
Abstract
Introduction: Cisplatin is used as a standard chemotherapeutic agent for head and neck cancer treatment. However, some head and neck cancers have cisplatin resistance, leading to difficulty in treatment and poor prognosis. Overcoming cisplatin resistance remains an important strategy to improve prognoses for head and neck cancer patients.
Objective: Elucidation of the mechanisms underlying cisplatin resistance can suggest novel targets to enhance the anticancer effects of cisplatin for treating head and neck cancers.
Material and methods: We used a cisplatin-resistant human maxillary cancer cell line, IMC-3CR to analyse the cisplatin resistance mechanisms. Cisplatin-induced genes were analysed in IMC-3CR cells using PCR array. Among the genes with expression increased by cisplatin, we specifically examined SESN1. SESN family reportedly regenerates peroxiredoxin and suppresses oxidative DNA injury by reactive oxygen species (ROS), which can be induced by chemotherapeutic agents such as cisplatin, radiation, and hyperthermia. The function of SESN1 in cisplatin resistance and ROS generation were analysed using specific RNAi.
Results: Results show that SESN1 was induced by cisplatin treatment in IMC-3CR cells. Suppression of SESN1 by RNAi induced apoptosis and reduced cell viability through enhancement of ROS after cisplatin treatment. Moreover, suppression of SESN1 enhanced the cell-killing effects of hyperthermia with increased ROS, but did not affect the cell-killing effects of radiation.
Conclusions: This study demonstrated the participation of SESN1 in cisplatin and hyperthermia resistance of human head and neck cancers. SESN1 is a novel molecular target to overcome cisplatin resistance and hyperthermia resistance and improve head and neck cancer treatment.
Introduction
Chemotherapy has been used widely to treat cancers of various kinds. Recently, it has been gaining importance especially for head and neck squamous cell carcinoma (HNSCC) because complete removal of cancer by surgery is difficult as a result of their smaller anatomical margin. Chemotherapy with cisplatin accompanied by radiation, an accepted standard strategy, improves the loco-regional control and disease-free survival of advanced HNSCC [Citation1]. Nevertheless, some HNSCC have resistance against cisplatin, leading to poor local control or distant metastasis. For cancers of some kinds, several earlier reports have described studies of cisplatin-resistance mechanisms. Glutathione inactivates or exports cisplatin through the putative ATP-dependent glutathione S-conjugate export pump in ovarian cancer cells [Citation2]. Induction of XRCC3 causes cisplatin resistance in breast cancer cells [Citation3]. AKT1 induces cisplatin resistance in lung cancer cells through mammalian target of rapamycin (mTOR) signalling pathway [Citation4]. Although similar molecules are expected to be involved in HNSCC, the underlying mechanisms of resistance remain unclear.
Hyperthermia has been acknowledged as an effective modality for many types of cancers including head and neck cancers. Hyperthermia can enhance the antitumour effect of chemotherapy or radiation without severe side effects for head and neck cancers and other various malignancies [Citation5–8]. Reports of the last decade have described the clinical efficacy of hyperthermia and the biological mechanisms of hyperthermia. Actually, hyperthermia induces many stress responses including induction of heat shock proteins (HSPs), aggregation of proteins, RNA damage, reactive oxygen species (ROS) production leading to DNA damage, cell growth arrest and cell death in mammalian cells [Citation9,Citation10]. However, cancer cells can become tolerant of hyperthermia through induction of HSPs [Citation11], thereby limiting its clinical use to date. Although an earlier report of the relevant literature described that coinhibition of HSP70/HSP90 synergistically enhanced hyperthermia effects on nasopharyngeal carcinoma cells [Citation12], the greater part of the underlying hyperthermia resistance mechanism remains unclear. A mechanism must be found to overcome resistance and support the practical use of hyperthermia, leading to a breakthrough in cancer treatment.
Sestrins (SESN) are included in a family of highly conserved stress-responsive proteins activated by p53 [Citation13]. The human SESN family consists of SESN1, 2 and 3, which are crucially important for ROS suppression and which protect cells from oxidative stress, transformation and genomic instability [Citation13,Citation14]. Reportedly, SESN1 and 2 regenerate peroxiredoxin and degenerate ROS, leading to reduction of oxygenation to DNA and to re-establishment of the antioxidant firewall [Citation15]. It has been demonstrated recently that overexpression of SESN2 protects cells from apoptosis induced by ischaemia or H2O2, and that SESN2 plays an important role in defence against ROS [Citation16]. SESN2 can protect cells against energetic stress-induced apoptosis through the pro-survival function of Akt and the negative regulation of mTOR [Citation17]. By contrast, SESN2 mediates radiation-induced AMPK expression and promotes radiosensitization of breast cancer cells [Citation18]. Reports describing the SESN family have been increasing, but fewer reports describe SESN1. Although SESN1 is induced by cellular stresses such as radiation, the greater part of the biological function of SESN1 remains unknown. This study was conducted to demonstrate that suppression of SESN1 reduces cisplatin resistance and hyperthermia resistance through increasing ROS production in head and neck cancer cells. SESN1 is a novel and promising target to overcome resistance in cisplatin-based chemotherapy accompanied by hyperthermia for the treatment of HNSCC.
Material and methods
Cell lines
This study used human maxillary squamous cell carcinoma IMC-3 cells and cisplatin-resistant IMC-3CR cells that were cultured in RPMI-1640 medium (Nissui Pharmaceutical Co. Ltd., Tokyo, Japan) supplemented with 10% heat-inactivated FCS (Thermo Fisher Scientific, Inc., Waltham, MA), 0.29 mg/ml glutamine, 100 units/ml penicillin and 100 µg/ml streptomycin, at 37 °C in a conventional humidified CO2 incubator. Regarding cisplatin resistance, the IC50 of cisplatin were, respectively, 8.4 and 41.0 (μg/ml) in IMC-3 and IMC-3CR cells [Citation19].
Cisplatin and heat treatment
Cisplatin was purchased from Bristol-Myers Squibb Co. (New York, NY). Cisplatin was added to the culture medium to the final concentration of 1 μg/ml. Cells were used for analyses after incubation at 37 °C. Regarding heat treatment, the cells were seeded in 25 cm2 screw-capped polystyrene flasks (Falcon; Becton, Dickinson and Company, Franklin Lakes, NJ) with 6 ml of medium for at least 48 h to reach a logarithmic stage of growth. After incubation, heat treatment was performed by immersing the flasks with fully tightened screw caps in a water bath (BK-43; Yamato Kagaku Co. Ltd., Tokyo, Japan) preset to 44 °C ± 0.05 °C. Immediately after heating, the treated cells were rinsed twice with medium and were re-fed fresh medium (pH 7.4) for additional incubation. Hyperthermia at 42 °C, which is used clinically, can induce thermo-tolerance in in vitro assays [Citation20]. To analyse the mechanisms of hyperthermia-induced cell-killing effects and their enhancement by suppressing SESN1 in vitro, we used hyperthermia at 44 °C [Citation21].
RNA extraction and reverse transcription
After cisplatin treatment, total RNA was isolated from IMC-3CR cells using an RNeasy Mini kit (Qiagen, Hilden, Germany). Then the cDNA was synthesised using a High-capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, Inc.) for quantitative real-time PCR according to the manufacturer’s instructions.
PCR array
The change of mRNA expressions after cisplatin treatment in IMC-3CR was analysed using a RT2 ProfilerTM PCR array (Human DNA Damage Signaling Pathway; Qiagen) according to the manufacturer's protocol [Citation22]. IMC-3CR cells (5 × 105) were prepared in RPMI 1640 supplemented with 10% heat-inactivated FCS. Cells were incubated with or without cisplatin (1 μg/ml) at 37 °C in a humidified atmosphere containing 5% CO2 for 6 h. After incubation, total RNA was extracted using RNeasy Mini Kit (Qiagen), and cDNA was synthesised through RT performed with an RT2 First Strand Kit (Qiagen) according to the manufacturer's protocol. The cDNA was used in the PCR array with a ABI PRISM® 7000 Sequence Detection System (Thermo Fisher Scientific, Inc.) using a PCR master mix (SA Biosciences RT2 qPCR Master Mix; Qiagen) for SYBR Green detection. Samples were amplified with a precycling hold at 95 °C for 10 min, followed by 40 cycles of denaturation at 95 °C for 15 s and annealing for 1 min at 60 °C. The PCR array results were uploaded to the RT2 Profiler PCR Array Data Analysis website (http://pcrdataanalysis.sabiosciences.com/pcr/arrayanalysis.php). The change of mRNA expression was analysed using the ΔΔCt method.
Quantitative real-time PCR
Quantification of mRNA of SESN1 was performed using real-time fluorescence detection TaqMan® technology and TaqMan® Universal PCR Master Mix (Thermo Fisher Scientific, Inc.) with a StepOnePlusTM real time PCR system (Thermo Fisher Scientific, Inc.). The Primers were from TaqMan® Gene Expression Assays (Thermo Fisher Scientific, Inc.), including Hs99999905-m1 for GAPDH, and Hs00902787-m1 for SESN1. Another set of primers (Hs00902782-m1; Thermo Fisher Scientific, Inc.) were used for validation of SESN1.
Western blot analysis
After cells were washed twice with ice-cold PBS, they were dissolved in solubilising buffer (pH 7.4, 1% Triton X-100, 1% deoxycholic acid sodium salt, 0.1% SDS, 20 mM Tris-HCl, 150 mM NaCl, 5 mM EDTA, 1 mM PMSF, 10 mg/ml pepstatin and 10 mg/ml leupeptin). Each aliquot of protein (50 μg) was subjected to Western blot analysis. After electrophoresis on Mini-PROTEAN® TGXTM Gel (Bio-Rad Laboratories, Inc., Hercules, CA), the protein was transblotted using a Trans-Blot® TurboTM Transfer Pack and Trans-Blot® TurboTM system (Bio-Rad Laboratories, Inc.). After the blots were blocked with 3% non-fat dry milk in pH 7.4 TBST, they were incubated with anti-SESN1 antibody (1:200; Santa Cruz Biotechnology, Inc., Dallas, TX), and were then incubated secondarily with a horse radish peroxidase-conjugated (HRP-conjugated) anti-rabbit or anti-mouse IgG antibody (Dako; Agilent Technologies, Inc., Santa Clara, CA). Subsequently, the blots were developed using chemiluminescent Western blot detection reagents (Amersham ECL Prime; GE Healthcare, Chicago, IL) according to the manufacturer’s instructions. The blot density was quantified using ImageJ software (National Institutes of Health, Bethesda, MD).
RNA interference
The siRNA against SESN1 and the non-specific control siRNA were designed and synthesised by Thermo Fisher Scientific, Inc. (Stealth RNAiTM siRNA). IMC-3CR cells were seeded, respectively, to a 96-well plate (1 × 104 cells/well) for MTT assay or six-well plate (1 × 105 cells/well) for flow cytometry and clonogenic cell survival assay without antibiotics. The siRNA was transfected using a reagent (Lipofectamine® 2000 Transfection Reagent; Thermo Fisher Scientific, Inc.). Cisplatin was added to the medium 24 h after transfection. Suppression of SESN1 protein was confirmed with Western blotting 3 h after cisplatin treatment.
MTT assay
Cell viability 48 h after cisplatin treatment (1 μg/ml, dissolved in water) and hyperthermia (44 °C, 30 min) in serum-free medium was analysed using 3–(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). The MTT (0.4 mg/ml; Sigma-Aldrich Co. LLC, St Louis, MO) was added to culture wells after washing with PBS. The converted dye was dissolved with dimethyl sulfoxide 2 h after incubation at 37 °C. The absorbance was measured at wavelengths of 540 nm and 630 nm using a microplate reader (Spectra Max 250; Molecular Devices, LLC, Sunnyvale, CA).
Clonogenic cell survival assay
Clonogenic surviving fractions of treated and control cultures were determined in colony-forming units. The values were corrected using the plating efficiency of the controls, as described previously [Citation21]. At 14 days after treatment with cisplatin (1 μg/ml) or hyperthermia (44 °C, 15 min or 30 min), visible colonies composed of more than 50 cells were counted as having grown from surviving cells.
Flow cytometric analysis of apoptosis
We analysed the apoptosis of IMC-3CR cells after cisplatin treatment (1 μg/ml) and hyperthermia (44 °C, 30 min) using BD FACSCantoTM II Flow cytometer (Becton, Dickinson and Company). Analysis of apoptotic cells was performed using Annexin V-FITC Kit System for Detection of Apoptosis (Beckman Coulter Inc., Brea, CA) and BD FACSDivaTM software according to manufacturer’s instructions.
ROS analyses
The ROS production was analysed using OxiselectTM Intracellular ROS Assay Kit (Cell Biolabs, Inc., San Diego, CA) according to the manufacturer’s protocol. Briefly, cells were cultured in a clear 96-well culture plate. After media was removed from all wells, the cells were washed with DPBS. The plate was incubated at 37 °C for 30–60 min with 1× DCFH-DA/media. After incubation, media was removed and cells were washed with DPBS. DCFH-DA loaded cells were treated with cisplatin (1 μg/ml) or hyperthermia (44 °C, 15 min). After washing cells, 100 μl of medium and 100 μl of the cell lysis buffer were added to each well. The lysate of 150 μl was transferred to a 96-well plate. Then the absorbance was measured using a microplate reader (Spectra Max 250; Molecular Devices, LLC) at wavelengths of 480 nm/530 nm.
Caspase 3 activity assay
The activation of apoptotic pathway after cisplatin or hyperthermia treatment was analysed using Caspase 3 Assay Kit (Colorimetric; Abcam, Milton, Cambridge, UK). IMC-3CR cells were collected 48 h after treatment with cisplatin (1 μg/ml) or hyperthermia (44 °C, 30 min). Cells were treated with lysis buffer and protein samples were harvested. Each aliquot of protein (50 μg) was subjected to caspase 3 activity assay. The protein samples mixed with Reaction Buffer including 10 mM DTT and 200 μM DEVD-p-NA substrate were incubated at 37 °C for 120 min. The output was measured using a microplate reader (Spectra Max 250; Molecular Devices, LLC.) at 405 nm wavelength.
Statistical analysis
Statistical analyses were applied using Wilcoxon signed rank tests. Results are shown as mean ± SD. Significance was inferred for p values of < .05 (two-tailed).
Results
Analyses of gene expression by PCR array
PCR array system was used to analyse the expression kinetics of mRNA after treatment with cisplatin in IMC-3CR cells. Results showed that the expressions of several kinds of mRNA had changed by 6 h after treatment with cisplatin at 1 μg/ml in IMC-3CR cells (). The genes with a ratio higher than 1.5 were selected. This study specifically examines those genes for which expression was increased by cisplatin (), and especially those genes related to the redox metabolism. We chose SESN1 as a candidate target gene to assess resistance against cisplatin because SESN1 played an important role in the antioxidant firewall [Citation15].
Table 1. Results of PCR array analysis.
Expression of SESN1 in IMC-3 and IMC-3CR cells
The expression of SESN1 mRNA was confirmed using quantitative real-time PCR.
Levels of SESN1 mRNA were significantly higher in IMC-3CR cells than in IMC-3 cells (). Expression of SESN1 mRNA was induced in both IMC-3CR and IMC-3 cells 6 h after treatment with cisplatin. The fold change of levels of SESN1 mRNA was significantly higher in IMC-3CR than in IMC-3 (39 vs. 1.4, , upper panel). Similar expression of SESN1 was confirmed using another set of primers for real-time PCR (6.7 vs. 1.5, Supplementary Figure S1).
Figure 1. Expression of SESN1 in IMC-3 and IMC-3CR cells. The mRNA expression of SESN1 was analysed using real-time PCR without cisplatin treatment (A) and after cisplatin treatment (B, upper panel) in IMC-3 and IMC-3CR cells. Cells were treated with cisplatin (1 μg/ml) for 6 h. *p < .05. Error bars ± SD. The protein expression of SESN1 was analysed using Western blotting in IMC-3 and IMC-3CR cells (B, lower left panel). The blot density was quantified (B, lower right panel). Cells were treated with cisplatin (1 μg/ml) for 6 h. GAPDH is used as a loading control.
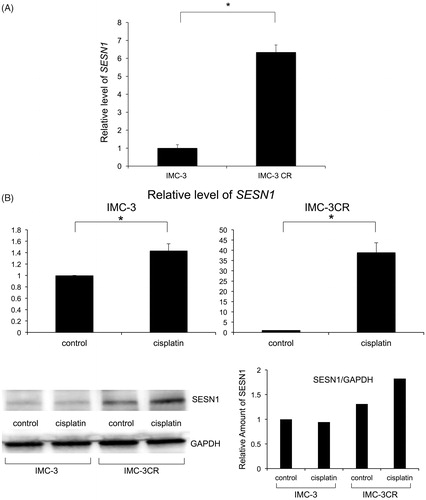
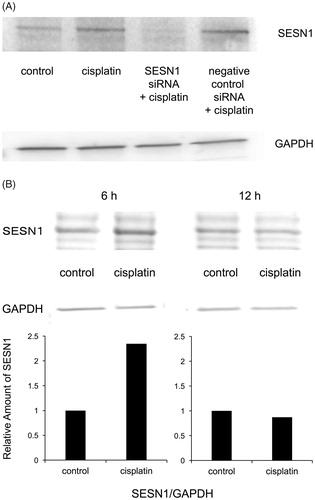
The protein accumulation of SESN1 was analysed using Western blotting in IMC-3 and IMC-3CR cells. SESN1 was expressed more dominantly in IMC-3CR than in IMC-3 cells without any stimulation (, lower left panel). The density of the Western blot was quantified (, lower right panel). The accumulation of SESN1 was enhanced after treatment with cisplatin only in IMC-3CR, but not in IMC-3 cells (, lower panel). To analyse the function of SESN1, we transfected small interfering RNA (siRNA) for SESN1 in IMC-3CR cells. Cisplatin-induced SESN1 was suppressed completely by specific siRNA for SESN1, although negative control siRNA did not affect it (). Western blot results demonstrated that cisplatin-enhanced SESN1 was confirmed at 6 h, but not at 12 h after treatment in IMC-3CR (, upper panel). The density of the Western blot was quantified (, lower panel). We confirmed that cisplatin induced SESN1 from 3 to 6 h after treatment only in IMC-3CR cells (Supplementary Figure S2), but that cisplatin did not induce SESN1 in IMC-3 cells (Supplementary Figure S3). These results suggest that cisplatin can induce the accumulation of SESN1 rapidly in IMC-3CR cells and that SESN1 can play an important role in resistance.
Figure 2. Western blot analyses of SESN1 in IMC-3CR cells. The protein expression of SESN1 was analysed by Western blotting in IMC-3CR cells (A) 3 h and (B) 6 or 12 h after treatment of cisplatin (1 μg/ml). GAPDH is used as a loading control. The blot density was quantified (lower panel).
Functions of SESN1 on cell viability and apoptosis
The functions of SESN1 related to cell viability and apoptosis after treatment with cisplatin in IMC-3CR cells were analysed using MTT assay and flow cytometry. MTT assay showed that cisplatin did not reduce cell viability 48 h after treatment in IMC-3CR cells (). By contrast, suppression of SESN1 by RNAi showed a 35% decrease of cell viability after treatment with cisplatin (). Negative control siRNA did not affect cell viability. Apoptotic cells were detected using flow cytometry with Annexin V labelling (). The induction of apoptosis was enhanced by treatment with cisplatin (2.8–10.5%). The suppression of SESN1 by RNAi enhanced cisplatin-induced apoptosis (10.5–19.5%) significantly 48 h after the treatment (). Negative control siRNA did not affect the induction of apoptosis. Cell viability was confirmed by clonogenic cell survival assay after treatment with cisplatin in IMC-3CR cells (). Suppression of SESN1 by RNAi significantly reduced the surviving fraction after treatment (81 vs. 24%, ). Negative control siRNA did not affect the surviving fraction.
Figure 3. SESN1 induce cisplatin resistance. (A) Cell viability was analysed using MTT assay in IMC-3CR cells after cisplatin treatment (1 μg/ml, 48 h). *p < .05. Error bars ± SD. (B) Apoptotic cells were analysed using flow cytometry with Annexin V labelling in IMC-3CR cells after cisplatin treatment (1 μg/ml, 48 h). *p < .05. Error bars ± SD. (C) Cell viability 14 days after cisplatin treatment (1 μg/ml) was confirmed by clonogenic cell survival assay. *p < .05. Error bars ± SD. (D) ROS production was analysed after cisplatin treatment (1 μg/ml) in IMC-3 and IMC-3CR cells. *p < .05. Error bars ± SD. (E) ROS production was measured 3 h after cisplatin treatment (1 μg/ml) in IMC-3CR cells. *p < .05. Error bars ± SD. (F) Caspase 3 activity was analysed 48 h after cisplatin treatment (1 μg/ml) in IMC-3CR cells. *p < .05. Error bars ± SD. (G) Cell viability was analysed using clonogenic cell survival assay with or without the specific siRNA for SESN1 in IMC-3CR cells.
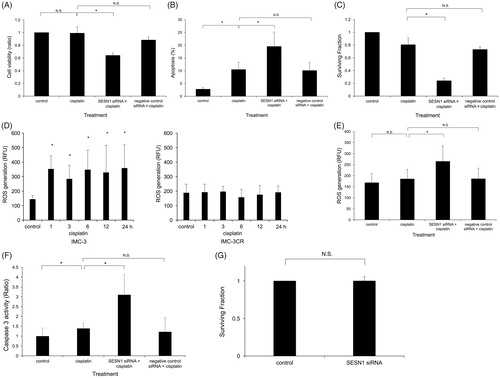
The ROS production was analysed after treatment with cisplatin in IMC-3 and IMC-3CR cells (). Although cisplatin significantly enhanced ROS production in IMC-3 cells, treatment with cisplatin did not evoke ROS production in IMC-3CR cells (). This result is attributable to differences of the IMC-3 and IMC-3CR cells’ SESN1 expression levels and response after cisplatin treatment. By contrast, suppression of SESN1 by RNAi induced ROS production significantly, by 1.4 times, from treatment with cisplatin in IMC-3CR cells (). Negative control siRNA did not affect the ROS production (). To investigate ROS function for cisplatin-induced apoptosis pathway, Caspase 3 activity was analysed in IMC-3CR (). Suppression of SESN1 enhanced caspase 3 activity significantly after cisplatin treatment compared to cisplatin alone in IMC-3CR cells (). The clonogenic cell survival assay showed that the specific siRNA for SESN1 alone did not have cell-killing effect in IMC-3CR cells (). These results suggest that suppression of SESN1 impaired the cisplatin resistance of IMC-3CR cells through reduction of the antioxidant firewall for ROS.
Cell-killing effect of heat treatment in IMC-3CR
Because, in general, stress response after heat-shock induces ROS, the involvement of SESN1 in hyperthermia resistance was investigated. Clonogenic cell survival assay showed that IMC-3CR cells possessed 10 times higher hyperthermic resistance than IMC-3 cells do (). The protein expression of SESN1 was analysed using Western blotting in IMC-3 and IMC-3CR cells. SESN1 was expressed more dominantly in IMC-3CR than in IMC-3 cells without any stimulation (, upper left panel). The density of the Western blot was quantified (, upper right panel). SESN1 was induced only in IMC-3CR, but not in IMC-3 cells after hyperthermia ( upper panel). Western blot results demonstrated that hyperthermia induced accumulation of SESN1 in IMC-3CR cells (, lower left panel). The density of the Western blot was quantified (, lower right panel), confirming hyperthermia-induced accumulation of SESN1 at 1–12 h after the treatment. By contrast, SESN1 was less expressed in IMC-3, and SESN1 was not induced at 1–12 h after hyperthermia in IMC-3 cells, suggesting that SESN1 can be involved in hyperthermia resistance (Supplementary Figure S3).
Figure 4. SESN1 induce hyperthermia resistance. (A) Colony formation was analysed by clonogenic cell survival assay after hyperthermia (44 °C, 15 min or 30 min) in IMC-3 and IMC-3CR cells. The ratio of colony numbers (survival fraction) is shown by log10. *p < .05. Error bars ± SD. (B) The protein expression of SESN1 was analysed using Western blotting in IMC-3 and IMC-3CR cells 6 h after hyperthermia (44 °C, 30 min, upper left panel). GAPDH is used as a loading control. The blot density was quantified (upper right panel). SESN1 expression was analysed using Western blotting at 1–12 h after hyperthermia (44 °C, 30 min) in IMC-3CR cells (lower left panel). GAPDH is used as a loading control. The blot density was quantified (lower right panel). (C) Cell viability was analysed by MTT assay in IMC-3CR cells 48 h after heat treatment (44 °C, 30 min). *p < .05. Error bars ± SD. (D) The apoptotic cells were analysed by flow cytometry with Annexin V labelling in IMC-3CR cells 48 h after heat treatment (44 °C, 30 min). *p < 0.05. Error bars ± SD. (E) Cell viability after heat treatment (44 °C, 15 min) was confirmed by clonogenic cell survival assay. *p < .05. Error bars ± SD. (F) ROS production was analysed after hyperthermia (44 °C, 15 min) in IMC-3 and IMC-3CR cells. *p < .05. Error bars ± SD. (G) ROS production was measured 1 h after hyperthermia (44 °C, 15 min) in IMC-3CR cells. *p < .05. Error bars ± SD. (H) Caspase 3 activity was analysed 48 h after hyperthermia (44 °C, 30 min) in IMC-3CR cells. *p < .05. Error bars ± s.d. (I) Cell viability was analysed by MTT assay in IMC-3CR cells 48 h after cisplatin (1 μg/ml) with heat treatment (44 °C, 30 min). *p < .05. Error bars ± SD.
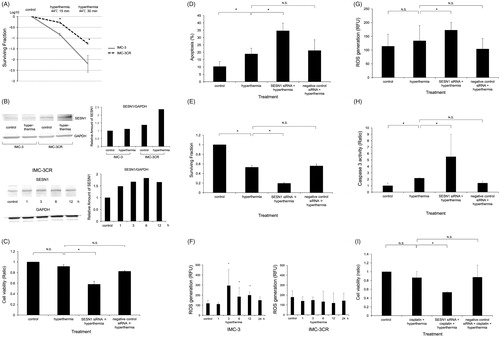
Cell viability of IMC-3CR was analysed after hyperthermia with siRNA for SESN1. MTT assay showed that suppression of SESN1 by RNAi reduced cell viability 48 h after hyperthermia significantly compared to hyperthermia alone (92 vs. 58%, ). Negative control siRNA did not affect cell viability ().
Flow cytometric analyses proved that suppression of SESN1 by RNAi enhanced apoptosis 48 h after hyperthermia compared to hyperthermia alone (18.9 vs. 34.6%, ). Negative control siRNA did not affect apoptosis ().
Clonogenic cell survival assay was performed to confirm the cell viability after hyperthermia with suppression of SESN1 (). IMC-3CR cells with RNAi for SESN1 had a significantly smaller surviving fraction after hyperthermia (52.9 vs. 19.5%, ). Negative control siRNA did not affect colony formation ().
We analysed ROS production in IMC-3 and IMC-3CR cells after hyperthermia (). Although hyperthermia significantly induced ROS production in IMC-3, ROS enhancement was not observed after hyperthermia in IMC-3CR cells (). By contrast, suppression of SESN1 significantly induced ROS production after hyperthermia in IMC-3CR cells (). Negative control siRNA did not affect the ROS production (). To confirm ROS function in hyperthermia-induced apoptosis pathway, Caspase 3 activity was analysed in IMC-3CR (). Suppression of SESN1 enhanced caspase 3 activity after hyperthermia significantly compared to hyperthermia alone in IMC-3CR cells (). These results suggest that suppression of SESN1 reduced hyperthermia resistance of IMC-3CR cells through release of the inhibition of ROS production by SESN1. Synergistic cytotoxicity of cisplatin and hyperthermia was analysed using MTT assay (). IMC-3CR cells demonstrated resistance against simultaneous cisplatin and hyperthermia treatment. Suppression of SESN1 enhanced synergistic cell-killing effects with cisplatin and hyperthermia in IMC-3CR cells (86.5 vs. 53.2%, ).
Discussion
The main mechanisms of cell-killing effects mutually differ for cisplatin, radiation, and hyperthermia. Cisplatin induces anticancer effects through DNA damage such as inter-strand and/or intra-strand cross linkage, followed by mitochondrial apoptosis [Citation23]. Radiation can induce cell-killing effects in cancers of various types through a direct effect, double and/or single strand breakage of DNA, as well as indirect DNA damage by free radicals [Citation24]. Regarding hyperthermia, functional loss of protein through unfolding and aggregation plays a central role in biological effects of the cell-killing effect. However, a large part of the cell-killing effect of hyperthermia remains unknown [Citation11]. Although cisplatin, radiation and hyperthermia kill cancer cells in different ways, a common stress response can be evoked: production of ROS [Citation14,Citation23,Citation25].
Enhanced production of intracellular ROS causes oxidative stress with or without impaired function of cellular antioxidant protection. It engenders downstream cellular events such as alterations of mitochondrial function and cell signalling pathways, resulting in apoptotic cell death [Citation26]. ROS activate the mitochondrial pathway by inducing the release of cytochrome c from the mitochondria and the formation of the apoptosome complex with Apaf-1 and pro-caspase-9 in the cytoplasm. Caspase-9 is activated at the apoptosome, activates caspase-3 and cleaves PARP, resulting in apoptosis () [Citation26,Citation27]. Various cancer cells have strong antioxidant defences to endure elevated ROS and to avoid apoptosis [Citation28]. Therefore, disturbing the redox system of cancer cells can be a feasible strategy in cancer therapy.
Figure 5. Hypothetical mechanism of cisplatin resistance and hyperthermia resistance via induction of SESN1. The cisplatin-induced or hyperthermia-induced ROS is degenerated by SESN1, resulting in resistance. Suppressing SESN1 can engender apoptosis through the ROS-Caspase pathway.
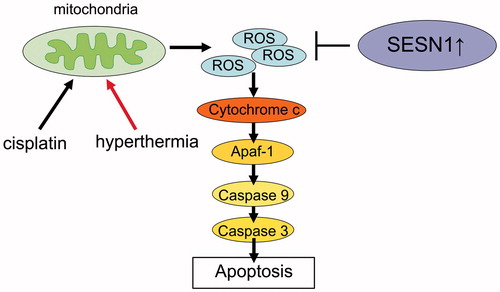
SESN1 was reported originally as a potential regulator of cellular growth, which can be induced by doxorubicin, UV radiation or γ radiation [Citation29]. SESN1 and SESN2 serve important roles for negative ROS regulation in different cancer cell lines under low-stress or oxidative-stress conditions [Citation15]. Reports suggest that SESN1 can play an important role as a protector in various situations of DNA damage. Our results suggest that SESN1 induced cisplatin and hyperthermia resistance through reduction of ROS-induced apoptotic pathway in head and neck cancer cells ().
Consequently, we investigated whether SESN1 might be involved in radiation resistance. The clonogenic cell survival assay showed no difference in the survival fraction of IMC-3 and IMC-3CR after radiation (Supplementary Figure S4). The MTT assay showed that SESN1 suppression did not significantly reduce cell viability after radiation in IMC-3CR (Supplementary Figure S5). Taken together, we conclude from these results that SESN1 cannot cause radiation resistance in IMC-3CR cells. The results suggest that the main cell-killing effect of radiation is attributable to double-strand or single-strand breakage of DNA, and that ROS is secondary in IMC-3CR cells. ROS can participate in apoptosis after irradiation to different degrees in cancers of various types. Further investigation must be done of SESN1 in radiation resistance.
In contrast to our results of SESN1 as a cancer protector from DNA damage, an earlier report described SESN2 as a tumour suppressor: silencing SESN2 accelerates tumour growth in lung carcinoma xenografts [Citation30]. This discrepancy can be derived from binary function of ROS. Cancer cells fundamentally involve increased ROS production and permanent oxidative stress [Citation31,Citation32]. The continuous accumulation of ROS can stimulate cancer cells into cellular proliferation, mutagenesis promotion and genomic instability [Citation31,Citation32]. Cancer cells maintain certain levels of intracellular ROS for normal cellular function and survival. Thereafter, chronic higher ROS level prolonged by silencing SESN can promote tumour growth. However, either extreme increasing or decreasing ROS levels can be lethal for cancer cells [Citation33]. Transient higher ROS levels evoked by cellular stress such as cisplatin can be cytocidal, even in cancer cells. In such situations, induced SESN1 can prevent cancer cells from following the apoptosis pathway. Chronic lower or higher ROS levels can impair normal cellular functions, resulting in side effects. Transient suppression of SESN1 followed by induction of ROS is more feasible for cancer treatment.
Indeed, cisplatin improves the efficacy of radiation, but the synergic effect is not infinite in advanced HNSCC [Citation1]. The development of chemosensitization without side effects is necessary for the strategy including cisplatin. Similarly, the insufficient effect of hyperthermia on cancer patients found in clinical studies, in addition to troublesome treatment practices, present obstacles to widespread use [Citation7]. Detection of new molecular targets to enhance the efficacy of hyperthermia is necessary for practical adaptation in head and neck cancers. Our data demonstrated that suppression of SESN1-sensitised cisplatin and hyperthermia, but it did not affect cell viability by itself. The observation unveiled the possibility of using SESN1 as a specific sensitiser of cisplatin and hyperthermia.
Conclusions
Suppressing SESN1 reduces cisplatin and hyperthermia resistance through enhancement of ROS-induced apoptosis. SESN1 can be a promising target to overcome cisplatin and hyperthermia resistance and to enhance cell-killing efficacy in cisplatin-based chemotherapy accompanying hyperthermia for the treatment of HNSCC.
Supplementary Figure S5
Download JPEG Image (1 MB){kind=link}
Supplementary Figure S4
Download JPEG Image (1.1 MB){kind=link}
Supplementary Figure S3
Download JPEG Image (1.5 MB){kind=link}
Supplementary Figure S2
Download JPEG Image (1.3 MB){kind=link}
Supplementary Figure S1
Download JPEG Image (1.1 MB){kind=link}
Acknowledgements
We thank Dr. S. Hayashi (Division of Oncology, Biomedical Imaging Research Center, Faculty of Medical Sciences, University of Fukui) for a critical review of this work. We are also grateful to Ms. Mika Masuyama, Yukako Ishikawa, Hiroko Tsuchiya, and Makiko Imamura for excellent technical assistance.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Cooper JS, Pajak TF, Forastiere AA, et al. Postoperative concurrent radiotherapy and chemotherapy for high-risk squamous-cell carcinoma of the head and neck. N Engl J Med. 2004;350:1937–1944.
- Hamaguchi K, Godwin AK, Yakushiji M, et al. Cross-resistance to diverse drugs is associated with primary cisplatin resistance in ovarian cancer cell lines. Cancer Res. 1993;53:5225–5232.
- Xu ZY, Loignon M, Han FY, et al. Xrcc3 induces cisplatin resistance by stimulation of Rad51-related recombinational repair, S-phase checkpoint activation, and reduced apoptosis. J Pharmacol Exp Ther. 2005;314:495–505.
- Liu LZ, Zhou XD, Qian G, et al. AKT1 amplification regulates cisplatin resistance in human lung cancer cells through the mammalian target of rapamycin/p70S6K1 pathway. Cancer Res. 2007;67:6325–6332.
- Huilgol NG, Gupta S, Sridhar CR. Hyperthermia with radiation in the treatment of locally advanced head and neck cancer: a report of randomized trial. J Cancer Res Ther. 2010;6:492–496.
- Van Der Zee J, De Bruijne M, Mens JW, et al. Reirradiation combined with hyperthermia in breast cancer recurrences: overview of experience in Erasmus MC. Int J Hyperthermia. 2010;26:638–648.
- Hurwitz MD. Today's thermal therapy: not your father's hyperthermia: challenges and opportunities in application of hyperthermia for the 21st century cancer patient. Am J Clin Oncol. 2010;33:96–100.
- Mitsudo K, Koizumi T, Iida M, et al. Thermochemoradiation therapy using superselective intra-arterial infusion via superficial temporal and occipital arteries for oral cancer with N3 cervical lymph node metastases. Int. Int J Radiat Oncol Biol Phys. 2012;83:e639–e645.
- Cook JA, Gius D, Wink DA, et al. Oxidative stress, redox, and the tumor microenvironment. Semin Radiat Oncol. 2004;14:259–266.
- Richter K, Haslbeck M, Buchner J. The heat shock response: life on the verge of death. Mol Cell. 2010;40:253–266.
- Kampinga HH. Cell biological effects of hyperthermia alone or combined with radiation or drugs: a short introduction to newcomers in the field. Int J Hyperthermia. 2006;22:191–196.
- Cui XB, Yu ZY, Wang W, et al. Co-inhibition of HSP70/HSP90 synergistically sensitizes nasopharyngeal carcinoma cells to thermotherapy. Integr Cancer Ther. 2012;11:61–67.
- Budanov AV, Lee JH, Karin M. Stressin' Sestrins take an aging fight. EMBO Mol Med. 2010;2:388–400.
- Budanov AV. Stress-responsive sestrins link p53 with redox regulation and mammalian target of rapamycin signaling. Antioxid Redox Signal. 2011;15:1679–1690.
- Budanov AV, Sablina AA, Feinstein E, et al. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science. 2004;304:596–600.
- Budanov AV, Shoshani T, Faerman A, et al. Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene. 2002;21:6017–6031.
- Ben-Sahra I, Dirat B, Laurent K, et al. Sestrin2 integrates Akt and mTOR signaling to protect cells against energetic stress-induced death. Cell Death Differ. 2013;20:611–619.
- Sanli T, Linher-Melville K, Tsakiridis T, et al. Sestrin2 modulates AMPK subunit expression and its response to ionizing radiation in breast cancer cells. PLoS One. 2012;7:e32035.
- Sugimoto C, Fujieda S, Seki M, et al. Apoptosis-promoting gene (bax) transfer potentiates sensitivity of squamous cell carcinoma to cisplatin in vitro and in vivo. Int J Cancer. 1999;82:860–867.
- Hayashi S, Kano E, Tsuji K, et al. Modification of thermosensitivity and chemosensitivity induced by combined treatments with hyperthermia and adriamycin. Int J Mol Med. 2001;8:417–422.
- Narita N, Fujieda S, Tokuriki M, et al. Inhibition of histone deacetylase 3 stimulates apoptosis induced by heat shock under acidic conditions in human maxillary cancer. Oncogene. 2005;24:7346–7354.
- Goto Y, Arigami T, Kitago M, et al. Activation of Toll-like receptors 2, 3, and 4 on human melanoma cells induces inflammatory factors. Mol Cancer Ther. 2008;7:3642–3653.
- Galluzzi L, Senovilla L, Vitale I, et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–1883.
- Baskar R, Lee KA, Yeo R, et al. Cancer and radiation therapy: current advances and future directions. Int J Med Sci. 2012;9:193–199.
- Boonstra J, Post JA. Molecular events associated with reactive oxygen species and cell cycle progression in mammalian cells. Gene. 2004;337:1–13.
- Yang YI, Kim JH, Lee KT, et al. Costunolide induces apoptosis in platinum-resistant human ovarian cancer cells by generating reactive oxygen species. Gynecol Oncol. 2011;123:588–596.
- Sawal HA, Asghar K, Bureik M, et al. Bystander signaling via oxidative metabolism. Onco Targets Ther. 2017;10:3925–3940.
- Sun C, Zhang H, Ma XF, et al. Isoliquiritigenin enhances radiosensitivity of HepG2 cells via disturbance of redox status. Cell Biochem Biophys. 2013;65:433–444.
- Velasco-Miguel S, Buckbinder L, Jean P, et al. PA26, a novel target of the p53 tumor suppressor and member of the GADD family of DNA damage and growth arrest inducible genes. Oncogene. 1999;18:127–137.
- Sablina AA, Budanov AV, Ilyinskaya GV, et al. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005;11:1306–1313.
- Polyak K, Xia Y, Zweier JL, et al. A model for p53-induced apoptosis. Nature. 1997;389:300–305.
- Behrend L, Henderson G, Zwacka RM. Reactive oxygen species in oncogenic transformation. Biochem Soc Trans. 2003;31:1441–1444.
- Tong L, Chuang CC, Wu S, et al. Reactive oxygen species in redox cancer therapy. Cancer Lett. 2015;367:18–25.