
Abstract
Background: As environmental and body temperatures vary, lung epithelial cells experience temperatures significantly different from normal core temperature. Our previous studies in human lung epithelium showed that: (i) heat shock accelerates wound healing and activates profibrotic gene expression through heat shock factor-1 (HSF1); (ii) HSF1 is activated at febrile temperatures (38–41 °C) and (iii) hypothermia (32 °C) activates and hyperthermia (39.5 °C) reduces expression of a subset of miRNAs that target protein kinase-Cα (PKCα) and enhance proliferation.
Methods: We analysed the effect of hypo- and hyperthermia exposure on Wnt signalling by exposing human small airway epithelial cells (SAECs) and HEK293T cells to 32, 37 or 39.5 °C for 24 h, then analysing Wnt-3a-induced epithelial–mesenchymal transition (EMT) gene expression by qRT-PCR and TOPFlash reporter plasmid activity. Effects of miRNA mimics and inhibitors and the HSF1 inhibitor, KNK437, were evaluated.
Results: Exposure to 39.5 °C for 24 h increased subsequent Wnt-3a-induced EMT gene expression in SAECs and Wnt-3a-induced TOPFlash activity in HEK293T cells. Increased Wnt responsiveness was associated with HSF1 activation and blocked by KNK437. Overexpressing temperature-responsive miRNA mimics reduced Wnt responsiveness in 39.5 °C-exposed HEK293T cells, but inhibitors of the same miRNAs failed to restore Wnt responsiveness in 32 °C-exposed HEK293T cells.
Conclusions: Wnt responsiveness, including expression of genes associated with EMT, increases after exposure to febrile-range temperature through an HSF1-dependent mechanism that is independent of previously identified temperature-dependent miRNAs. This process may be relevant to febrile fibrosing lung diseases, including the fibroproliferative phase of acute respiratory distress syndrome (ARDS) and exacerbations of idiopathic pulmonary fibrosis (IPF).
Introduction
Pulmonary epithelia perform multiple essential functions required for homeostasis and response to infection and injury, including secretion of mucins [Citation1], antimicrobial peptide [Citation2], surfactant [Citation3], gas exchange [Citation3] and detoxification of xenobiotics [Citation4]. Epithelial cells engage in bidirectional communication with lung fibroblasts, which is essential to lung development and lung healing, but may also contribute to fibrosis [Citation5]. Epithelial dysfunction is associated with reversible and chronic obstructive lung disease [Citation6], pulmonary fibrosis [Citation7], recurrent infections [Citation8] and acute lung injury [Citation9], and most lung cancers are epithelial in origin [Citation10]. Potential mechanisms linking epithelial injury, lung fibrosis and neoplasia includes: loss of antifibrotic factors such as hepatocyte growth factor (HGF), keratinocyte growth factor (KGF) and prostaglandin (PG)-E2, epithelial to mesenchymal transition (EMT), and expression of profibrotic factors such as TGFβ [Citation11]. Wnt signalling is central to many of these disease pathways [Citation12].
We [Citation13–16] and others [Citation17,Citation18] have shown that modest deviations in temperature within the clinically relevant range impact cell survival and function of lung epithelium, including regulation of the heat shock response. Specifically, exposure to febrile-range temperature (∼39.5 °C) sensitises lung epithelium to Fas-associated protein with death domain (FADD)/caspase-8-dependent apoptosis [Citation13,Citation17] and stimulates submaximal heat shock factor-1 (HSF1) activation and heat shock protein (HSP)-72 expression [Citation16]. We have also shown that activation of HSF1 in human lung epithelial cells accelerates wound healing and modifies gene expression, including activation of several profibrotic genes [Citation15]. We recently showed that expression of a small subset of microRNAs in human lung epithelium is temperature-dependent, induced by hypothermia (32 °C) and reduced at febrile-range temperature (39.5 °C) [Citation14]. Three of the hypothermia-induced miRNAs-targeted protein kinase-Cα (PKCα), reduced PKCα protein levels, and enhanced proliferation by releasing cells from a PKCα-mediated G1/S proliferation block. Since the lung epithelium is exposed to a temperature gradient between ambient and core temperatures [Citation19], the epithelium may be exposed to temperatures well below or well above normal core temperature, depending on ambient and core temperature, minute ventilation, location within the bronchoalveolar tract, and deviations in core temperature during fever and hypothermia.
In this article, we have extended our previous study by analysing the effects of clinically relevant hypo- and hyperthermic exposures on Wnt signalling in primary cultured human small airway epithelial cells (SAECs) and HEK293T cells. We demonstrate that that exposure to 39.5 °C increases responsiveness to Wnt ligand and subsequent expression of genes that drive EMT through a mechanism that is independent of our previously identified temperature-dependent miRNAs but requires activated HSF1.
Materials and methods
Cell culture and treatment
Primary cultured human SAECs (lot no. 58704924) isolated from a 37-year-old Caucasian male were purchased from the ATCC (Manassas, VA, USA) and maintained in airway epithelial cell basal medium supplemented with the SAEC growth kit (ATCC) at 37 °C and 5% CO2, in automatic CO2 incubators. BEAS2B cells were obtained from ATCC and cultured in DMEM with high glucose (Gibco Thermo Fisher) supplemented with 2 mM l-glutamine, 1 mM sodium pyruvate, 10 mM HEPES buffer, pH 7.3 and 5% heat-inactivated foetal bovine serum (FBS). HEK293T cells were obtained from ATCC and were maintained at 37 °C in Dulbecco’s Minimal Essential Medium (DMEM) with 4.5 g/L glucose and supplemented with 5% FBS and 1% penicillin–streptomycin. Recombinant human Wnt-3a (Wnt-3a) was purchased from R&D Biosystems (cat# 5036-WN-010). KNK437 was purchased from EMD Millipore (Billerica, MA, USA) and cells were pre-treated for 60 min at 30 μM final concentration in 0.1% DMSO.
Immunoblotting
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer containing protease and phosphatase inhibitors and resolved by SDS–PAGE. Proteins were transferred to polyvinylidene fluoride membrane (Immobilon-FL; Millipore) blocked with Odyssey™ Blocking Buffer (LI-COR; Lincoln, NE, USA), probed with primary mouse antibodies against β-catenin (BD Biosciences; San Jose, CA, USA; cat#610154), transcription factor-7 (TCF7) (Novus Biologicals LLC; Littleton, CO, USA; cat#H00006932-M01 and H00006932-M02) and HSP27 (Cell Signaling Technology; Danvers, MA, USA; cat#2402) and β-tubulin (Millipore; Cat#MAB3408), then secondary IRDye 680RD Goat anti-Mouse IgG (LI-COR). The immunoblots were imaged on a LI-COR Odyssey infra-red imaging system and band densities were quantified using the LI-COR software.
TOPFlash reporter assay
HEK293T cells were seeded at 70 000 cells/well in 24-well plates and pairs of monolayers were transfected 24 h later with 500 ng/well reporter plasmid TOPFlash driven by TCF/lymphoid enhancer-binding factor (LEF) response elements or the control FOPFlash reporter plasmid in which the TCF/LEF sequences have been mutated (Addgene; Cambridge, MA, USA) and 5 ng the renilla luciferase expression plasmid, pRL (Promega; Madison, WI, USA) using lipofectamine 2000 (Life Technologies) as per manufacturer instructions. The cells were allowed to recover at 37 °C for 24 h, then were incubated at 32, 37 or 39.5 °C for an additional 24 h. The cells were then switched back to 37 °C and treated with 200 ng/ml Wnt-3a and lysed 24 h later using passive lysis buffer (Promega). Dual luciferase assay was performed using the dual luciferase assay kit (Promega) as per standard protocol. The firefly luciferase activities of TOPFlash and FOPFlash constructs were corrected for transfection efficiency by normalising to renilla luciferase activity. The normalised TOPFlash was expressed as a ratio to the normalised FOPFlash activity in corresponding monolayers.
Quantitative RT-PCR analysis of baseline and wnt-3a responsive gene expression
Primary-cultured SAEC monolayers were incubated at 32, 37 or 39.5 °C for 24 h, then switched back to 37 °C and treated with 200 ng/ml recombinant human Wnt-3a for 3 h for early Wnt-responsive genes or 24 h for EMT genes. Cells were lysed and total RNA was extracted using Trizol™ (Thermo Fisher) as per the manufacturer’s instructions. The purity and concentration of RNA samples were determined by absorbance at 260 and 280 nm (Nanodrop; Thermo-Fisher Scientific). cDNA was generated from 1 μg of total RNA by reverse transcription using iScript™ cDNA Synthesis Kit (Bio-Rad; Hercules, CA, USA; cat#1708891). qRT-PCR was performed using SsoFast™ EvaGreen® Supermix (Bio-Rad cat#1725204) on a Biorad CFX96 or StepOnePlus™ Real-Time PCR System (Thermo Fisher) as per the manufacturer’s instructions and the primer sequences listed in . Ct values were normalised to glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and fold-change vs. 37 °C was calculated using the delta-delta method [Citation16].
Table 1. PCR primers used for quantitative RT-PCR.
Transfection with miRNA mimics and inhibitors
HEK293T cells were transfected with miRNA mimics and inhibitors as we have previously described [Citation14]. Briefly, cells were seeded in six-well cell culture plates (BD Biosciences) and transfected 24 h later. For experiments with miRNA mimics, the cells were transfected with a combination of synthetic miRNA mimics for hsa-miR-92a1-5p (50 nM), hsa-miR-27b-5p (50 nM) and hsa-miR-1260a (25 nM) or 125 nM scrambled sequence control MISSION® microRNA mimics (Sigma-Aldrich; St. Louis, MO, USA). For experiments with miRNA inhibitors, the cells were transfected with pEZX-AM03 plasmid-based miRNA inhibitors from Genecopoeia (Rockville, MD, USA) against hsa-miR-92a-1-5p (cat# HmiR-AN0831-AM03-B), hsa-miR-27b-5p (cat# HmiR-AN0360-AM03-B) and hsa-miR-1260a (cat# HmiR-AN0100-AM03-B) or a control plasmid with scrambled sequence (cat# CmiR-AN0001-AM03-B). All transfections were performed using lipofectamine 2000 (Thermo Fisher). Cells were recovered at 37 °C for 24 h, then incubated for 24 h at 32, 37 or 39.5 °C, lysed in RIPA, and immunoblotted for TCF7, ß-catenin and ß-tubulin.
Immunofluorescence microscopy
Human SAECs were seeded at 10 000 cells/well in Millicell EZ SLIDE four-well glass slides (EMD Millipore) and incubated at 37 °C for 24 h, then switched to 32, 37 or 39.5 °C for the next 24 h. The cells were then treated with 200 ng/ml Wnt-3a and switched back to 37 °C for 3 h. Cells were washed twice with ice-cold phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde in PBS. Cells were blocked for 1 h at room temperature (RT) in blocking buffer [PBS, 0.03% Triton, 1% bovine serum albumin (BSA) and 1% donkey serum], incubated overnight at 4 °C with mouse anti-β-catenin (BD Biosciences cat#610154) in blocking buffer. Cells were washed twice with washing buffer (PBS, 0.03% Triton). Cells were incubated with donkey anti-mouse CY3-conjugate (Jackson Immunoresearch; West Grove, PA, USA) for 1 h at RT, washed twice with washing buffer, and stained with 4,6-diamidine-2-phenylindole, dihydrochloride (DAPI) (Kirkegaard & Perry Laboratories Inc.; Gaithersburg, MD, USA; cat# 710301) for 1 min at RT. Cells were washed once with PBS and the slides were sealed with coverslips. To analyse HSF1 activation, triplicate SAEC monolayers were exposed to 39.5 °C for 1, 3 or 24 h or to 42 °C for 2 h. Some monolayers were recovered at 37 °C for 3 h after 24 h exposure to 39.5 °C. Cells were immunostained as described above using rabbit anti-HSF1 primary antibody (Santa Cruz Biotechnology; Santa Cruz, CA, USA; cat# sc9144) and donkey anti-rabbit Cy3-conjugate (Jackson Immunoresearch). Cells were visualised using Olympus (Tokyo, Japan) confocal microscope and Olympus Fluo View software.
Statistics
For quantitative RT-PCR and immunoblot data, differences between treatment groups were analysed by repeat Mann–Whitney test using JMP 9 (SAS Institute Inc.; Cary, NC, USA). Differences among more than two treatment conditions were analysed by one-way analysis of variance (ANOVA). In some cases, we applied a post hoc Tukey’s Honestly Significant Difference test to compare pairs of treatments.
Results
Exposure to clinically relevant hypothermia and hyperthermia modifies canonical Wnt-3a-dependent gene expression and transactivation
To assess the impact of hypo- and hyperthermia exposure on canonical Wnt signalling in human lung epithelium, we analysed the responsiveness of SAECs to the soluble Wnt ligand, Wnt-3a, and measured expression of Wnt-dependent genes. After 24 h pre-exposure to 32, 37 or 39.5 °C, all cells were incubated with 200 ng/ml Wnt-3a for 3 h at 37 °C and expression levels of four Wnt-dependent genes, axin-2, cyclin D2 (CCND2), matrix metallopeptidase (MMP)-2 and avian v-myc myelocytomatosis viral oncogene homologue (c-Myc), were analysed by qRT-PCR (). All four genes exhibited the same temperature-dependent expression pattern, 44–74% reduction in expression at 32 °C and 1.5–2-fold increase at 39.5 °C, consistent with reduced canonical Wnt/β-catenin signalling after exposure to hypothermia and enhanced signalling after exposure to hyperthermia. Importantly, none of these four genes are predicted to be direct targets of our previously identified temperature-sensitive miRNAs [Citation14]. Wnt-induced expression of a fifth gene, MMP7, which is a predicted direct target of the temperature-responsive miRNA, has-miR-92a-5p, was reduced by 11% and increased by 18-fold in 32 and 39.5 °C cells, respectively ().
Figure 1. Effects of clinically relevant hypothermia and hyperthermia on canonical Wnt-3a signalling capacity. (A–E) SAECs were incubated at 32, 37 or 39.5 °C for 24 h, switched back to 37 °C, treated with 200 ng/ml recombinant human Wnt-3a for 3 h, total RNA was collected and mRNA levels of AXIN2 (A), CCND2 (B), c-Myc (C), MMP2 (D) and MMP7 (E) were measured by qRT-PCR and expressed as fold-change vs. levels in 37 °C cells. (F) HEK293T cells treated similarly and Axin2 mRNA levels were measured. (G) HEK293T cells were co-transfected with TOPFlash or FOPFlash and pRL renila luciferase control plasmid, recovered for 24 h at 37 °C, incubated at 32, 37 or 39.5 °C for 24 h, then activated with 200 ng/ml Wnt-3a for 24 h at 37 °C, and firefly and renilla luciferase measured. Firefly luciferase activity was normalised to renilla activity and the ratio of TOPFlash:FOPFlash calculated and expressed as fold-change vs. 37 °C. Mean ± standard error (SE) of three experiments for qRT-PCR and six experiments for TOPFlash. Asterisk (*) denotes p < 0.05 vs. 37 °C and †p < 0.01 vs. 32 °C.
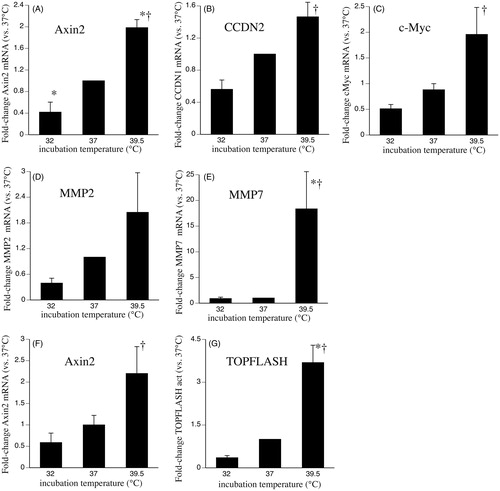
Wnt-3a activates gene expression through the canonical β-catenin pathway by stimulating the nuclear translocation of β-catenin where it complexes with intranuclear TCF7/LEF1 to form a transcription factor that transactivates promoters containing TCF-binding sites [Citation20]. Activation of the canonical pathway was measured in HEK293T cells using the TOPFlash luciferase reporter plasmid, which contains multiple TCF-binding sites in its promoter region. To confirm that HEK293T cells exhibit the same temperature-dependent gene expression as SAECs, we quantified axin-2 mRNA in HEK293T cells treated with same hypo- and hyperthermia exposure and Wnt-3a stimulation protocol as the SAECs. Levels of axin-2 mRNA were reduced by 42% in 32 °C-exposed HEK293T cells and increased 2.2-fold in cells 39.5 °C-exposed cells (). In HEK293T cells transfected with the TOPFlash reporter plasmid, Wnt-3a-induced TOPFlash activity was reduced by 65% in 32 °C-exposed cells and increased by 270% in 39.5 °C-exposed cells compared with 37 °C cells ().
Effect of hypothermia and hyperthermia on levels and distribution of TCF7 and β-catenin
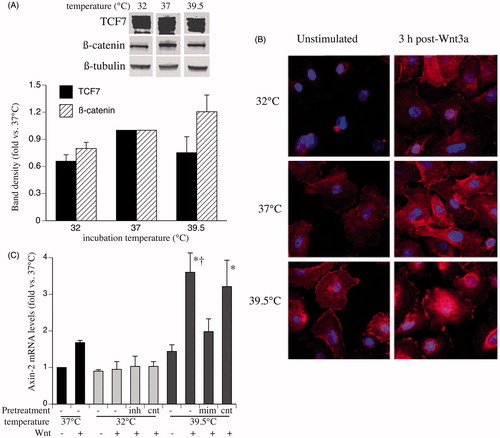
Since Wnt-induced gene expression and TOPFlash activity is driven by increased intranuclear levels of TCF7 and ß-catenin, we compared whole-cell levels of TCF7 and ß-catenin protein and Wnt-induced nuclear translocation of ß-catenin in SAECs incubated at 32, 37 and 39.5 °C. Immunoblot analysis showed similar levels of the two proteins in cells lysates from SAECs incubated at the three temperatures (). To assess whether pre-exposure to 32 or 39.5 °C altered Wnt-induced nuclear translocation of ß-catenin, we analysed cellular distribution of ß-catenin in SAECs after 24 h pre-exposure to 32, 37 or 39.5 °C followed by 3 h stimulation with 200 ng/ml Wnt-3a at 37 °C. Immunofluorscent confocal imaging of immunostained cells showed greater Wnt-induced nuclear ß-catenin translocation in 39.5 °C pre-exposed cells and reduced ß-catenin translocation in 32 °C pre-exposed cells compared with 37 °C cells ().
Figure 2. Effects of clinically relevant hypothermia and hyperthermia on cell levels and distribution of TCF7 and β-catenin. (A) SAECs were incubated at 32, 37 or 39.5 °C for 24 h and whole-cell lysate was immunoblotted for β-catenin, TCF7 and ß-tubulin. The band densities were quantified, normalised to ß-tubulin band density and expressed as fold-change vs. 37 °C. A representative immunoblot and a graph summarising the mean ± standard error (SE) of four experiments is shown. (B) SAECs grown on chamber slides were incubated at 32, 37 or 39.5 °C for 24 h and either fixed (left panels) or switched back to 37 °C and stimulated with 200 ng/ml Wnt-3a for 3 h and fixed. The fixed monolayers were immunostained for β-catenin (red) and DAPI (blue) and analysed by confocal immunofluorescence microscopy. Representative fields from three similar experiments are shown. (C) HEK293T cells were transfected with either inhibitors (inh) or mimics (mim) of the three temperature-responsive miRNAs (miR ×3), scrambled sequence controls for each (cnt) or nothing (−), recovered for 24 h, incubated at the indicated temperature for 24 h, then stimulated with 200 ng/ml Wnt-3a at 37 °C for 3 h and Axin2 mRNA levels were measured by qRT-PCR and expressed as fold-change vs. unstimulated 37 °C cells. Mean ± SE of three experiments. Asterisk (*) and dagger (†) denote p < 0.05 vs. unstimulated 39.5 °C and mimic-pre-treated cells, respectively.
Analysis of potential targets of temperature-dependent miRNAs
An in silico search for predicted gene targets of our previously identified temperature-dependent miRNAs using miRTar algorithm [Citation21] () identified Wnt signalling as the pathway most likely to be modified by these miRNAs. Wnt signalling comprises three overlapping pathways, the canonical, planar cell polarity (PCP), and Wnt/Ca2+ pathways [Citation20]. Of the 182 genes known to participate in Wnt signalling, 43 were targeted by at least one of the following temperature-sensitive miRNAs, hsa-miR-92a-1–5p, 24, hsa-miR-1260a or hsa-miR-27b-5p. The 43 genes participate in one or more Wnt pathways, including 26 that participate in the canonical pathway, 17 in the PCP pathway and 14 in the Wnt/Ca2+ pathway (). Since these miRNAs are induced at 32 °C and reduced at 39.5 °C and are predicted to target Wnt-signalling elements, we reasoned that they may mediate the effects of hypo- and hyperthermia exposure on Wnt-signalling capacity.
Table 2. Pathways targeted by a representative temperature sensitive miRNA hsa-miR-92a-1–5pTable Footnotea.
Table 3. Wnt-signalling genes targeted by temperature-sensitive miRNAsTable Footnotea.
To analyse the potential contribution of our previously identified temperature-dependent miRNAs to temperature-dependent Wnt-signalling capacity, we measured Wnt-3a-induced axin-2 expression in HEK293T cells after transfection with inhibitors of hsa-miR-92a1–5p (50 nM), hsa-miR-27b-5p (50 nM) and hsa-miR-1260a and incubation at 32 °C for 24 h or in HEK293T cells after transfection with mimics of these three miRNAs and incubation at 39.5 °C for 24 h. Wnt-3a-induced axin-2 expression was increased in the 39.5 °C pre-exposed cells and tended to be decreased in the 32 °C pre-exposed cells compared with 37 °C cells. Treating 39.5 °C-exposed cells with miRNA mimics reduced Wnt-3a-induced axin-2, but treating 32 °C-exposed cells with miRNA inhibitors failed to increase Wnt-3a-induced axin-2 expression ().
Impact of temperature-dependent Wnt signalling on genes involved in EMT and fibrosis
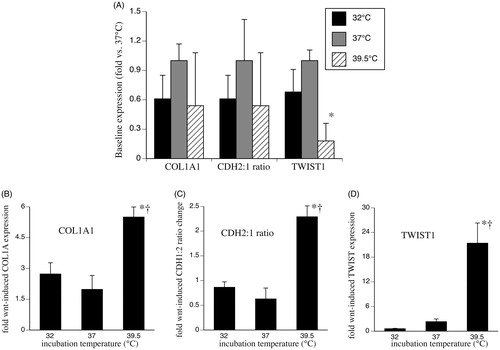
Wnt signalling stimulates EMT gene expression, including increasing levels of collagen type I (COL1A) [Citation12] and twist family basic helix–loop–helix transcription factor-1 (TWIST1) [Citation22], and increasing the ratio of N-cadherin (CDH2) to E-cadherin (CDH1) [Citation23]. To determine whether the temperature-dependent changes in Wnt signalling are associated with EMT gene expression, we analysed baseline and Wnt-3a-induced expression of these EMT markers in human SAECs after 24 h pre-incubation at 32, 37 or 39.5 °C. Wnt-induced EMT gene expression was measured after treating the cells with 200 ng/ml Wnt-3a for 24 h at 37 °C. Incubating SAECs at 32 °C for 24 h had no significant effect on either baseline or Wnt-3a-stimulated expression of COL1A1 or TWIST1 or CDH2:1 ratio (). Incubating SAECs at 39.5 °C for 24 h reduced baseline TWIST1 expression by 80% and increased Wnt-3a-stimulated expression of COL1A1 and TWIST1 by 2.8-fold and 9.2-fold, respectively and increased the CDH2:1 ratio by 3.6-fold compared with Wnt-3a-stimulated 37 °C cells.
Figure 3. Effect of temperature-dependent Wnt signalling on genes involved in EMT and fibrosis. SAECs were incubated at 32, 37 or 39.5 °C for 24 h and total RNA was collected for analysis of basal EMT gene expression (A) or the cells were stimulated with 200 ng/ml Wnt-3a for 24 h at 37 °C and total RNA collected for analysis of Wnt-3a-stimulated gene expression (B–D). Type I collagen-α (COL1A1) (B), ratio of N-cadherin (CDH-2) to E-cadherin (CDH-1) (C) and TWIST1 (D) were quantified by qRT-PCR and expressed as fold-change vs. unstimulated 37 °C cells. Mean ± standard error (SE) of four experiments are shown. Asterisk (*) denotes p < 0.005 vs. 37 °C and †p < 0.02 vs. 32 °C.
Contribution of HSF1 to hyperthermia effects on Wnt-3a signalling
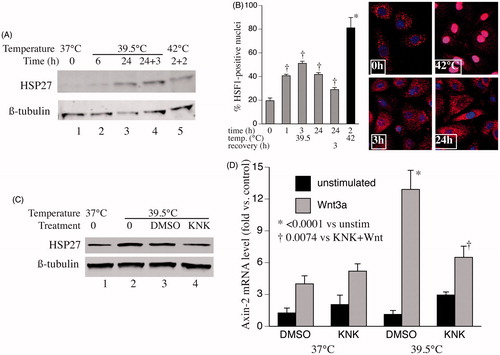
While hypothermia may reduce Wnt-signalling capacity, in part through increased expression of one or more temperature-responsive miRNAs, the inability of miRNA inhibitors to increase Wnt responsiveness in the 32 °C-exposed cells suggests the participation of other temperature-dependent mechanisms. The heat/stress-activated transcription factor, HSF1 is activated at 38–39.5 °C [Citation16] and can regulate expression of many genes that are not classic HSPs [Citation24], including >500 genes in cancer and cancer stromal cells [Citation25,Citation26]. Whether HSF1 modifies expression of any genes that participate in Wnt signalling is unknown. To assess whether HSF1 contributes to the increased Wnt-signalling capacity in 39.5 °C pre-exposed SAECs, we analysed HSF1 activation by measuring expression of a known HSF1-dependent gene, HSP27 (), and by analysing HSF1 nuclear translocation () in SAECs incubated at 37 or 39.5 °C. Levels of HSP27 protein increased after 6 h exposure to 39.5 °C. Nuclear translocation of HSF1 was evident in 39.5 °C-exposed cells after 1 h and persisted for 24 h, but less so than occurred during 42 °C exposure. To evaluate whether HSF1 was required for the increased Wnt responsiveness in the 39.5 °C-exposed cells, we used KNK-437, a pharmacologic inhibitor of HSF1-dependent transcription [Citation27]. Pretreating SAECs with 30 μM KNK-437 prior to initiating the 39.5 °C exposure blocked both HSP27 expression () and the increase in Wnt-3a-induced axin-2 expression ().
Figure 4. Contribution of HSF1 to hyperthermia-enhanced Wnt-signalling capacity. (A) SAECs were incubated at 39.5 °C for 6 or 24 h, 39.5 °C for 24 h then 37 °C for 3 h, or 42 °C for 2 h then 37 °C for 2 h. Cells were lysed and immunoblotted for HSP27 and ß-tubulin (representative of three similar blots). (B) SAECs were grown on cover slips and incubated at 39.5 °C for 0, 1, 3 or 24 h, at 39.5 °C for 24 h then 37 °C for 3 h, or 42 °C for 2 h. Cells were fixed, immunostained for HSF1 and DAPI, and imaged by confocal immunofluorescence. Representative images for 0, 3 and 24 h at 39.5 °C and 2 h at 42 °C. Percent of cells with nuclear HSF1 staining calculated and mean ± standard error (SE) from three experiments shown. Dagger (†) denotes p < 0.009 vs. time 0, *p < 0.0001 vs. all other treatments. (C) SAECs were pre-treated for 30 min with nothing (0), vehicle (1% DMSO) or 30 μM KNK437 then incubated at 39.5 °C for 24 h, lysed and immunoblotted for HSP27 and ß-tubulin (representative of three similar blots). (D) SAECs were pre-treated with DMSO or 30 μM KNK437 for 30 min, then at 37 or 39.5 °C for 24 h, then with or without 200 ng/ml Wnt-3a for 3 h. Cells were lysed, total RNA collected and axin mRNA quantified by qRT-PCR and expressed as percent of untreated 37 °C control cells. Mean ± SE of six experiments. Asterisk (*) denotes p < 0.0001 vs. all unstimulated cells; †p = 0.007 vs. DMSO-treated, Wnt-stimulated 39.5 °C cells.
Discussion
We analysed the effects of exposure to hypo- and hyperthermia on basal levels of Wnt-dependent genes and the capacity for canonical Wnt signalling, utilising the canonical soluble Wnt ligand, Wnt-3a [Citation28,Citation29]. Prior studies of how hypothermia [Citation30] and hyperthermia [Citation31] affect Wnt signalling focussed on the temperature-dependent modification of ß-catenin levels, but at a lower hypothermic temperature (30 °C) and higher hyperthermic temperatures (43–48 °C) than the clinically relevant temperatures used in our study. The temperatures and the exposure time used in the present study were based on our previous study [Citation14] and are clinically relevant to accidental and therapeutic hypothermia exposures and febrile illnesses. Since the experiments were designed to determine how exposure to hypo- and hyperthermia modifies Wnt-signalling capacity, all cells were stimulated with Wnt-3a at 37 °C following hypo- or hyperthermia exposure. We used two measures of canonical Wnt signal activation, mRNA expression levels of five known early Wnt-dependent genes and Wnt-induced activation of the TOPFlash reporter plasmid. TOPFlash is a firefly luciferase reporter driven by an artificial promoter containing three copies of the TCF-binding sequence upstream of the thymidine kinase promoter and is activated when TCF7 and ß-catenin co-localize within the nucleus [Citation32]. Although the magnitude of the temperature-dependent changes in expression levels varied among the five Wnt-dependent genes, the pattern of expression, lower in 32 °C-exposed cells and higher in 39.5 °C cells was consistent and paralleled the effects of hypo- and hyperthermia on Wnt-induced TOPFlash activity. Expression of one of the genes, MMP7, was increased by hyperthermia exposure much more than the other four genes. It is possible that since MMP7 is the only one of these Wnt-responsive genes to be directly targeted by the temperature-responsive miRNAs, it may be modified by multiple temperature-dependent mechanisms.
The increased Wnt-dependent gene expression and TOPFlash activity in HEK293T cells exposed to 39.5 °C suggests higher Wnt-3a-induced intranuclear levels of TCF7 and ß-catenin after cells are exposed to 39.5 °C. Although ß-catenin and TCF7 are predicted to be targets of the temperature-responsive miRNAs [Citation14] (), immunoblotting failed to detect a significant effect of hypo- and hyperthermia exposure on whole cell levels of either protein. However, confocal immunofluorescence imaging showed that treatment with Wnt-3a induced greater ß-catenin nuclear translocation after 39.5 °C exposure and less after 32 °C exposure, suggesting that exposure to hypo- and hyperthermia modifies one or more elements in the canonical Wnt-signalling pathway.
Consistent with the canonical Wnt-signalling pathway containing 26 potential temperature-dependent miRNA targets, treatment with mimics of these miRNAs reduced Wnt responsiveness in 39.5 °C-exposed cells. However, treating 32 °C-exposed cells with inhibitors of the same miRNAs failed to increase Wnt-3a responsiveness to the levels in 37 or 39.5 °C cells. These data demonstrate that the temperature-responsive miRNAs can reduce Wnt-signalling capacity, but suggest that other factors are responsible for the changes in Wnt responsiveness following exposure to clinically relevant hypo- and hyperthermia.
We have shown that exposure to temperature within the usual febrile range activates a submaximal heat shock response [Citation16]. Chou et al. [Citation33] demonstrated that phosphorylation of HSF1 on serine 326 by mTOR stabilises β-catenin expression through the RNA binding protein HuR, but we found that hyperthermia increased Wnt-stimulated ß-catenenin nuclear translocation rather than whole cell levels of ß-catenin. Zhang et al. [Citation34] demonstrated that treating rat cardiac stem cells with low dose LPS increased total cell and nuclear levels of ß-catenin and the effect was blocked by the HSP90 inhibitor geldanamycin. Cordonnier et al. [Citation35] demonstrated that HSP27 depletion in prostate cancer cells reduced EGF-stimulated interaction of ß-catenin with GSK3ß, which is required for its phosphorylation and nuclear translocation, and EMT gene expression. HSP27 overexpression had the opposite effects, demonstrating a potential mechanism for HSF1 activation to increase Wnt-signalling capacity. Cytoplasmic HSF1 trimerizes and translocates to nucleus in response to stress [Citation36], including, exposure to febrile-range hyperthermia [Citation16]. In this study, exposing SAECs to 39.5 °C stimulated a submaximal nuclear translocation of HSF1 that occurred within 1 h and persisted for 24 h, and an increase in expression of HSP27. Pretreatment with the HSF1 transcriptional inhibitor, KNK437 [Citation27], blocked both HSP27 expression and the increase in Wnt-induced axin2 expression. Although the SAECs used in this study were obtained from a single individual, the SAECs and HEK293T cells had similar responses to shifts in temperature, suggesting the effect of hypo- and hyperthermia on Wnt-signalling capacity may be a common property of human cells. These results implicate gene expression driven by submaximal HSF1 activation in the increased Wnt-signalling capacity following exposure to febrile-range hyperthermia. Moreover, the data suggest that the hypo- and hyperthermia modify Wnt-signalling capacity through distinct mechanisms.
Canonical Wnt-dependent changes in EMT gene regulation, including a switch from E-cadherin to N-cadherin expression [Citation23], activation of the transcriptional repressor, TWIST1 [Citation22], and increase in type I collagen expression [Citation12], contributes to embryogenesis, tissue repair, fibrosis and cancer metastasis in multiple organs, including lung [Citation37–41]. Consistent with the effect of hyperthermia on Wnt responsiveness, Wnt-3a-induced expression of type I collagen and TWIST1 and switch from E-cadherin to N-cadherin expression in SAECs was increased after exposure to 39.5 °C. These gene products and MMP7, which was also expressed at higher levels in 39.5 °C-exposed SAECs, are associated with fibrosing disorders, including pulmonary fibrosis [Citation42–44]. The association of Wnt-signalling activation and human idiopathic pulmonary fibrosis (IPF) [Citation45] and the effectiveness of pharmacological Wnt-signalling inhibitors in reducing fibrosis in the rodent bleomycin-induced lung injury [Citation46] underscores the importance of understanding the modifiable factors that regulate Wnt signalling. To that end, we note that fever is often associated with certain fibrosing lung processes, including the Acute Respiratory Distress Syndrome (ARDS) [Citation47] and exacerbations of IPF [Citation48].
Conclusion
We have shown that exposure to clinically relevant hypothermia reduces and fever-range hyperthermia increases Wnt responsiveness and Wnt-stimulated EMT gene expression in human lung epithelium. These results support the importance of clinical studies to determine whether fever is profibrotic in these clinical settings and whether hypothermia or fever prevention, or inhibition of HSF1-dependent transcription will be protective against post-ARDS fibrosis and irreversible loss of lung function following IPF exacerbations.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Additional information
Funding
References
- Munkholm M, Mortensen J. (2014). Mucociliary clearance: pathophysiological aspects. Clin Physiol Funct Imaging 34:171–7.
- Hiemstra PS. (2007). The role of epithelial beta-defensins and cathelicidins in host defense of the lung. Exp Lung Res 33:537–42.
- Whitsett JA, Weaver TE. (2015). Alveolar development and disease. Am J Respir Cell Mol Biol 53:1–7.
- Hukkanen J, Pelkonen O, Hakkola J, Raunio H. (2002). Expression and regulation of xenobiotic-metabolizing cytochrome P450 (CYP) enzymes in human lung. Crit Rev Toxicol 32:391–411.
- Sakai N, Tager AM. (2013). Fibrosis of two: epithelial cell-fibroblast interactions in pulmonary fibrosis. Biochim Biophys Acta 1832:911–21.
- Crosby LM, Waters CM. (2010). Epithelial repair mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol 298:L715–31.
- Selman M, King TE, Pardo A, et al. (2001). Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med 134:136–51.
- Schaffer K. (2015). Epidemiology of infection and current guidelines for infection prevention in cystic fibrosis patients. J Hosp Infect 89:309–13.
- Edelson JD. (1995). Epithelial dysfunction in acute lung injury. New Horiz 3:229–39.
- Cheung WK, Nguyen DX. (2015). Lineage factors and differentiation states in lung cancer progression. Oncogene 34:5771–80.
- Thompson HG, Mih JD, Krasieva TB, et al. (2006). Epithelial-derived TGF-beta2 modulates basal and wound-healing subepithelial matrix homeostasis. Am J Physiol Lung Cell Mol Physiol 291:L1277–85.
- Akhmetshina A, Palumbo K, Dees C, et al. (2012). Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis. Nat Commun 3:735.
- Nagarsekar A, Tulapurkar ME, Singh IS, et al. (2012). Hyperthermia promotes and prevents respiratory epithelial apoptosis through distinct mechanisms. Am J Respir Cell Mol Biol 47:824–33.
- Potla R, Singh IS, Atamas SP, Hasday JD. (2015). Shifts in temperature within the physiologic range modify strand-specific expression of select human microRNAs. RNA 21:1261–73.
- Scheraga RG, Thompson C, Tulapurkar ME, et al. (2016). Activation of heat shock response augments fibroblast growth factor-1 expression in wounded lung epithelium. Am J Physiol Lung Cell Mol Physiol 311:L941–55.
- Tulapurkar ME, Asiegbu BE, Singh IS, Hasday JD. (2009). Hyperthermia in the febrile range induces HSP72 expression proportional to exposure temperature but not to HSF-1 DNA-binding activity in human lung epithelial A549 cells. Cell Stress Chaperones 14:499–508.
- Lipke AB, Matute-Bello G, Herrero R, et al. (2010). Febrile-range hyperthermia augments lipopolysaccharide-induced lung injury by a mechanism of enhanced alveolar epithelial apoptosis. J Immunol 184:3801–13.
- Lipke AB, Matute-Bello G, Herrero R, et al. (2011). Death receptors mediate the adverse effects of febrile-range hyperthermia on the outcome of lipopolysaccharide-induced lung injury. Am J Physiol Lung Cell Mol Physiol 301:L60–70.
- McFadden ER Jr, Pichurko BM. (1985). Intraairway thermal profiles during exercise and hyperventilation in normal man. J Clin Invest 76:1007–10.
- Rao TP, Kuhl M. (2010). An updated overview on Wnt signaling pathways: a prelude for more. Circ Res 106:1798–806.
- Hsu JB, Chiu CM, Hsu SD, et al. (2011). miRTar: an integrated system for identifying miRNA-target interactions in human. BMC Bioinformatics 12:300.
- Reinhold MI, Kapadia RM, Liao Z, Naski MC. (2006). The Wnt-inducible transcription factor Twist1 inhibits chondrogenesis. J Biol Chem 281:1381–8.
- Wheelock MJ, Shintani Y, Maeda M, et al. (2008). Cadherin switching. J Cell Sci 121:727–35.
- Singh IS, Shah NG, Almutairy EA, Hasday JD. (2009). Role of HSF1 in infectious disease. In: Pockley AG, Santoro MG, Calderwood SK, editors. Prokaryotic and eukaryotic heat shock proteins in infectious disease. Dordrecht Heidelberg London New York: Springer, 1–32.
- Mendillo ML, Santagata S, Koeva M, et al. (2012). HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 150:549–62.
- Scherz-Shouval R, Santagata S, Mendillo ML, et al. (2014). The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell 158:564–78.
- Yokota S, Kitahara M, Nagata K. (2000). Benzylidene lactam compound, KNK437, a novel inhibitor of acquisition of thermotolerance and heat shock protein induction in human colon carcinoma cells. Cancer Res 60:2942–8.
- Baarsma HA, Konigshoff M, Gosens R. (2013). The WNT signaling pathway from ligand secretion to gene transcription: molecular mechanisms and pharmacological targets. Pharmacol Ther 138:66–83.
- Gujral TS, MacBeath G. (2010). A system-wide investigation of the dynamics of Wnt signaling reveals novel phases of transcriptional regulation. PLoS One 5:e10024.
- Zhang H, Ren C, Gao X, et al. (2008). Hypothermia blocks beta-catenin degradation after focal ischemia in rats. Brain Res 1198:182–7.
- Olkku A, Leskinen JJ, Lammi MJ, et al. (2010). Ultrasound-induced activation of Wnt signaling in human MG-63 osteoblastic cells. Bone 47:320–30.
- Veeman MT, Slusarski DC, Kaykas A, et al. (2003). Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr Biol 13:680–5.
- Chou SD, Murshid A, Eguchi T, et al. (2015). HSF1 regulation of β-catenin in mammary cancer cells through control of HuR/elavL1 expression. Oncogene 34:2178–88.
- Zhang L, Geng WR, Hu J, et al. (2016). Lipopolysaccharide pretreatment promotes cardiac stem cell migration through heat shock protein 90-dependent beta-catenin activation. Life Sci 153:132–40.
- Cordonnier T, Bishop JL, Shiota M, et al. (2015). Hsp27 regulates EGF/β-catenin mediated epithelial to mesenchymal transition in prostate cancer. Int J Cancer 136:E496–507.
- Voellmy R. (2004). On mechanisms that control heat shock transcription factor activity in metazoan cells. Cell Stress ChaperonesChaperones 9:122–33.
- MacDonald BT, Tamai K, He X. (2009). Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell 17:9–26.
- Kalluri R, Weinberg RA. (2009). The basics of epithelial-mesenchymal transition. J Clin Invest 119:1420–8.
- Takebe N, Warren RQ, Ivy SP. (2011). Breast cancer growth and metastasis: interplay between cancer stem cells, embryonic signaling pathways and epithelial-to-mesenchymal transition. Breast Cancer Res 13:211.
- Thiery JP, Acloque H, Huang RY, Nieto MA. (2009). Epithelial-mesenchymal transitions in development and disease. Cell 139:871–90.
- Yang J, Weinberg RA. (2008). Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell 14:818–29.
- Hung C, Linn G, Chow YH, et al. (2013). Role of lung pericytes and resident fibroblasts in the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med 188:820–30.
- Kim KK, Kugler MC, Wolters PJ, et al. (2006). Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci USA 103:13180–5.
- Villar J, Cabrera NE, Valladares F, et al. (2011). Activation of the Wnt/beta-catenin signaling pathway by mechanical ventilation is associated with ventilator-induced pulmonary fibrosis in healthy lungs. PLoS One 6:e23914.
- Chilosi M, Poletti V, Zamo A, et al. (2003). Aberrant Wnt/beta-catenin pathway activation in idiopathic pulmonary fibrosis. Am J Pathol 162:1495–502.
- Henderson WR Jr, Chi EY, Ye X, et al. (2010). Inhibition of Wnt/beta-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc Natl Acad Sci USA 107:14309–14.
- Netzer G, Dowdy DW, Harrington T, et al. (2013). Fever is associated with delayed ventilator liberation in acute lung injury. Ann Am Thorac Soc 10:608–15.
- Ambrosini V, Cancellieri A, Chilosi M, et al. (2003). Acute exacerbation of idiopathic pulmonary fibrosis: report of a series. Eur Respir J 22:821–6.