
Abstract
Purpose: The aims of the present study were 2-fold: first, to test the hypothesis that heat stress induces MET and EGFR signalling in hepatocellular carcinoma (HCC) cells and inhibition of this signalling decreases HCC clonogenic survival; and second, to identify signalling pathways associated with heat stress induced MET signalling.
Materials and Methods: MET+ and EGFR+ HCC cells were pre-treated with inhibitors to MET, EGFR, PI3K/mTOR or vehicle and subjected to heat stress or control ± HGF or EGF growth factors and assessed by colony formation assay, Western blotting and/or quantitative mass spectrometry. IACUC approved partial laser thermal or sham ablation was performed on orthotopic N1S1 and AS30D HCC tumours and liver/tumour assessed for phospho-MET and phospho-EGFR immunostaining.
Results: Heat-stress induced rapid MET and EGFR phosphorylation that is distinct from HGF or EGF in HCC cells and thermal ablation induced MET but not EGFR phosphorylation at the HCC tumour ablation margin. Inhibition of the MET and EGFR blocked both heat stress and growth factor induced MET and EGFR phosphorylation and inhibition of MET decreased HCC clonogenic survival following heat stress. Pathway analysis of quantitative phosphoproteomic data identified downstream pathways associated with heat stress induced MET signalling including AKT, ERK, Stat3 and JNK. However, inhibition of heat stress induced MET signalling did not block AKT signalling.
Conclusions: Heat-stress induced MET and EGFR signalling is distinct from growth factor mediated signalling in HCC cells and MET inhibition enhances heat stress induced HCC cell killing via a PI3K/AKT/mTOR-independent mechanism.
Introduction
Hepatocellular carcinoma (HCC) is a deadly malignancy with an increasing incidence worldwide highlighting the urgent need for better treatments [Citation1–4]. Thermal ablative therapies are important interventional oncologic therapies in the multidisciplinary care of patients with early stage HCC, achieving short-term outcomes similar to surgery with less morbidity [Citation5–7]. However, as tumour size increases beyond 2–3 cm, thermal ablation of HCC is plagued by high local recurrence and intrahepatic tumour progression rates and the overall survival of these patients remains poor [Citation5–7]. The ablation margin is the most common site for HCC local tumour recurrence, suggesting incomplete treatment resulting from sublethal heat stress or increased tumoural resistance [Citation8–10]. Currently, no adjuvant therapies exist to prevent HCC local recurrence to thermal ablative therapies [Citation2]. However, recent studies suggest that inhibition of PI3K/AKT/mTOR signalling may enhance thermal ablation induced HCC cell killing [Citation11]. As such, fundamental basic and translational questions remain unanswered regarding alterations in HCC oncogenic signalling that may mediate HCC survival in response to heat stress [Citation12]. Consequently, there remains a critical need to delineate molecular mechanisms that mediate HCC local recurrence, particularly at the ablation margin, to identify adjuvant therapeutic targets for enhancing thermal ablation-induced HCC cell killing.
Receptor tyrosine kinases (RTKs) are key integrators of extracellular signals and induce a wide-range of intracellular signalling pathways [Citation13]. As a general model, binding of specific ligands (growth factors, cytokines) to the extracellular RTK binding domain induces receptor homodimerization or heterodimerization and subsequent auto(trans)phosphorylation of the c-terminal kinase domain, thereby activating numerous downstream signalling pathways [Citation14]. Growth factor signalling mediated by RTKs is frequently dysregulated in cancer, including HCC [Citation12,Citation15,Citation16]. In particular, MET and EGFR have been shown to have biological, therapeutic and prognostic significance in HCC [Citation17–23]. However, it is not known if mechanisms beyond binding of growth factor ligands such as heat stress can induce MET and EGFR signalling in HCC.
The aims of the present study were 2-fold: first, to test the hypothesis that heat stress induces MET and EGFR signalling in HCC cells and inhibition of this signalling decreases HCC clonogenic survival; and second, to identify signalling pathways associated with heat stress induced MET signalling.
Materials and methods
Cell lines
Clone9, N1S1, Hep3B, HepG2, PLC/PRF/5, SNU-387, SNU-423, SNU-449, SNU-475 (ATCC, Manassas, VA), AS30D (DSMZ, Braunschweig, Germany) and HuH-7 (JCRB Cell Bank, Japan) cell lines were cultured according to supplier recommendations.
Small molecule inhibitors
Erlotinib, PF-042179030 and NVP-BEZ235 were purchased from Selleck Chemicals (Houston, TX); SU11274 was from EMD Millipore (Calbiochem®, Billerica, MA).
Growth factors
Recombinant rat EGF (3214-EG) and recombinant mouse/rat HGF (2207-HG) were purchased from R&D Systems (Minneapolis, MN).
Antibodies
Antiphosphotyrosine (P-Tyr-1000; #8954), Antiphospho-Tyr1234/1235-MET (#3077), MET (#3127; #8198), antiphospho-Tyr1068-EGFR (#3777), EGFR (#2646; #4267), antiphospho-Ser473-AKT (#4060, #9271), AKT (#4691), antiphospho-Thr202/Tyr204-p44/42 MAPK (Erk1/2) (#9101; #4370), p44/42 MAPK (Erk1/2) (#9102, #4695), PRAS40 (#2691), antiphospho-Thr246-PRAS40, 4E-BP1 (#9644), antiphospho-Thr37/46–4E-BP1, SAPK/JNK (#9252), antiphospho-Thr183/Tyr185-SAPK/JNK, ATF-2 (#9226), antiphospho-Thr71-ATF-2, antiphospho-Ser63-c-Jun (#2361), antiphospho-Ser73-c-Jun (#3270), c-Jun (#9165), antiphospho-Ser897-EphA2 (#6347), EphA2 (#6997) and Vimentin (#5741) antibodies were purchased from Cell Signalling Technology (Danvers, MA); anti-Beta Actin antibody (sc-1615) was purchased from Santa Cruz Biotechnology (Dallas, TX). Peroxidase-labelled affinity purified anti-rabbit IgG (074–1506), anti-mouse IgG (074–1806) and anti-goat IgG (14–13-06) antibodies were purchased from KPL (Gaithersburg, MD).
Cellular heat stress protocol
The indicated cell lines were suspended in complete media in 1.5 ml microcentrifuge tubes and allowed to equilibrate to 37 °C in an incubator. Next, the cells were placed in a 45 °C isothermic water bath for 10 min (heat stress) or 37 °C (physiologic control) [Citation11,Citation24,Citation25]. Treatment temperature was monitored continuously using an Omega HH41 digital thermometer (Omega Engineering, Stamford, CT) and maintained to within ±0.05 °C. 45 °C for 10 min was chosen as the thermal dose for heat stress based on prior experiments which empirically determined in vitro heat stress conditions (45 °C for 10 min) that induce a significant reduction in cell survival but still result in colony formation whereas 50 °C for 10 min resulted in complete cell killing with no colony formation and 37 °C for 10 min had no effect on cell survival or colony formation [Citation11]. This in vitro model recapitulates the thermal injury experienced at the margin of the ablation zone in vivo where both neoplastic and parenchymal cells experience heat stress that may or may not induce complete cell killing. As such, 45 °C for 10 min was used as the thermal dose based on the incomplete HCC cell killing.
For all experiments using a small molecule inhibitor, cells were pre-treated with the designated small molecule inhibitor (SU11274, PF-04217903, Erlotinib or BEZ235) or vehicle control (0.1% DMSO) for one hour prior to heat stress. For experiments utilising recombinant growth factors, HGF or EGF was administered concurrently at the time of heat stress for 10 min after the 1-h pre-treatment with small molecule inhibitor or vehicle control. Cells were harvested immediately after the 10-min heat stress, rinsed in PBS and snap frozen in liquid N2 for Western blot or mass spectrometry analysis or analysed using a colony formation assay.
For the dose titration experiments in , N1S1 and AS30D HCC cells were treated with a dose-titration of the MET inhibitor SU11274 (0.04–10 μM), EGFR inhibitor erlotinib (0.08–20 μM) or vehicle control (0.1% DMSO) and assessed for viability at 72 h using a WST-1 assay. For the combination small molecule inhibitor plus heat stress experiments in , N1S1 and AS30D HCC were cells pre-treated with SU11274 (1 and 10 μM), erlotinib (1 and 10 μM) or vehicle control (0.1% DMSO) for 1 h, heat stressed (45 °C) or control (37 °C) for 10 min, harvested immediately post-heat stress and whole-cell lysates were subjected to Western blotting. For the combination small molecule inhibitor and growth factor experiments in , N1S1 cells were pre-treated for 1 h with SU11274 (5 μM) or vehicle control (0.1% DMSO) followed by heat stress (45 °C) or control (37 °C) ± concomitant treatment with recombinant HGF (50 ng/ml) or vehicle control for 10 min. Immediately post-heat stress whole-cell lysates were subjected to Western blotting. For the combination small molecule inhibitor and growth factor experiments in , AS30D cells were pre-treated for 1 h with erlotinib (5 μM) or vehicle control (0.1% DMSO) followed by heat stress (45 °C) or control (37 °C) ± concomitant treatment with recombinant EGF (50 ng/ml) or vehicle control for 10 min. Immediately post-heat stress whole-cell lysates were subjected to Western blotting. For the combination, small molecule inhibitor plus heat stress experiments in , N1S1 HCC cells were pre-treated with a dose-titration of the MET inhibitor PF-042179030 (0.1–10 μM) or vehicle control (0.1% DMSO) and AS30D HCC cells were pre-treated with a dose-titration of the EGFR inhibitor erlotinib (0.1–10 μM) or vehicle control (0.1% DMSO) for 1 h and subjected to heat stress (45 °C) or control (37 °C) for 10 min and immediately plated for a colony formation assay. For the combination, small molecule inhibitor plus heat stress experiments in , N1S1 cells were pre-treated with a dose-titration of the MET inhibitor PF-042179030 (0.1–10 μM) or vehicle control (0.1% DMSO) for 1 h followed by heat stress (45 °C) or control (37 °C) for 10 min. Immediately post-heat stress whole-cell lysates were subjected to Western blotting. For the combination, small molecule inhibitor plus heat stress experiments in , N1S1 cells were pre-treated with the PI3K/mTOR inhibitor BEZ235 (1 μM) or vehicle control (0.1% DMSO) for 1 h followed by heat stress (45 °C) or control (37 °C) for 10 min. Immediately post-heat stress whole-cell lysates were subjected to Western blotting.
Cell viability
WST-1 assay (Roche) was performed per manufacturer instruction as previously described [Citation25]. Non-linear regression curve fitting was used to calculate an IC50 using Prism 5.0 (GraphPad Software, Inc., La Jolla, CA).
Soft agar colony formation assay (clonogenic survival)
Soft agar colony formation assay was performed as previously described [Citation11]. Two to four technical replicates and three biological replicates were used per experimental condition. Per cent colony formation per cell number plated was calculated and the data normalised to the non-heat stressed, 37 °C vehicle control to calculate per cent colony formation.
Western blotting
Cells were washed with ice cold calcium- and magnesium-free PBS, lysed in cell lysis buffer (#9803; Cell Signalling Technology) per manufacturer instructions and protein concentration determined using Bradford assay (ThermoScientific). Whole-cell lysates were separated by SDS-PAGE and electrophoretically transferred to PVDF or nitrocellulose, blotted for the indicated antigens using the indicated primary antibodies per manufacturer’s instruction followed by incubation with corresponding secondary antibodies (1:8000 or 1:10 000) and developed using a LI-COR infra-red imaging system or incubated with enhanced chemiluminesence (ECL, Pierce, Thermo Fisher, Minneapolis, MN), exposed to film (Kodak/Carestream, Rochester, NY) and developed using a Kodak X-Omat M20 processor. Digital images were captured using a Kodak 440CF gel documentation system.
PVDF membranes were used for all Western blots in whereas nitrocellulose was used for the Western blots in . Given that both total and phospho-MET and -EGFR were the principal proteins of interest in these experiments and these are higher molecular weight proteins (140–175 kDa) with relatively lower abundance, PVDF membranes were chosen for their increased sensitivity and improved detection of these higher MW proteins. For the Western blots in , nitrocellulose was used due to improved detection of smaller molecular weight proteins (<20–30 kDa) and the relatively higher abundance of the proteins examined.
Kinomeview® profiling
Per Cell Signalling Technology (CST; Danvers, MA) protocol, 30 µg of total protein was run in each lane for Western blotting using a phosphotyrosine pY-1000 antibody (#8803) as previously described [Citation11,Citation26].
Quantitative mass spectrometry (LC-MS/MS) and phosphoproteomic pathway analysis
Per Cell Signalling Technology (CST; Danvers, MA) protocol, samples were analysed using the PTMScan method as previously described [Citation26–28]. The Phosphotyrosine pY-1000 antibody (#8803), Multipathway Reagent [Citation27] and Basophilic Motif Antibody Mix (#32948) were used for peptide enrichment. The protein interaction networks were generated from the Ingenuity Pathway Analysis (IPA) (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis) as previously described [Citation28,Citation29]. Please see supplemental methods for further details on the LC-MS/MS methods and phosphoproteomic pathway analysis.
Tumour ablation in orthotopic N1S1 and AS30D HCC models
All animal studies were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of the Mayo Clinic (Protocol Number: A31010). The N1S1 orthotopic HCC model was developed and tumour-bearing rats were imaged using non-contrast enhanced 3 T magnetic resonance imaging (MRI; GE Healthcare, Milwaukee, WI) to measure tumour size and confirm tumour location as previously described [Citation11,Citation24,Citation30,Citation31]. Adult male Sprague–Dawley rats (400–500 g) were randomised to percutaneous thermal ablation (N = 6) or sham ablation (N = 3) using an FDA-approved 980-nm laser generator (Visualase, Houston, TX) as previously described [Citation11,Citation24,Citation25,Citation30,Citation31]. Under ultrasound-guidance using an L8–18i transducer (logiq E9 Ultrasound, GE Healthcare, Milwaukee, WI), a bare 400 μm core optical laser fibre with a 1.0 cm diffusing tip was percutaneously inserted through a 22-gauge introducer sheath at the tumour margin and a 22-gauge needle with a 25-gauge wire thermocouple (Valleylab, Boulder, CO) was inserted 4–5 mm from the laser fibre tip within the tumour for continuous intraprocedural temperature monitoring. For the ablation group, tumours were ablated at a power setting of 3 watts (W) under continuous US-monitoring and the ablation stopped when the thermocouple reached 45 °C in order to generate an intentional partial ablation. The laser was not turned on for sham-ablated animals. Rats were euthanized at 6 or 24 h post-ablation to assess early liver/tumour response to thermal injury. Experiments were repeated in the AS30D HCC model using methods previously described [Citation32].
Gross and microscopic pathology
Liver/tumour tissue was removed and 2-mm cross-sections cut encompassing the ablation-zone. All specimens were formalin-fixed paraffin embedded (FFPE) and sectioned for immunohistochemical analysis. FFPE sections were stained with antiphospho-antibodies against the indicated antigens per manufacturer instruction as previously described [Citation11,Citation24,Citation25,Citation30,Citation32].
Statistical analyses
Statistical analyses were performed using Prism 5.0 (GraphPad Software, Inc., La Jolla, CA). For the colony formation experiments combining MET and EGFR inhibition plus heat stress, differences between vehicle plus heat stress and inhibitor plus heat stress groups were compared using one-way analysis of variance (ANOVA) followed by post-hoc pairwise comparison using an unpaired t test. All p values were two-tailed and p < 0.05 was considered statistically significant.
Results
Heat stress induces rapid changes in kinome-wide protein tyrosine phosphorylation in HCC cells and hepatocytes
Using an experimental model of post heat stress in vitro recurrence, we hypothesised that heat stress induces rapid changes in cell signalling [Citation11]. A panel of hepatocyte and HCC cell lines were heat stressed (45 °C–10 min) or control (37 °C) and assessed by Western blotting using a Phospho-Motif antibody that has broad immunoreactivity for tyrosine as previously described [Citation11]. Western blot analysis identified significant changes in global tyrosine phosphorylation across all cell lines () suggesting that heat stress induces rapid changes in the global tyrosine phosphorylation in both hepatocytes and HCC cells.
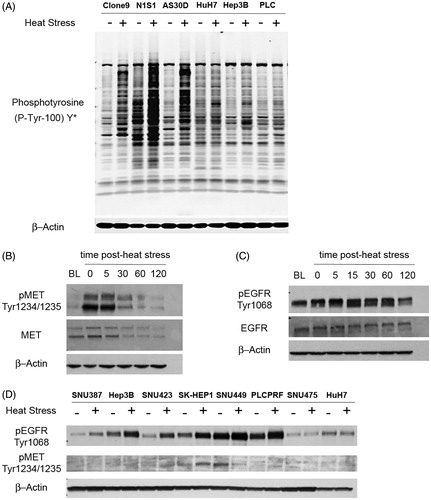
Figure 1. Heat stress induces rapid MET and EGFR signalling in HCC cells in vitro. (A) Western blot analysis of kinome-wide changes in protein tyrosine phosphorylation in response to heat stress in hepatocytes and HCC cells. A panel of rat hepatocyte (Clone 9) and HCC (N1S1, AS30D) cell lines and human HCC cell lines (HuH7, Hep3B, PLC/PRF/5) were heat stressed (45 °C) or control (37 °C) for 10 min, harvested immediately post-heat stress and whole-cell lysates were subjected to Western blotting using a phosphotyrosine (P-Tyr-100) Y* antibody with broad reactivity to tyrosine phosphorylated proteins and peptides. The antibody does not cross-react with phospho-serine or phospho-threonine residues. β-Actin was used as a loading control. (B) N1S1 and (C) AS30D HCC cell lines cells were heat stressed (45 °C for 10 min), recovered up to 2-h post heat stress and whole-cell lysates were subjected to Western blotting for phospho and total MET and EGFR. β-actin was used as a loading control. BL: baseline, non-heat stress control; t = time post heat stress in minutes. For example, t = 0 indicates immediate post-heat stress. (D) A panel of human HCC cell lines were heat stressed (45 °C) or control (37 °C) for 10 min, harvested immediately post-heat stress and whole-cell lysates were subjected to Western blotting using phospho-specific antibodies against EGFR and MET. β-actin was used as a loading control. Representative images from n ≥ 3 independent experiments.
Heat stress induces rapid MET and EGFR phosphorylation in vitro and thermal ablation induces MET but not EGFR phosphorylation at the tumour ablation margin in vivo
Having established that heat stress induces rapid, global changes in tyrosine phosphorylation in the absence of exogenous ligand in both hepatocytes and HCC cells, we sought to determine if heat stress induces autophosphorylation of receptor tyrosine kinases (RTKs). Prior gene expression data suggested that the N1S1 and AS30D HCC cell lines express the MET (hepatocyte growth factor receptor) and EGFR (epidermal growth factor receptor) RTKs, respectively [Citation11]. N1S1 and AS30D cells were heat stressed and Western blotting demonstrated that the N1S1 and AS30D cell lines express MET () and EGFR () proteins and that heat stress induces a rapid, time-dependent increase in both phosphotyrosine-MET1234/1235 and phosphotyrosine-EGFR1068. Furthermore, Western blot analysis demonstrated that heat stress induced rapid, increased phosphorylation of EGFR in six of eight human HCC cell lines while there was evidence of minimal heat stress induced MET phosphorylation in the SK-HEP1 cell line only (). Taken together, these data demonstrate that heat stress rapidly induces RTK and cell-line dependent auto-phosphorylation of MET and EGFR in the absence of exogenous ligand.
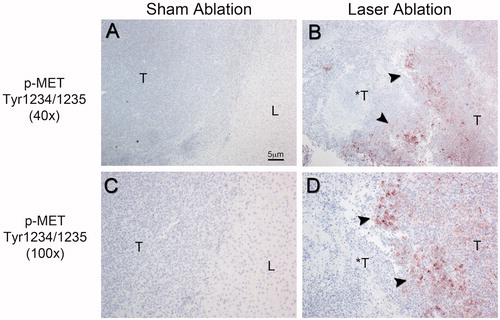
Moreover, immunohistochemical analysis of the ablation zone from the laser ablated MET+ N1S1 tumours demonstrated markedly increased immunostaining for phospho-MET at the tumour ablation margin () but not the liver ablation margin (data not shown). Additionally, there was decreasing phospho-MET tumour immunostaining further from the tumour ablation margin in the non-ablated regions of the tumour. There was no evidence of increased immunostaining for phospho-MET in the tumour or at the margin between liver and tumour in the sham ablation group (). Repeat ablation experiments in the EGFR+ AS30D HCC tumour model demonstrated no appreciable increase in phospho-EGFR at the tumour ablation margin (Supplementary Figure S1). In short, these data suggest that thermal ablation induces MET phosphorylation but not EGFR phosphorylation at the tumour ablation margin in vivo.
Figure 2. Representative phospho-MET immunohistochemical staining of the ablation zone 24-h post-ablation in the orthotopic N1S1 HCC model. Low power (40×; A,B) and high power (100×; C,D) photomicrographs of phospho-MET immunostained sections. (A,C) Sham ablated tumour. No evidence of MET phosphorylation in the tumour (T), background liver (L) or at the tumour-liver margin in the sham-ablated tumour. (B,D) Laser ablated tumour. Markedly increased MET phosphorylation at the tumour-ablation margin (black arrowheads) in the laser-ablated tumour (*T) with decreased MET phosphorylation further from the ablation margin toward the non-ablated tumour (T). T: non-ablated tumour; *T: ablated tumour; L: normal background liver.
Heat stress and growth factors differentially increase MET and EGFR signalling and inhibition of the MET and EGFR cytoplasmic kinase domain blocks heat stress and growth factor induced MET and EGFR signalling
To investigate the dependence of the MET+ N1S1 and EGFR+ AS30D cell lines on basal MET and EGFR signalling, both cell lines were treated with a dose-titration of small molecule MET and EGFR inhibitors and assessed for cell viability [Citation33,Citation34]. Inhibition of MET resulted in a dose-dependent decrease in N1S1 cell viability with an IC50 of ∼4.1 μM () whereas there was minimal effect on the AS30D cell line (IC50>10 μM; Supplementary Figure S2A). Conversely, inhibition EGFR had no effect on the viability of AS30D cells (IC50>20 μM; ) or N1S1 cells (IC50>20 μM; Supplementary Figure S2B), thereby suggesting that the MET+ N1S1 cell line is in part dependent on MET signalling under basal conditions whereas the AS30D cell line is not dependent on EGFR signalling under basal conditions in vitro.
Figure 3. Effect of MET and EGFR inhibition on heat stress and/or growth factor induced MET and EGFR signalling in HCC cells. (A) N1S1 and (B) AS30D HCC cells were treated with a dose-titration of the MET inhibitor SU11274 (0.04–10 μM), EGFR inhibitor erlotinib (0.08–20 μM) or vehicle control (0.1% DMSO) and assessed for viability at 72 h using WST-1 assay. Data were normalised to vehicle control and the IC50 estimated using non-linear regression curve fitting. Data are presented as mean ± SEM of three independent experiments. (C) N1S1 and (D) AS30D HCC cells pre-treated with SU11274 (1 and 10 μM), erlotinib (1 and 10 μM) or vehicle control (0.1% DMSO) for 1 h, heat stressed (45 °C) or control (37 °C) for 10 min, harvested immediately post-heat stress and whole-cell lysates were subjected to Western blotting using phospho-specific and total antibodies against MET and EGFR. β-actin was used as a loading control. (E) N1S1 cells pre-treated for 1 h with SU11274 (5 μM) or vehicle control (0.1% DMSO) followed by heat stress (45 °C) or control (37 °C) ± concomitant treatment with recombinant HGF (50 ng/ml) or vehicle control for 10 min. Immediately post-heat stress whole-cell lysates were subjected to Western blotting for phospho and total MET. β-Actin was used as a loading control. (F) AS30D cells pre-treated for 1 h with erlotinib (5 μM) or vehicle control (0.1% DMSO) followed by heat stress (45 °C) or control (37 °C) ± concomitant treatment with recombinant EGF (50 ng/ml or vehicle control for 10 min. Immediately post-heat stress whole-cell lysates were subjected to Western blotting for phospho and total EGFR. β-actin was used as a loading control.
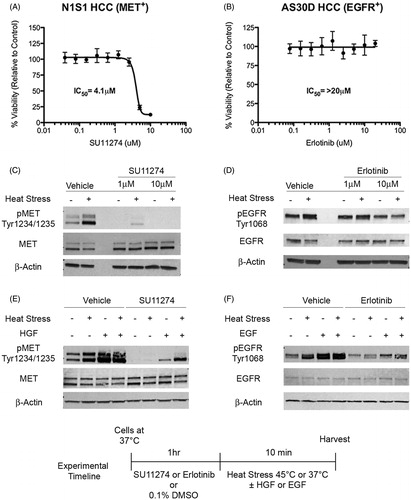
Next, we sought to determine if inhibition of the MET and EGFR cytoplasmic domain blocks heat stress induced MET and EGFR signalling. Western blotting showed that inhibition of MET decreased both constitutive and heat stress induced MET phosphorylation in the N1S1 cell line () and inhibition of EGFR partially decreased heat stress induced EGFR phosphorylation in AS30D cells ().
To determine if heat stress induced autophosphorylation of MET and EGFR is distinct from their respective growth factor ligands, HGF (hepatocyte growth factor) and EGF (epidermal growth factor), each cell line was grown in reduced serum media and pre-treated with the indicated small molecule inhibitor toward MET or EGFR followed by treatment with heat stress, growth factor (HGF or EGF) or both. Western blot analysis demonstrated that heat stress and growth factor differentially induce MET () and EGFR () autophosphorylation and that inhibition of the cytoplasmic kinase domain decreased both constitutive, heat stress and growth factor induced MET and EGFR autophosphorylation.
Inhibition of MET and EGFR enhances heat stress induced HCC cell killing
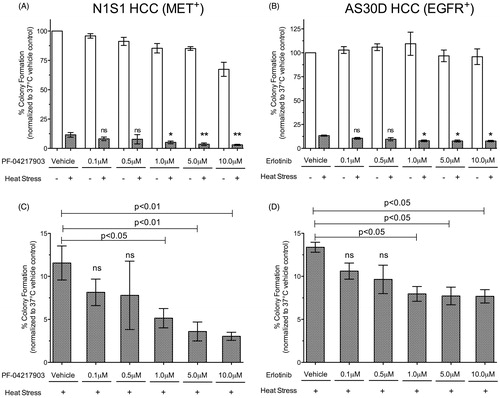
The observation that heat stress induces rapid MET and EGFR signalling and that inhibition of the cytoplasmic kinase domain blocks this heat stress induced signalling raises the question whether heat stress induced MET or EGFR signalling mediates HCC survival to heat stress. To determine if inhibition of heat stress induced MET or EGFR signalling decreases HCC survival to heat stress we used a clonogenic survival assay as an in vitro model of recurrence. The small molecule MET inhibitor PF-04217903 was selected for further MET inhibition experiments because it is more potent than SU11274 and has oral bioavailability [Citation35]. Inhibition of MET plus heat stress resulted in a dose-dependent 55–74% decrease in N1S1 clonogenic survival compared to vehicle plus heat stress alone (p < 0.03; ). On the other hand, inhibition of EGFR plus heat stress resulted in only a small (<20%) dose-dependent decrease in AS30D clonogenic survival compared to vehicle plus heat stress alone (p < 0.05; ).
Figure 4. Effect of MET and EGFR inhibition on HCC clonogenic survival following heat stress. (A) N1S1 HCC cells were pre-treated with a dose-titration of the MET inhibitor PF-042179030 (0.1–10 μM) or vehicle control (0.1% DMSO) and (B) AS30D HCC cells were pre-treated with a dose-titration of the EGFR inhibitor erlotinib (0.1–10 μM) or vehicle control (0.1% DMSO) for 1 h, subjected to heat stress (45 °C) or control (37 °C) for 10 min and plated for the colony formation assay. Per cent colony formation was calculated relative control to 37 °C control. Data are presented as mean ± SEM of three independent experiments and were analysed using one-way ANOVA followed by post-hoc pairwise comparison using an unpaired t-test. ns p > 0.05; *p ≤ 0.05; **p ≤ 0.01. White bars = drug only; Grey bars = drug plus heat stress. (C, D) Graphs of drug plus heat stress data only from (A) and (B) for (C) N1S1 and (D) AS30D, respectively.
Heat stress induced PI3K/mTOR-dependent AKT signalling is independent of MET signalling
Having established (i) that heat stress induces rapid MET phosphorylation in HCC cells in vitro and at the tumour ablation margin in vivo and (ii) that inhibition of cytoplasmic kinase domain of MET blocks heat stress induced MET signalling and enhances heat stress induced HCC cell killing, we sought to investigate heat stress induced signalling pathways downstream of MET. N1S1 cells were heat stressed, harvested, digested to peptides and enriched using three different reagents: a phosphotyrosine antibody, a mix of Ser/Thr antibodies corresponding to basophilic kinase substrates and a multiplexed pathway enrichment cocktail containing site-specific antibodies on beads (Multipathway reagent). The enriched samples were analysed by LC-MS/MS and relative abundance of peptides in each sample was determined. Pathway analysis from this quantitative phosphoproteomic data showed that heat stress induced MET signalling is associated with numerous downstream signalling pathways including PI3K/AKT, Stat3, ERK and JNK (; see Supplementary Figure S3). Prior experiments demonstrated that inhibition of heat stress induced PI3K/mTOR dependent AKT signalling but not MEK-dependent ERK signalling enhances heat stress induced HCC cell killing [Citation11]. As such, this raised the question, is heat stress induced AKT and ERK signalling MET dependent? Western blot analysis showed that inhibition of heat stress induced MET signalling does not block heat stress induced AKT and ERK signalling (), suggesting that heat stress induced AKT and ERK signalling are independent of MET.
Figure 5. Phosphoproteomic pathway analysis of heat stress induced MET signalling. The protein interaction network was generated from the Ingenuity Pathway Analysis (IPA) (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis) [Citation28,Citation29]. Ingenuity pathway analysis of quantitative phosphoproteomic data identifies numerous signalling pathways associated with heat stress induced MET signalling (green = increased phosphorylation, red = decreased phosphorylation, grey = neutral, yellow = both increases and decreases).
![Figure 5. Phosphoproteomic pathway analysis of heat stress induced MET signalling. The protein interaction network was generated from the Ingenuity Pathway Analysis (IPA) (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis) [Citation28,Citation29]. Ingenuity pathway analysis of quantitative phosphoproteomic data identifies numerous signalling pathways associated with heat stress induced MET signalling (green = increased phosphorylation, red = decreased phosphorylation, grey = neutral, yellow = both increases and decreases).](/cms/asset/a31b7b0f-0d7d-43a4-bb7a-39c446a61757/ihyt_a_1385859_f0005_c.jpg)
Figure 6. Heat stress induced PI3K/mTOR dependent AKT signalling is independent of MET signalling. (A) N1S1 cells pre-treated with a dose-titration of the MET inhibitor PF-042179030 (0.1–10 μM) or vehicle control (0.1% DMSO) for 1 h followed by heat stress (45 °C) or control (37 °C) for 10 min. Immediately post-heat stress whole-cell lysates were subjected to Western blotting for phospho-MET, AKT and ERK and total MET. β-actin was used as a loading control. (C) N1S1 cells pre-treated with the PI3K/mTOR inhibitor BEZ235 (1 μM) or vehicle control (0.1% DMSO) for 1 h followed by heat stress (45 °C) or control (37 °C) for 10 min. Immediately post-heat stress whole-cell lysates were subjected to Western blotting for phospho and total MET, AKT, PRAS40, 4EBP1, ERK, SAPK/JNK, cJUN, ATF2 and EphA2. Vimentin was used as a loading control. See Supplementary Figure S3 for corresponding quantitative mass spectrometry (LCMS) data.
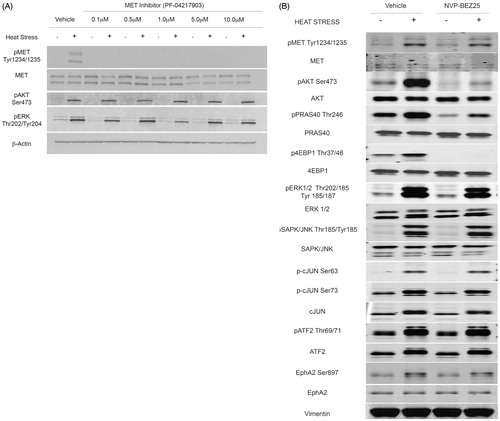
Given that prior studies demonstrated that inhibition of PI3K/mTOR blocks heat stress induced AKT signalling and that this signalling is independent of MET, we investigated the effect PI3K/mTOR inhibition on heat stress induced MET signalling using the same peptide enrichments (phosphotyrosine, basophilic mix, multipathway) followed by quantitative mass spectrometry. Mass spectrometry data were validated using Western blotting. Inhibition of PI3K/mTOR blocks heat stress induced AKT signalling (4EBP1, PRAS40) but not heat stress induced MET, ERK or JNK (c-Jun, ATF2) signalling (; see Supplementary Figure S3 for corresponding quantitative mass spectrometry data). As such, the mechanisms whereby PI3K/mTOR and MET inhibition enhance heat stress induced HCC cell killing are likely independent of one another. Additionally, given that numerous upstream signalling pathways converge on PI3K/AKT/mTOR/signalling, it is possible that other upstream kinases are activated by heat stress to induce PI3K/AKT/mTOR/signalling and may explain why MET inhibition does not abolish heat stress induced PI3K/AKT/mTOR signalling. LCMS and Western blotting showed that heat stress induced phosphorylation of the EphA2 receptor tyrosine kinase (), known to signal via PI3K-AKT [Citation36,Citation37].
Discussion
Thermal ablative therapies are potentially curative in the treatment of early stage HCC. Nonetheless, risk of local tumour recurrence increases with tumours beyond 2–3 cm and remains a challenging clinical problem. The tumour ablation margin is a common site for HCC local tumour recurrence, suggesting incomplete treatment resulting from sublethal heat stress [Citation8–10]. The present experiments sought to elucidate molecular signalling pathways mediating HCC cell survival to heat stress in order to identify candidate adjuvant therapeutic targets for enhancing thermal-ablation induced HCC cell killing and prevent HCC local recurrence.
Prior experiments have shown that heat stress at 45 °C for 10 min induces PI3K/mTOR-dependent AKT and MEK-dependent ERK signalling in HCC cells in vitro and at the tumour ablation margin in vivo, thereby raising the question of what upstream mechanism(s) initiate these signalling pathways [Citation11]. Receptor tyrosine kinases (RTKs) are key integrators of extracellular growth factors and induce a wide-range of intracellular signalling pathways, including PI3K/AKT/mTOR and RAS–RAF–MEK–ERK and are frequently dysregulated in cancer, including HCC [Citation13,Citation15,Citation16]. Mechanistically, binding of specific ligands (growth factors, cytokines) to the extracellular RTK binding domain induces receptor homodimerization or heterodimerization and subsequent auto(trans)phosphorylation of the c-terminal kinase domain, thereby activating numerous downstream signalling pathways [Citation14]. However, it is not known if mechanisms beyond extracellular binding growth factor ligands such as heat stress can induce RTK signalling.
Utilizing an in vitro model for HCC recurrence following heat stress, the initial kinome screen demonstrated that heat stress induces rapid changes in global tyrosine phosphorylation in hepatocytes and HCC cells, thereby suggesting that heat stress may alter numerous cell-signalling pathways. The MET and EGFR RTKs were selected for further evaluation given their biological and prognostic significance in HCC and regulation of numerous downstream growth, proliferation, survival and metastasis pathways [Citation17–19,Citation21,Citation38–41]. The in vitro experiments demonstrated that heat stress induces rapid auto-phosphorylation of both MET and EGFR that is distinct from growth factor induction by HGF and EGF. Moreover, the in vivo ablation experiments demonstrated that MET phosphorylation is preferentially localised to the tumour ablation margin in the MET+ N1S1 HCC model. Interestingly, there was no evidence of EGFR phosphorylation at the tumour ablation margin in the EGFR+ AS30D model. Taken together, these data suggest that heat stress induces MET and EGFR auto-phosphorylation in the absence of their growth factor ligands in a cell-line/tumour model and RTK-specific manner and provide evidence that heat stress induces MET signalling in HCC cells at the tumour ablation margin in vivo, the region of the ablation zone at highest risk for tumour recurrence and progression. Given the known pro-invasive and pro-metastatic effects of MET signalling, these findings support the hypothesis that induction MET signalling may promote HCC tumour progression at the ablation margin [Citation42].
Next, we sought to determine if inhibition of MET and EGFR blocks heat stress induced MET and EGFR auto-phosphorylation and enhances heat stress induced HCC cell killing. Blockade of the MET and EGFR c-terminal kinase domain using ATP-competitive small molecule inhibitors abolished or decreased constitutive and heat stress induced MET and EGFR auto-phosphorylation. Importantly, inhibition of MET significantly reduced HCC clonogenic survival following heat stress whereas inhibition EGFR had minimal effect on enhancing heat stress induced HCC cell killing. Overall, given the greater decrease in HCC clonogenic survival following heat stress with MET inhibition and induction of MET phosphorylation at the tumour ablation margin, MET but not EGFR signalling may in part mediate HCC cell survival to heat stress. Of interest, a subset of HCCs driven by MET has been identified that have a more aggressive phenotype and worse overall prognosis [Citation20]. While identifying patients with MET positive HCC tumours would require biopsy or molecular imaging, addition of neo-adjuvant MET inhibition with small molecule inhibitors or monoclonal antibodies prior to thermal ablation may decrease HCC cell survival to thermal ablation, thereby preventing local recurrence [Citation43]. Conversely, although the data suggest that heat stress induced EGFR signalling does not regulate HCC cell survival to heat stress, other cellular functions beyond survival warrant further investigation.
Given the findings that inhibition of MET enhances heat stress induced HCC cell killing in vitro, we sought to identify signalling pathways associated with heat stress induced MET signalling. The quantitative phosphoproteomic, Western blot and pathway analysis data showed that heat stress induced MET signalling is associated with numerous downstream signalling pathways including PI3K/AKT, Stat3, ERK and JNK. However, inhibition of MET did not block heat stress induced AKT and ERK signalling, thereby suggesting that heat stress induced AKT and ERK signalling is independent of MET. Given that prior studies demonstrated that inhibition of PI3K/mTOR blocks heat stress induced AKT signalling and the present findings that this signalling is independent of MET, we investigated the effect PI3K/mTOR inhibition on heat stress induced MET signalling using quantitative mass spectrometry and Western blotting. Inhibition of PI3K/mTOR blocks heat stress induced AKT signalling (4EBP1, PRAS40) but not heat stress induced MET, ERK or JNK (c-Jun, ATF2) signalling. As such, the mechanisms whereby PI3K/mTOR and MET inhibition enhance heat stress induced HCC cell killing are likely independent of one another. Additionally, given that numerous upstream signalling pathways converge on PI3K/AKT/mTOR signalling, it is possible that other upstream kinases are activated by heat stress to induce PI3K/AKT/mTOR/signalling and may explain why MET inhibition does not abolish heat stress induced PI3K/AKT/mTOR signalling. LCMS and Western blotting showed that heat stress induced phosphorylation of the EphA2 RTK which is known to signal via PI3K-AKT [Citation36,Citation37]. MET and EGFR signalling pathways may in part mediate HCC cellular functions including survival following heat stress in a cell-type dependent manner. However, other receptor and non-receptor tyrosine kinases (TK) may be involved in activating downstream signalling pathways and cellular function. Thermal ablation induced RTK signalling including MET, EGFR and other TKs warrants further investigation as a candidate mechanism for recurrence and progression following thermal ablation of HCC.
There are limitations to this study. These data support a mechanism of heat stress induced, MET and EGFR signalling that is distinct of growth factor ligands HGF and EGF, however, we have yet to determine the biochemical mechanism for heat stress induced auto-phosphorylation. One hypothesis is that heat stress changes cell membrane fluidity and/or RTK activation energy to overcome auto-inhibition at the c-terminal kinase domain of MET and EGFR, thereby allowing auto-phosphorylation. Moreover, if the mechanism whereby heat stress induced RTKs is non-specific, other RTKs and cytoplasmic tyrosine kinases (CTKs) may also be induced. As such, targeting upstream mechanisms may be insufficient if multiple RTKs or CTKs converge on key common downstream cell growth, survival and proliferation pathways. This may in part explain why heat stress induced AKT and ERK signalling was identified as associated with MET signalling from the proteomics experiments but that inhibition of MET did not block either AKT or ERK signalling. Further experiments identified EphA2 RTK as one in the N1S1 model, which is known to signal via PI3K/AKT, but additional RTKs warrant further investigation. Additionally, not all cell lines that express MET or EGFR were induced by heat stress and the presence of receptor expression does not mean that the receptor is a key driver of onocogenesis. For example, EGFR phosphorylation was not induced by thermal ablation in vivo in these studies and inhibition of EGFR had minimal effect on enhancing heat stress induced HCC cell killing. There may be changes in RTK density or RTK dependence from in vitro to in vivo, thereby highlighting a limitation of in vitro models. The findings need to be expanded to different HCC models and cancer types to further understand the potential generalizability of heat stress induced RTK signalling. Finally, cell survival was assessed as the endpoint in these experiments but heat stress induced MET and EGFR signalling may mediate other cellular functions such as transcription/translation, cell growth/proliferation and motility/invasion that merit further investigation.
Conclusion
Heat stress induced MET and EGFR signalling represents a potentially novel mechanism whereby heat stress is transduced from the extracellular environment to the intracellular environment via ligand-independent activation of RTK signalling. Translationally, heat stress induced MET signalling may in part mediate HCC cell survival at the thermal ablation margin in MET-dependent HCCs and adjuvant MET inhibition may be a potential therapeutic strategy to enhance thermal ablation induced HCC cell killing. Nonetheless, basic cellular and molecular questions remain regarding how heat stress mechanistically induces auto-phosphorylation of these RTKs at the HCC cell membrane and what cellular functions beyond survival are altered.
Supplemental File
Download Zip (2.2 MB)Acknowledgements
Dr Thompson thanks the Mayo Clinic Medical Scientist Training Program (MSTP) for fostering an outstanding environment for physician-scientist training. The contents herein are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Disclosure statement
Dr Stokes and Ms. Jia are paid employees of Cell Signalling Technology, Inc. The remaining authors report no conflicts of interest related to the contents of this manuscript.
Additional information
Funding
Related Research Data
References
- Ferlay J, Soerjomataram I, Dikshit R, et al. (2015). Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 136:E359–86.
- Bruix J, Gores GJ, Mazzaferro V. (2014). Hepatocellular carcinoma: clinical frontiers and perspectives. Gut 63:844–55.
- Yang JD, Kim B, Sanderson SO, et al. (2012). Hepatocellular carcinoma in olmsted county, Minnesota, 1976-2008. Mayo Clin Proc 87:9–16.
- Yang JD, Roberts LR. (2010). Hepatocellular carcinoma: a global view. Nat Rev Gastroenterol Hepatol 7:448–58.
- Tiong L, Maddern GJ. (2011). Systematic review and meta-analysis of survival and disease recurrence after radiofrequency ablation for hepatocellular carcinoma. Br J Surg 98:1210–24.
- Wang JH, Wang CC, Hung CH, et al. (2012). Survival comparison between surgical resection and radiofrequency ablation for patients in BCLC very early/early stage hepatocellular carcinoma. J Hepatol 56:412–18.
- Cho YK, Kim JK, Kim MY, et al. (2009). Systematic review of randomized trials for hepatocellular carcinoma treated with percutaneous ablation therapies. Hepatology 49:453–9.
- Harrison LE, Koneru B, Baramipour P, et al. (2003). Locoregional recurrences are frequent after radiofrequency ablation for hepatocellular carcinoma. J Am Coll Surg 197:759–64.
- Kei SK, Rhim H, Choi D, et al. (2008). Local tumor progression after radiofrequency ablation of liver tumors: analysis of morphologic pattern and site of recurrence. AJR Am J Roentgenol 190:1544–51.
- Liu CH, Arellano RS, Uppot RN, et al. (2010). Radiofrequency ablation of hepatic tumours: effect of post-ablation margin on local tumour progression. Eur Radiol 20:877–85.
- Thompson SM, Callstrom MR, Jondal DE, et al. (2016). Heat stress-induced PI3K/mTORC2-dependent AKT signaling is a central mediator of hepatocellular carcinoma survival to thermal ablation induced heat stress. PLoS One 11:e0162634.
- Dhanasekaran R, Venkatesh SK, Torbenson M, Roberts LR. (2016). Clinical implications of basic research in hepatocellular carcinoma. J Hepatol 64:736–45.
- Lemmon MA, Schlessinger J. (2010). Cell signaling by receptor tyrosine kinases. Cell 141:1117–34.
- Casaletto JB, McClatchey AI. (2012). Spatial regulation of receptor tyrosine kinases in development and cancer. Nat Rev Cancer 12:387–400.
- Breuhahn K, Longerich T, Schirmacher P. (2006). Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene 25:3787–800.
- Blume-Jensen P, Hunter T. (2001). Oncogenic kinase signalling. Nature 411:355–65.
- Hoshida Y, Toffanin S, Lachenmayer A, et al. (2010). Molecular classification and novel targets in hepatocellular carcinoma: recent advancements. Semin Liver Dis 30:35–51.
- You H, Ding W, Dang H, et al. (2011). c-Met represents a potential therapeutic target for personalized treatment in hepatocellular carcinoma. Hepatology 54:879–89.
- Wu F, Wu L, Zheng S, et al. (2006). The clinical value of hepatocyte growth factor and its receptor-c-met for liver cancer patients with hepatectomy. Dig Liver Dis 38:490–7.
- Kaposi-Novak P, Lee JS, Gomez-Quiroz L, et al. (2006). Met-regulated expression signature defines a subset of human hepatocellular carcinomas with poor prognosis and aggressive phenotype. J Clin Invest 116:1582–95.
- Blivet-Van Eggelpoel MJ, Chettouh H, Fartoux L, et al. (2012). Epidermal growth factor receptor and HER-3 restrict cell response to sorafenib in hepatocellular carcinoma cells. J Hepatol 57:108–15.
- Berasain C, Nicou A, Garcia-Irigoyen O, et al. (2012). Epidermal growth factor receptor signaling in hepatocellular carcinoma: inflammatory activation and a new intracellular regulatory mechanism. Dig Dis 30:524–31.
- Ezzoukhry Z, Louandre C, Trecherel E, et al. (2012). EGFR activation is a potential determinant of primary resistance of hepatocellular carcinoma cells to sorafenib. Int J Cancer 131:2961–9.
- Thompson SM, Callstrom MR, Butters KA, et al. (2014). Heat stress induced cell death mechanisms in hepatocytes and hepatocellular carcinoma: in vitro and in vivo study. Lasers Surg Med 46:290–301.
- Thompson SM, Callstrom MR, Butters KA, et al. (2014). Role for putative hepatocellular carcinoma stem cell subpopulations in biological response to incomplete thermal ablation: in vitro and in vivo pilot study. Cardiovasc Intervent Radiol 37:1343–51.
- Rush J, Moritz A, Lee KA, et al. (2005). Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat Biotechnol 23:94–101.
- Stokes MP, Farnsworth CL, Moritz A, et al. (2012). PTMScan direct: identification and quantification of peptides from critical signaling proteins by immunoaffinity enrichment coupled with LC-MS/MS. Mol Cell Proteomics 11:187–201.
- Stokes MP, Farnsworth CL, Gu H, et al. (2015). Complementary PTM profiling of drug response in human gastric carcinoma by immunoaffinity and IMAC methods with total proteome analysis. Proteomes 3:160–83.
- Kramer A, Green J, Pollard J, Jr, Tugendreich S. (2014). Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 30:523–30.
- Thompson SM, Callstrom MR, Knudsen B, et al. (2012). Development and preliminary testing of a translational model of hepatocellular carcinoma for MR imaging and interventional oncologic investigations. J Vasc Interv Radiol 23:385–95.
- Thompson SM, Callstrom MR, Knudsen BE, et al. (2013). Molecular bioluminescence imaging as a noninvasive tool for monitoring tumor growth and therapeutic response to MRI-guided laser ablation in a rat model of hepatocellular carcinoma. Invest Radiol 48:413–21.
- Thompson SM, Callstrom MR, Knudsen B, et al. (2013). AS30D model of hepatocellular carcinoma: tumorigenicity and preliminary characterization by imaging, histopathology, and immunohistochemistry. Cardiovasc Intervent Radiol 36:198–203.
- Sattler M, Pride YB, Ma P, et al. (2003). A novel small molecule met inhibitor induces apoptosis in cells transformed by the oncogenic TPR-MET tyrosine kinase. Cancer Res 63:5462–9.
- Bulgaru AM, Mani S, Goel S, Perez-Soler R. (2003). Erlotinib (Tarceva): a promising drug targeting epidermal growth factor receptor tyrosine kinase. Expert Rev Anticancer Ther 3:269–79.
- Zou HY, Li Q, Lee JH, et al. (2012). Sensitivity of selected human tumor models to PF-04217903, a novel selective c-Met kinase inhibitor. Mol Cancer Ther 11:1036–47.
- Pandey A, Lazar DF, Saltiel AR, Dixit VM. (1994). Activation of the Eck receptor protein tyrosine kinase stimulates phosphatidylinositol 3-kinase activity. J Biol Chem 269:30154–7.
- Efazat G, Novak M, Kaminskyy VO, et al. (2016). Ephrin B3 interacts with multiple EphA receptors and drives migration and invasion in non-small cell lung cancer. Oncotarget 7:60332–47.
- Ishikawa T, Factor VM, Marquardt JU, et al. (2012). Hepatocyte growth factor/c-met signaling is required for stem-cell-mediated liver regeneration in mice. Hepatology 55:1215–26.
- Huh CG, Factor VM, Sanchez A, et al. (2004). Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc Natl Acad Sci USA 101:4477–82.
- Ueki T, Fujimoto J, Suzuki T, et al. (1997). Expression of hepatocyte growth factor and its receptor, the c-met proto-oncogene, in hepatocellular carcinoma. Hepatology 25:619–23.
- Tavian D, De Petro G, Benetti A, et al. (2000). u-PA and c-MET mRNA expression is co-ordinately enhanced while hepatocyte growth factor mRNA is down-regulated in human hepatocellular carcinoma. Int J Cancer 87:644–9.
- Goyal L, Muzumdar MD, Zhu AX. (2013). Targeting the HGF/c-MET pathway in hepatocellular carcinoma. Clin Cancer Res 19:2310–18.
- Jagoda EM, Lang L, Bhadrasetty V, et al. (2012). Immuno-PET of the hepatocyte growth factor receptor Met using the 1-armed antibody onartuzumab. J Nucl Med 53:1592–600.