl Cancer Inst 89:480–7.
- Los G, Mutsaers PH, Lenglet WJ, et al. (1990). Platinum distribution in intraperitoneal tumors after intraperitoneal cisplatin treatment. Cancer Chemother Pharmacol 25:389–94.
- Heldin CH, Rubin K, Pietras K, Ostman A. (2004). High interstitial fluid pressure – an obstacle in cancer therapy. Nat Rev Cancer 4:806–13.
- Young EW. (2013). Cells, tissues, and organs on chips: challenges and opportunities for the cancer tumor microenvironment. Integr Biol (Camb) 5:1096–109.
- Li H, Fan X, Houghton J. (2007). Tumor microenvironment: the role of the tumor stroma in cancer. J Cell Biochem 101:805–15.
- De Wever O, Mareel M. (2003). Role of tissue stroma in cancer cell invasion. J Pathol 200:429–47.
- Jamin Y, Boult JK, Li J, et al. (2015). Exploring the biomechanical properties of brain malignancies and their pathologic determinants in vivo with magnetic resonance elastography. Cancer Res 75:1216–24.
- Zaleska-Dorobisz U, Kaczorowski K, Pawlus A, et al. (2014). Ultrasound elastography – review of techniques and its clinical applications. Adv Clin Exp Med 23:645–55.
- Baker AM, Bird D, Lang G, et al. (2013). Lysyl oxidase enzymatic function increases stiffness to drive colorectal cancer progression through FAK. Oncogene 32:1863–8.
- Paszek MJ, Zahir N, Johnson KR, et al. (2005). Tensional homeostasis and the malignant phenotype. Cancer Cell 8:241–54.
- Sun Z, Guo SS, Fassler R. (2016). Integrin-mediated mechanotransduction. J Cell Biol 215:445–56.
- Butcher DT, Alliston T, Weaver VM. (2009). A tense situation: forcing tumour progression. Nat Rev Cancer 9:108–22.
- Chauhan VP, Lanning RM, Diop-Frimpong B, et al. (2009). Multiscale measurements distinguish cellular and interstitial hindrances to diffusion in vivo. Biophys J 97:330–6.
- Esquis P, Consolo D, Magnin G, et al. (2006). High intra-abdominal pressure enhances the penetration and antitumor effect of intraperitoneal cisplatin on experimental peritoneal carcinomatosis. Ann Surg 244:106–12.
- Jacquet P, Stuart OA, Chang D, Sugarbaker PH. (1996). Effects of intra-abdominal pressure on pharmacokinetics and tissue distribution of doxorubicin after intraperitoneal administration. Anticancer Drugs 7:596–603.
- Facy O, Al Samman S, Magnin G, et al. (2012). High pressure enhances the effect of hyperthermia in intraperitoneal chemotherapy with oxaliplatin: an experimental study. Ann Surg 256:1084–8.
- Tempfer CB, Celik I, Solass W, et al. (2014). Activity of pressurized intraperitoneal aerosol chemotherapy (PIPAC) with cisplatin and doxorubicin in women with recurrent, platinum-resistant ovarian cancer: preliminary clinical experience. Gynecol Oncol 132:307–11.
- Solass W, Kerb R, Murdter T, et al. (2014). Intraperitoneal chemotherapy of peritoneal carcinomatosis using pressurized aerosol as an alternative to liquid solution: first evidence for efficacy. Ann Surg Oncol 21:553–9.
- Solass W, Giger-Pabst U, Zieren J, Reymond MA. (2013). Pressurized intraperitoneal aerosol chemotherapy (PIPAC): occupational health and safety aspects. Ann Surg Oncol 20:3504–11.
- Blanco A, Giger-Pabst U, Solass W, et al. (2013). Renal and hepatic toxicities after pressurized intraperitoneal aerosol chemotherapy (PIPAC). Ann Surg Oncol 20:2311–16.
- Venkatasubramanian R, Henson MA, Forbes NS. (2008). Integrating cell-cycle progression, drug penetration and energy metabolism to identify improved cancer therapeutic strategies. J Theor Biol 253:98–117.
- Kyle AH, Huxham LA, Yeoman DM, Minchinton AI. (2007). Limited tissue penetration of taxanes: a mechanism for resistance in solid tumors. Clin Cancer Res 13:2804–10.
- Toley BJ, Tropeano Lovatt ZG, Harrington JL, Forbes NS. (2013). Microfluidic technique to measure intratumoral transport and calculate drug efficacy shows that binding is essential for doxorubicin and release hampers Doxil. Integr Biol (Camb) 5:1184–96.
- De Smet L, Ceelen W, Remon JP, Vervaet C. (2013). Optimization of drug delivery systems for intraperitoneal therapy to extend the residence time of the chemotherapeutic agent. ScientificWorldJournal 2013:720858.
- Van Oudheusden TR, Grull H, Dankers PY, De Hingh IH. (2015). Targeting the peritoneum with novel drug delivery systems in peritoneal carcinomatosis: a review of the literature. Anticancer Res 35:627–34.
- Dakwar GR, Shariati M, Willaert W, et al. (2017). Nanomedicine-based intraperitoneal therapy for the treatment of peritoneal carcinomatosis – mission possible? Adv Drug Deliv Rev 108:13–24.
- Sen A, Capitano ML, Spernyak JA, et al. (2011). Mild elevation of body temperature reduces tumor interstitial fluid pressure and hypoxia and enhances efficacy of radiotherapy in murine tumor models. Cancer Res 71:3872–80.
- Hauck ML, Coffin DO, Dodge RK, et al. (1997). A local hyperthermia treatment which enhances antibody uptake in a glioma xenograft model does not affect tumour interstitial fluid pressure. Int J Hyperthermia 13:307–16.
- Klaver YLB, Hendriks T, Lomme RMLM, et al. (2011). Hyperthermia and intraperitoneal chemotherapy for the treatment of peritoneal carcinomatosis: an experimental study. Ann Surg 254:125–30.
- Los G, Sminia P, Wondergem J, et al. (1991). Optimisation of intraperitoneal cisplatin therapy with regional hyperthermia in rats. Eur J Cancer 27:472–7.
- Zeamari S, Floot B, van der Vange N, Stewart FA. (2003). Pharmacokinetics and pharmacodynamics of cisplatin after intraoperative hyperthermic intraperitoneal chemoperfusion (HIPEC). Anticancer Res 23:1643–8.
- Facy O, Radais F, Ladoire S, et al. (2011). Comparison of hyperthermia and adrenaline to enhance the intratumoral accumulation of cisplatin in a murine model of peritoneal carcinomatosis. J Exp Clin Cancer Res 30:4.
- Moen I, Tronstad KJ, Kolmannskog O, et al. (2009). Hyperoxia increases the uptake of 5-fluorouracil in mammary tumors independently of changes in interstitial fluid pressure and tumor stroma. BMC Cancer 9:446.
- Leunig M, Goetz AE, Gamarra F, et al. (1994). Photodynamic therapy-induced alterations in interstitial fluid pressure, volume and water content of an amelanotic melanoma in the hamster. Br J Cancer 69:101–3.
- Perentes JY, Wang Y, Wang X, et al. (2014). Low-dose vascular photodynamic therapy decreases tumor interstitial fluid pressure, which promotes liposomal doxorubicin distribution in a murine sarcoma metastasis model. Transl Oncol 7:393–9.
- Osborne EM, Briere TM, Hayes-Jordan A, et al. (2016). Survival and toxicity following sequential multimodality treatment including whole abdominopelvic radiotherapy for patients with desmoplastic small round cell tumor. Radiother Oncol 119:40–4.
- Znati CA, Rosenstein M, Boucher Y, et al. (1996). Effect of radiation on interstitial fluid pressure and oxygenation in a human tumor xenograft. Cancer Res 56:964–8.
- Davies Cde L, Lundstrom LM, Frengen J, et al. (2004). Radiation improves the distribution and uptake of liposomal doxorubicin (caelyx) in human osteosarcoma xenografts. Cancer Res 64:547–53.
- Khosrawipour V, Bellendorf A, Khosrawipour C, et al. (2016). Irradiation does not increase the penetration depth of doxorubicin in normal tissue after pressurized intra-peritoneal aerosol chemotherapy (PIPAC) in an ex vivo model. In Vivo 30:593–7.
- Sassaroli E, O’Neill BE. (2014). Modulation of the interstitial fluid pressure by high intensity focused ultrasound as a way to alter local fluid and solute movement: insights from a mathematical model. Phys Med Biol 59:6775–95.
- O’Neill BE, Vo H, Angstadt M, et al. (2009). Pulsed high intensity focused ultrasound mediated nanoparticle delivery: mechanisms and efficacy in murine muscle. Ultrasound Med Biol 35:416–24.
- Li T, Wang YN, Khokhlova TD, et al. (2015). Pulsed high-intensity focused ultrasound enhances delivery of doxorubicin in a preclinical model of pancreatic cancer. Cancer Res 75:3738–46.
- Wang S, Shin IS, Hancock H, et al. (2012). Pulsed high intensity focused ultrasound increases penetration and therapeutic efficacy of monoclonal antibodies in murine xenograft tumors. J Control Release 162:218–24.
- Carmeliet P, Jain RK. (2011). Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov 10:417–27.
- Fuso Nerini I, Cesca M, Bizzaro F, Giavazzi R. (2016). Combination therapy in cancer: effects of angiogenesis inhibitors on drug pharmacokinetics and pharmacodynamics. Chin J Cancer 35:61.
- Gremonprez F, Descamps B, Izmer A, et al. (2015). Pretreatment with VEGF(R)-inhibitors reduces interstitial fluid pressure, increases intraperitoneal chemotherapy drug penetration, and impedes tumor growth in a mouse colorectal carcinomatosis model. Oncotarget 6:29889–900.
- Skliarenko JV, Lunt SJ, Gordon ML, et al. (2006). Effects of the vascular disrupting agent ZD6126 on interstitial fluid pressure and cell survival in tumors. Cancer Res 66:2074–80.
- Lindner P, Heath D, Howell S, et al. (1996). Vasopressin modulation of peritoneal, lymphatic, and plasma drug exposure following intraperitoneal administration. Clin Cancer Res 2:311–17.
- Tsiftsis D, de Bree E, Romanos J, et al. (1999). Peritoneal expansion by artificially produced ascites during perfusion chemotherapy. Arch Surg 134:545–9.
- Mahteme H, Sundin A, Larsson B, et al. (2005). 5-FU uptake in peritoneal metastases after pretreatment with radioimmunotherapy or vasoconstriction: an autoradiographic study in the rat. Anticancer Res 25:917–22.
- Duvillard C, Benoit L, Moretto P, et al. (1999). Epinephrine enhances penetration and anti-cancer activity of local cisplatin on rat sub-cutaneous and peritoneal tumors. Int J Cancer 81:779–84.
- Guardiola E, Chauffert B, Delroeux D, et al. (2010). Intraoperative chemotherapy with cisplatin and epinephrine after cytoreductive surgery in patients with recurrent ovarian cancer: a phase I study. Anticancer Drugs 21:320–5.
- Royer B, Kalbacher E, Onteniente S, et al. (2012). Intraperitoneal clearance as a potential biomarker of cisplatin after intraperitoneal perioperative chemotherapy: a population pharmacokinetic study. Br J Cancer 106:460–7.
- Oman M, Lundqvist S, Gustavsson B, et al. (2005). Phase I/II trial of intraperitoneal 5-Fluorouracil with and without intravenous vasopressin in non-resectable pancreas cancer. Cancer Chemother Pharmacol 56:603–9.
- Podobnik B, Sersa G, Miklavcic D. (2001). Effect of hydralazine on interstitial fluid pressure in experimental tumours and in normal tissue. In Vivo 15:417–24.
- Olsson PO, Gustafsson R, In ’t Zandt R, et al. (2016). The tyrosine kinase inhibitor imatinib augments extracellular fluid exchange and reduces average collagen fibril diameter in experimental carcinoma. Mol Cancer Ther 15:2455–64.
- Provenzano PP, Cuevas C, Chang AE, et al. (2012). Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 21:418–29.
- Hingorani SR, Harris WP, Beck JT, et al. (2016). Phase Ib study of PEGylated recombinant human hyaluronidase and gemcitabine in patients with advanced pancreatic cancer. Clin Cancer Res 22:2848–54.
- Kato M, Hattori Y, Kubo M, Maitani Y. (2012). Collagenase-1 injection improved tumor distribution and gene expression of cationic lipoplex. Int J Pharm 423:428–34.
- Eikenes L, Bruland OS, Brekken C, Davies Cde L. (2004). Collagenase increases the transcapillary pressure gradient and improves the uptake and distribution of monoclonal antibodies in human osteosarcoma xenografts. Cancer Res 64:4768–73.
- Brown E, McKee T, diTomaso E, et al. (2003). Dynamic imaging of collagen and its modulation in tumors in vivo using second-harmonic generation. Nat Med 9:796–800.
- Perentes JY, McKee TD, Ley CD, et al. (2009). In vivo imaging of extracellular matrix remodeling by tumor-associated fibroblasts. Nat Methods 6:143–5.
- Chauhan VP, Martin JD, Liu H, et al. (2013). Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun 4:2516.
- Diop-Frimpong B, Chauhan VP, Krane S, et al. (2011). Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc Natl Acad Sci U S A 108:2909–14.
- Kumar V, Boucher Y, Liu H, et al. (2016). Noninvasive assessment of losartan-induced increase in functional microvasculature and drug delivery in pancreatic ductal adenocarcinoma. Transl Oncol 9:431–7.
- Olive KP, Jacobetz MA, Davidson CJ, et al. (2009). Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324:1457–61.
- Ko AH, LoConte N, Tempero MA, et al. (2016). A phase I study of FOLFIRINOX Plus IPI-926, a hedgehog pathway inhibitor, for advanced pancreatic adenocarcinoma. Pancreas 45:370–5.
- Kim EJ, Sahai V, Abel EV, et al. (2014). Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin Cancer Res 20:5937–45.
- Sherman MH, Yu RT, Engle DD, et al. (2014). Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 159:80–93.
- Shany S, Sigal-Batikoff I, Lamprecht S. (2016). Vitamin D and myofibroblasts in fibrosis and cancer: at cross-purposes with TGF-beta/SMAD signaling. Anticancer Res 36:6225–34.
- Moschetta M, Pretto F, Berndt A, et al. (2012). Paclitaxel enhances therapeutic efficacy of the F8-IL2 immunocytokine to EDA-fibronectin-positive metastatic human melanoma xenografts. Cancer Res 72:1814–24.
- Taghian AG, Abi-Raad R, Assaad SI, et al. (2005). Paclitaxel decreases the interstitial fluid pressure and improves oxygenation in breast cancers in patients treated with neoadjuvant chemotherapy: clinical implications. J Clin Oncol 23:1951–61.
- Geretti E, Leonard SC, Dumont N, et al. (2015). Cyclophosphamide-mediated tumor priming for enhanced delivery and antitumor activity of her2-targeted liposomal doxorubicin (MM-302). Mol Cancer Ther 14:2060–71.
- Hylander BL, Sen A, Beachy SH, et al. (2015). Tumor priming by Apo2L/TRAIL reduces interstitial fluid pressure and enhances efficacy of liposomal gemcitabine in a patient derived xenograft tumor model. J Control Release 217:160–9.
- Lu Z, Tsai M, Lu D, et al. (2008). Tumor-penetrating microparticles for intraperitoneal therapy of ovarian cancer. J Pharmacol Exp Ther 327:673–82.
- Sugahara KN, Teesalu T, Karmali PP, et al. (2010). Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science 328:1031–5.
- Sugahara KN, Teesalu T, Karmali PP, et al. (2009). Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell 16:510–20.
- Akashi Y, Oda T, Ohara Y, et al. (2014). Anticancer effects of gemcitabine are enhanced by co-administered iRGD peptide in murine pancreatic cancer models that overexpressed neuropilin-1. Br J Cancer 110:1481–7.
- Sugahara KN, Scodeller P, Braun GB, et al. (2015). A tumor-penetrating peptide enhances circulation-independent targeting of peritoneal carcinomatosis. J Control Release 212:59–69.
- Simon-Gracia L, Hunt H, Scodeller P, et al. (2016). iRGD peptide conjugation potentiates intraperitoneal tumor delivery of paclitaxel with polymersomes. Biomaterials 104:247–57.
- Patel KJ, Lee C, Tan Q, Tannock IF. (2013). Use of the proton pump inhibitor pantoprazole to modify the distribution and activity of doxorubicin: a potential strategy to improve the therapy of solid tumors. Clin Cancer Res 19:6766–76.
- Yu M, Lee C, Wang M, Tannock IF. (2015). Influence of the proton pump inhibitor lansoprazole on distribution and activity of doxorubicin in solid tumors. Cancer Sci 106:1438–47.