
Abstract
Background: Microscopic residual tumor often occurs after thermal ablation for medium-large hepatocellular carcinoma (HCC), leading to early aggressive recurrence or late relapse during follow-up. The mechanism how microscopic residual HCC cells survive sublethal heat stress and develop rapid outgrowth remains poorly understood.
Methods: HCC cells were exposed to sublethal heat treatment and co-cultured with conditioned media from activated HSCs (HSC-CM). Changes of cell proliferation, parameters of cell autophagy and activation of signaling pathways in heat-treated residual HCC cells were analyzed. An HCC orthotopic model was subjected to partial thermal ablation and antitumor effects of a combined treatment regimen were studied.
Results: HCC cells survived sublethal heat stress via activation of autophagy. HSC-CM enhanced autophagic survival within 24 h and then promoted proliferation of heat-treated residual HCC cells through HGF/c-Met signaling. Inhibition of autophagy or c-Met increased apoptosis of heat-treated residual HCC cells and reversed the protective effect of HSC-CM. HGF modulated biological status in autophagic survival or proliferation of heat-treated residual HCC through HGF/c-Met/ERK signaling and downstream components of ATG5/Beclin1 or cyclinD1. In an animal model, inhibiting autophagy in combination with c-Met inhibitor significantly thwarted tumor progression of residual HCC after incomplete thermal ablation via the suppressed autophagy, the decreased proliferation and the increased apoptosis.
Conclusions: Activated HSCs promote progression of residual HCC cells after sublethal heat treatment from autophagic survival to proliferation via HGF/c-Met signaling. A combined treatment regimen of inhibiting autophagy and c-Met signaling could be used to suppress tumor progression of residual HCC after incomplete thermal ablation.
Introduction
Thermal ablation, especially radiofrequency ablation (RFA), represents a potentially curative therapy for unresectable small hepatocellular carcinoma (HCC) [Citation1–5]. With the increasing use of RFA in the treatment for medium- large HCC, minimal or microscopic residual HCC often occurs after incomplete RFA and leads to tumor recurrence as high as 60%, significantly impairing the survival of patients [Citation6–9]. Two distinct patterns of recurrence can be observed in clinical scenarios. One is occult where an initial complete response (no identifiable tumors on radiological imaging after treatment) is followed by eventual tumor relapse after adequate follow-up period. The other is residual disease detected early by radiographic imaging to display a more aggressive behavior. These two types of phenomena are likely connected. How microscopic residual HCC cells survive sublethal heat stress and eventually develop rapid outgrowth remains poorly understood.
HCC often arises in the context of fibrotic environment enriched with activated hepatic stellate cells (HSCs). In analogy to fibroblasts in the wound healing, activated HSCs (myofibroblasts) are recruited to the perimeter of ablation zone after RFA [Citation10]. This provides the physical and functional connections between surviving HCC cells and activated HSCs. Reciprocal interactions of HCC cells and HSCs play a significant role in HCC progression [Citation11–13]. This led us to hypothesize that activated HSCs could modulate biological aspects of heat-treated residual HCC cells from sustaining cell survival to an aggressive tumor progression.
Autophagy is an evolutionarily conserved protective mechanism to recycle energy for cell homeostasis in response to various stresses including anticancer therapies (e.g., chemotherapy, radiation, heat) [Citation14–18]. Therefore, it could be speculated that autophagy of HCC cells took place under thermal treatment, leading to survival of microscopic residual HCC cells undetected by radiological imaging but eventually converted to active growth under appropriate conditions. This will explain the phenomenon of cancer returning after ‘successful’ initial RFA treatment. However, the exact molecular mechanism how this dynamic process is fine-tuned remains unclear.
Here, we showed the evidences that activated HSCs play a critical role in biphasic modulation of biological status of heat-treated residual HCC cells as the following: (1) HCC survived sublethal heat treatment via autophagy; (2) conditioned media from activated HSCs enhanced autophagy within 24 h and then promoted proliferation of heat-treated residual HCC cells through HGF; (3) HGF modulated biological activities of autophagic survival or proliferation of heat-treated residual HCC via HGF/c-Met signaling and downstream component of ATG5/Beclin1 or cyclinD1; (4) Inhibition of autophagy and c-Met synergistically thwarted tumor progression of residual HCC after incomplete RFA in an animal model via the suppressed autophagy, the decreased proliferation and the increased apoptosis.
From a new perspective, we demonstrate the importance of activated HSCs in modulating biological activities of heat-treated surviving HCC cells, and propose that dual inhibition of autophagy and c-Met as a potential treatment could block progression of minimal or microscopic residual HCC after incomplete thermal ablation, which ultimately improve the survival outcome after RFA.
Materials and methods
Cell culture, cell proliferation, cell apoptosis and cell cycle and animal experiments
The detailed protocols were provided in Supplementary material Doc S1.
Reagents and antibodies
Chloroquine (CQ) and PHA665752 (PHA) were purchased from Sigma-Aldrich. Recombinant human HGF was obtained from Peprotech (Rocky Hill, NJ, USA). U0126 was purchased from Cell Signaling Technology (CST, Danvers, MA). The used antibodies were listed in Supplementary material Table S1.
Quantitative reverse transcription polymerase chain reaction (qRT-PCR), Western blotting, Immunohistochemistry, immunofluorescence, cytokine antibody array and enzyme-linked immunosorbent assay (ELISA) analysis
The detailed procedures of qRT-PCR, Western blotting, Immunohistochemistry, Immunofluorescence and ELISA were provided in the Supplementary material Doc S1. The sequences of the primers are shown in Supplementary material Table S2.
Autophagy analysis, microarray analysis and TCGA dataset
The detailed protocol of autophagy analysis, microarray analysis and TCGA dataset were provided in Supplementary material Doc S1.
Statistical analysis
Statistical analysis was performed using the GraphPad Prism version 5.0 (GraphPad Software, CA, US). Quantitative results were expressed as mean and SD and significance was evaluated by Student’s t test between two groups or by one-way ANOVA for multiple groups. Categorical data were analyzed using the χ2 or Fisher’s exact tests.
Results
HCC cells survived sublethal heat treatment via autophagy
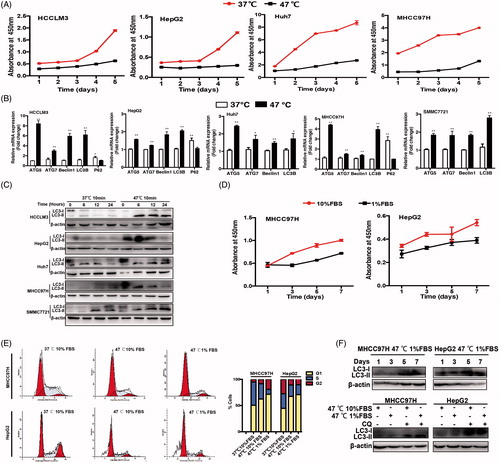
As we described previously [Citation19,Citation20], HCC cells were exposed to sublethal heat stress (IT50, 47 °C for 10 min) to simulate the incomplete RFA. Cell growth curves of heat-treated residual HCC cells showed an obvious latency growth and slower proliferation compared with control cells treated by 37 °C for 10 min (). To investigate whether HCC cells survived sublethal heat treatment via autophagy, we detected the changes of autophagy-associated genes in heat-treated surviving HCC cells. Increased mRNA expression of LC3B, Beclin1, ATG5 and ATG7, and decreased expression of autophagy substrate P62 were detected in heat-treated residual HCC cells compared with control cells (). The changes of those molecules in a time-dependent manner were confirmed by Western blotting (). RFA treatment not only induces tumor necrosis, but also disrupts the blood supply [Citation21]. To mimic the poorly vascular microenvironment following RFA [Citation21], heat-treated residual HCC cells were incubated in culture media with 1% FBS representative of ischemic condition. As shown in , growth kinetics of heat-treated residual HCC cells were further slow-downed in ischemic condition (1% FBS) compared with standard condition (10% FBS). Consistently, an obvious increase in the proportion of G0/G1 phase of cell cycles was observed in heat-treated residual HCC cells cultured under ischemic versus standard condition (MHCC97H, 71.5% vs. 62.5%; HepG2, 70.9% vs. 67.6%) (). Ischemic condition promoted the autophagic status of heat-treated surviving HCC cells lasting for one week as indicated by increased LC3-II expression, and a significant increase of LC3-II expression was observed when the cells were incubated with CQ (), suggesting increased autophagic flux in heat-treated surviving cells in ischemia condition.
Figure 1. HCC cells survived sublethal heat treatment via autophagy. (A) Cell proliferation of heat-treated (47 °C for 10 min) residual HCC cells (Huh7, MHCC97H, HepG2 and HCCLM3) was significantly suppressed compared with that of control cells treated by 37 °C for 10 min. (B) The mRNA expression of autophagy-related genes (ATG5, ATG7, Beclin1, LC3B, P62) were examined by quantitative RT-PCR. (C) The expression of LC3-II in heat-treated (47 °C for 10 min) residual HCC cells was significantly higher than that in control cells treated by 37 °C for 10 min, as indicated by Western blotting. The zero time represents the end of heat shock (47 °C for 10 min). (D) The growth latency of heat-treated residual HCC cells under ischemic (1% FBS) vs standard (10% FBS) condition was shown. (E) Compared with control cells treated by 37 °C for 10 min, an increase of the proportion of G0/G1phase in heat-treated (47 °C for 10 min) residual HCC cells cultured under ischemic (1% FBS) vs standard (10% FBS) condition. (F) LC3-II expression in heat-treated (47 °C for 10 min) residual HCC cells under ischemic (1% FBS) condition was increased gradually and maintained for one week, as indicated by Western blotting. LC3-II level in heat-treated (47 °C for 10 min) residual HCC cells cultured under ischemic (1% FBS) vs standard (10% FBS) condition was further upregulated in the presence of CQ (10 μM). **, p < .01; *, p < .05.
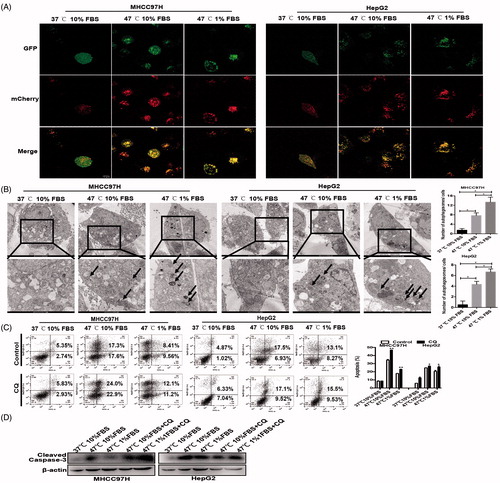
Next, we constructed MHCC97H and HepG2 cells stably expressing mCherry-GFP-LC3 and found that sublethal heat treatment induced a significant increase of autophagic flux in HCC cells as evidenced by profound LC3-II dot or puncta (membrane-associated, PE-conjugated form of LC3) (). Increased autophagy in heat-treated surviving HCC cells was confirmed by transmission electron microscopy (TEM) showing an increase of cytosolic autophagic vesicles (AVs), which was enhanced by ischemic condition (). CQ as an autophagy inhibitor significantly decreased autophagic survival of heat-treated residual HCC cells as evidenced by the increased fractions of early apoptotic and late apoptotic, which was confirmed by the increased expression of cleaved caspase-3 ()). Taken together, these results indicate that HCC cells survive sublethal heat treatment to avoid apoptosis via the increased autophagy.
Figure 2. Autophagy was activated in heat-treated surviving HCC cells. Representative images of an increase of autophagic flux indicated by punctate mRFP-EGFP-LC3 fusion protein under confocal microscopy (A) and AVs (black arrows) by TEM and the bar charts indicate quantification (B). Cell apoptosis of heat-treated (47 °C for 10 min) residual HCC cells cultured under ischemic (1% FBS) vs standard (10% FBS) condition in the presence of CQ (10 μM) was detected using flow cytometric analysis (C) and cleaved-caspase-3 (D).
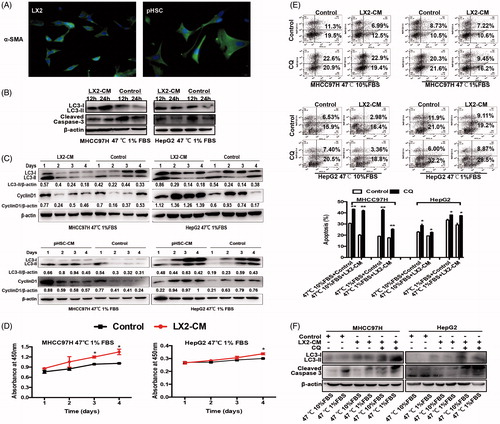
HSC-CM promoted proliferation of heat-treated residual HCC cells via decreasing autophagy
Immunofluorescence staining of α-SMA showed that HSCs were highly activated, displaying fibroblastic morphology (). Next, we co-cultured heat-treated surviving HCC cells with HSC-CM and examined the changes of autophagy. Compared with control media, HSC-CM increased LC3-II expression and decreased cleaved caspase-3 within 24 h, suggesting HSC-CM promotes autophagic survival of heat-treated residual HCC cells. After then, HSC-CM gradually down-regulated LC3-II expression and slowly increased cyclinD1 expression in heat-treated surviving HCC cells ()). This dynamic change was paralleled with the shortened growth latency to proliferation induced by HSC-CM (). CQ inhibited autophagy and increased apoptosis of heat-treated surviving HCC cells cultured with HSC-CM, as evidenced by an increase of LC3-II and cleaved caspase-3 expression ()). LX2-CM-mediated apoptotic changes in MHCC97H cells appeared more significant than those in Hep G2 (). The differences might be due to the different properties of HCC cell lines. Up-regulation of cleaved caspase-3 expression by CQ was confirmed by Western blotting (). These data suggest that HSC-CM promotes proliferation of heat-treated residual HCC cells via the decreased autophagy.
Figure 3. HSC-CM decreased cell apoptosis and promoted proliferation of heat-treated residual HCC cells via regulating autophagy. (A) Immunofluorescence staining of α-SMA in activated HSCs cells LX2 and pHSCs. (B) HSC-CM promoted autophagic survival within 24 h and decreased apoptosis in heat-treated (47 °C for 10 min) residual HCC cells cultured under ischemic (1% FBS) condition, as indicated by LC3-II and cleaved Caspase-3. (C) HSC-CM decreased autophagy and promoted proliferation of heat-treated (47 °C for 10 min) residual HCC cells cultured under ischemic (1% FBS) condition, as indicated by LC3-II and CyclinD1. Expression levels of LC3-II and CyclinD1 were normalized to β-actin. (D) Analysis of growth curves of heat-treated (47 °C for 10 min) residual HCC cells under ischemic (1% FBS) condition cultured with HSC-CM, as indicated by WST-1 assay. (E) Apoptosis of heat-treated (47 °C for 10 min) residual HCC cells under ischemic (1% FBS) vs standard (10% FBS) condition and cultured with HSC-CM in the presence of CQ (10 μM) was determined using Annexin V/PI staining assay. (F) The protein expression of autophagy (LC3-II) and apoptosis (cleaved Caspase-3) in heat-treated (47 °C for 10 min) residual HCC cells under ischemic (1% FBS) vs standard (10% FBS) condition and cultured with HSC-CM in the presence of CQ (10 μM) were determined by Western blotting. LX2-CM: conditioned media from LX2 cells; pHSC-CM: conditioned media from primary HSCs. **, p < .01; *, p < .05, bar = 1 mm.
HGF mediated the HSC-CM proliferation-promoting effects via regulation of autophagy
To identify the main mediator involved in HSC-CM promoting proliferation of heat-treated residual HCC cells, we compared cytokine profiles of HSC-CM with control media. The level of HGF in HSC-CM was ∼2.53-fold higher than that in control (). The secreted HGF in HSC-CM was confirmed by ELISA (). HGF, secreted by fibroblast-like cells including activated HSCs, is implicated in tumor survival and autophagic regulation [Citation22–24]. COL1A1 (collagen, type I, α 1) is a marker of HSCs activation. Correlation analysis in HCC cohorts from TCGA showed that the expression of HGF was positively correlated with that of COL1A1 (r = 0.5976, p < .0001) (), indicating HSCs appear to be the main producers of HGF in HCC tissues.
Figure 4. HGF mediated the protective effects of HSC-CM via regulating autophagy. The presence of HGF in HSC-CM was identified by cytokine profiles (A) and ELISA assay (B). The correlation between of HGF and COL1A1 expression in HCC cohorts from TCGA (C). (D, E) The alterations of cell growth and c-Met phosphorylation, LC3B-II, P62, CyclinD1 and cleaved caspases-3 protein expression in heat-treated (47 °C for 10 min) residual HCC cells under ischemic (1% FBS) condition cultured with HSC-CM, or in the presence of HGF-neutralizing antibody or c-Met inhibitor (PHA). (F) Autophagy (LC3B, P62, ATG5, Beclin1), CyclinD1 and cleaved caspases-3 protein expression in heat-treated (47 °C for 10 min) residual HCC cells cultured under ischemic (1% FBS) condition in response to HGF (0, 2, 10, 50 ng/ml) was detected by Western blotting. Cell growth curve (G) and decrease of the proportion of G0/G1phase (H) and cell apoptosis (I) of heat-treated (47 °C for 10 min) residual HCC cells cultured under ischemic (1% FBS) condition in response to HGF in response to HGF (0, 2, 10, 50 ng/mL). LX2-CM: conditioned media from LX2 cells; pHSC-CM: conditioned media from primary HSCs. **, p < .01; *, p < .05.
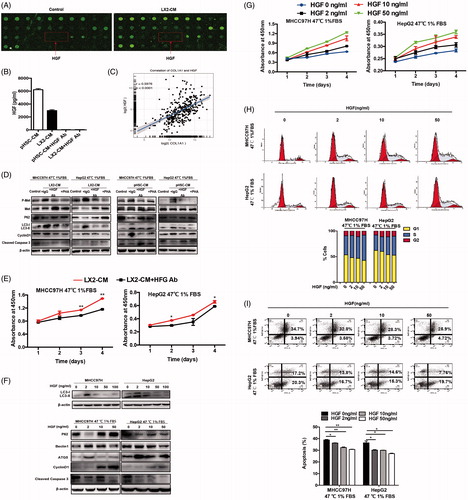
To define the role of HGF in HSC-CM in regulating cell autophagy, HGF-neutralizing antibody was added to HSC-CM to block HGF activity. The effects of HSC-CM on autophagy (LC3-II and P62), proliferation (cyclinD1) and apoptosis (cleaved caspase-3) on heat-treated residual HCC cells were significantly reversed after withdrawal of HGF in HSC-CM using HGF-neutralizing antibody (). This was paralleled by a delayed cell growth (). On the other hand, administration of exogenous recombinant HGF mimicked the effects of HSC-CM. Interestingly, LC3B-II expression in heat-treated residual HCC was significantly suppressed by HGF at the higher concentrations (10, 50, 100 ng/mL) in a dose-dependent manner except that LC3B-II was elevated by HGF at the low concentration (2 ng/mL) (), suggesting biphasic regulation of autophagy by HGF. This means that autophagy is induced by HGF at low concentration and suppressed by high concentration of HGF. In parallel, proliferation (cyclinD1) was increased and apoptosis (cleaved caspase-3) was decreased in heat-treated surviving HCC cells in response to HGF (). Decreased fractions of cells in the G0/G1 phase, reduced apoptosis and increased proliferation of heat-treated residual HCC were promoted by HGF stimulation in a dose-dependent manner ()). The above findings suggest that HGF mediates proliferation-promoting effects of HSC-CM via regulation of autophagy.
HGF/c-Met signaling modulated the biological status of autophagy or proliferation
c-Met as the only receptor of HGF is often over-expressed in cancers [Citation25]. HGF/c-Met signaling is strongly associated with survival of cancer cells [Citation26]. In addition, ERK1/2 is the direct downstream effector of c-Met [Citation27]. From analysis of TCGA-HCC data, we also found the close association between MAPK1 (ERK2) and cyclinD1 (r = 0.1292, p = .0125) (). The levels of c-Met, ERK1/2 phosphorylation, LC3B, P62, ATG5, Beclin1, cyclinD1 and cleaved caspase-3 in heat-treated surviving HCC cells were significantly altered by HSC-CM in a time and dose-dependent manners whereas their expressions were reversed after pre-adding with HGF-neutralizing antibody, c-Met inhibitor (PHA) or ERK1/2 inhibitor to HSC-CM ( and )). On the other hand, exogenous recombinant HGF increased c-Met and ERK1/2 phosphorylation and p62 expression, and reduced the corresponding LC3B-II, ATG5, Beclin1 and cleaved caspases-3 expression in heat-treated residual HCC cells in dose and time-dependent manners ( and Citation5(D,E)). These pro-autophagic changes induced by 2 ng/mL HGF were reversed by c-Met or ERK1/2 inhibitor ()).
Figure 5. HGF/c-Met signaling in autophagy and proliferation of heat-treated residual HCC cells. (A) The correlation analysis between MAPK1 (ERK2) and CyclinD1 expression in HCC data from TCGA. c-Met phosphorylation (at tyrosine residues 1234 and 1235) and ERK1/2 phosphorylation of heat-treated (47 °C for 10 min) residual HCC cells under ischemic (1% FBS) condition were significantly up-regulated by HSC-CM (B) or HGF in time and dose-dependent manners (D, E). Addition of c-Met inhibitor (PHA, 0.1 μM) or U0126 (25 μM) reversed the HSC-CM- (C) or HGF-mediated (F, G) alterations of c-Met and ERK1/2 phosphorylation, CyclinD1, cleaved caspase-3, autophagic protein (LC3-II, P62, ATG5 and Beclin1). ATG5- (H) or Beclin1-knockdown (I) suppressed heat-treated (47 °C for 10 min) residual HCC cells under ischemic (1% FBS) condition in response to HGF (0, 2, 10 ng/mL), as indicated by the alterations of LC3-II, P62, cleaved caspase-3 and bcl-2.
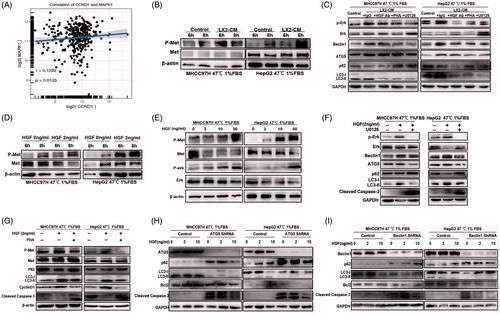
To identify downstream autophagic proteins that participate in HGF/c-Met signaling networks, we performed gene microarrays of heat-treated residual HCC cells in response to HGF (0, 2 or 10 ng/mL). Six types of dynamic expression patterns for all differential genes were categorized by hierarchical clustering, and 185 genes were identified as the common differentially expressed genes (Supplementary material Figure S1). In addition, 13 autophagy-related genes displayed dynamic alterations of increased expression induced by 2 ng/mL HGF and suppressed expression by 10 ng/mL HGF (Supplementary materials Table S3 and Figure S1). Elevated ATG5 and Beclin1 were identified as the mediators of autophagic activation in heat-exposed residual HCC cells treated with 2 ng/mL HGF. Autophagy was suppressed and converted to proliferation via down-regulation of autophagy-related molecules (LC3B, Beclin1 and ATG5) and up-regulation of anti-apoptosis protein bcl-2 when heat-exposed residual HCC cells were treated by 10 ng/mL HGF ( and Citation5(H,I)). Knockdown of ATG5 or Beclin1 gene expression promoted cell apoptosis by reduction of 2 ng/mL HGF-induced autophagy (LC3B and P62) and anti-apoptotic protein bcl-2 ()), suggesting that expression of ATG5 and Beclin1 promotes 2 ng/mL HGF-induced autophagy. These results show that HGF switches autophagic survival to proliferation of heat-treated residual HCC cells via modulation of HGF/c-Met/ERK1/2 signaling from downstream autophagic axis of ATG5/Beclin1 to proliferative axis of cyclinD1.
Incomplete RFA induced autophagy in residual HCC
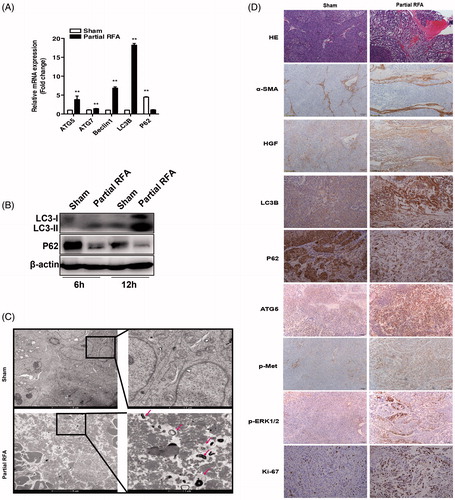
To examine whether autophagy was activated in residual HCC after incomplete RFA, we used a HCC orthotopic xenograft model that was subjected to partial RFA. In residual HCC foci at the periablational zone, ATG5, Beclin1, LC3B expression was significantly increased and P62 was markedly decreased (). The changes of autophagic markers (LC3B, P62) were confirmed by Western blotting (). In addition, an apparent increase of double-membrane-bound autophagic vacuoles was observed in surviving residual HCC cells in partial RFA group while autophagic marker was less found in sham ablation group (). Immunohistochemistry staining of α-SMA showed that the accumulation of activated HSCs surrounded residual HCC cells, and the upregulation of HGF in fibrotic stroma, indicating a clue to the possible source of HGF from activated HSCs (). Consistent with the above-mentioned, expression of LC3B, ATG5, phosphorylated c-Met, phosphorylated ERK1/2 and Ki-67 were increased and P62 expression was decreased in the residual HCC tissues after partial RFA as revealed by immunohistochemistry staining (). These data suggest that incomplete RFA promotes autophagy of residual HCC cells and imply the interaction between accumulation of activated HSCs and residual HCC cells.
Figure 6. Incomplete RFA induced autophagy in residual HCC in an orthotopic HCC model. The mRNA expression (A) of ATG5, ATG7, Beclin1, LC3B, P62 and protein expression (B) of LC3B, P62 were assessed in residual HCC tissues after incomplete RFA. (C) AVs (red arrows) in the tissue sections of post-RFA residual HCC were detected by electron microscopy. (D) Immunohistochemical staining for LC3B, P62, ATG5, Ki-67, α-SMA, HGF, phosphorylated c-Met and phosphorylated ERK1/2 in residual HCC foci after incomplete RFA. Accumulation of activated HSCs (α-SMA, a marker of activated HSCs) surrounded the residual HCC foci. **, p < .01; *, p < .05, bar = 1 mm.
CQ combined with c-Met inhibitor suppressed tumor progression of residual HCC
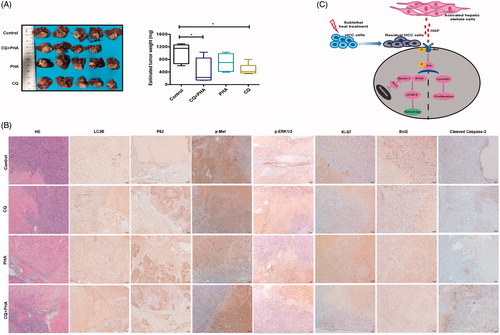
Finally, we tested whether autophagy inhibitor CQ combined with c-Met inhibitor (PHA) would effectively suppress tumor progression of residual HCC after incomplete RFA. Tumor size of post-RFA residual HCC were much smaller in combined treatment group than that in the other groups (control, CQ treatment, PHA treatment) (), except one mouse in combined treatment group died on day 1 after RFA due to intestinal perforation. In comparison with the other groups, CQ combined with PHA much suppressed autophagy (increased LC3B and p62 expression), c-Met phosphorylation, ERK1/2 phosphorylation and proliferation (decreased Ki-67 expression), and increased apoptosis (low Bcl2 and high cleaved-caspases-3) in residual HCC foci after incomplete RFA (). These data demonstrate that combined treatment of CQ and c-Met inhibitor could thwart tumor progression of residual HCC after incomplete RFA via the suppressed autophagy, the decreased proliferation and the increased apoptosis.
Figure 7. A combination of autophagy inhibitor CQ and c-Met inhibitor (PHA) suppressed tumor progression of residual HCC after incomplete RFA. (A) Tumor size of residual HCC after incomplete RFA was significantly smaller in combined treatment (CQ + PHA) group than that in the other groups (control, CQ, or PHA treatment), except one mouse in combined treatment group died on day 1 after RFA due to intestinal perforation. The box-plots indicate quantification. (B) CQ combined with PHA significantly suppressed autophagy (increased LC3B and P62 expression), ERK1/2 and c-Met phosphorylation and proliferation (decreased Ki-67 expression), and increased apoptosis (low Blc-2 and high cleaved caspases-3) in residual HCC tissues after incomplete RFA. (C) A schematic diagram of activated HSCs promoting tumor progression of heat-treated residual surviving HCC from autophagic survival to proliferation via HGF/c-Met signaling. **, p < .01; *, p < .05, bar = 50 μm/1 mm.
Discussion
Microscopic residual HCC at marginal zone of thermal ablation frequently occurs when RFA is used for treating medium-sized or large HCC. One study reported that viable HCC cells were found on the RFA needle immediately after local ablation even in 26.7% of patients [Citation28]. Recurrence after insufficient RFA significantly impairs the survival outcome of patients. Some relapses are detected during the period of follow-up and the others recur early showing more aggressive phenotype [Citation21,Citation29–31]. However, the molecular mechanism underlying these biological behaviors has not been elucidated. Herein, we provided new evidence that activated HSCs modulated from autophagic survival to proliferation of heat-treated residual HCC cells via HGF/c-Met signaling pathway, indicating the importance of cross-talk between activated HSCs and post-heat residual surviving HCC cells. This unwanted (‘off-target’) effect reminds us that we need notice the impact of tumor microenvironment on impairing the treatment response of tumor when we treat cancer locally. Notably, we proposed that combined treatment targeting autophagy and HGF/c-Met signaling synergistically suppressed residual tumor progression after incomplete RFA. This finding may have clinical implications in improving the therapeutic effects of suboptimal RFA and the survival outcome of patients.
In this study, we showed that biological status of heat-treated residual HCC cells from autophagic survival towards proliferation was modulated by HGF from activated HSCs. Analyses of HCC cohorts in TCGA indicate that activated HSCs are the main source of HGF expression in HCC tissues. HCC cells survived sublethal heat treatment by an elevation of autophagy indicated as the expression of LC3B-II, the formation of autophagic vacuoles and a punctate fluorescence pattern with mCherry-GFP-LC3 protein, all markers for cellular autophagy. Moreover, we showed that ischemic condition mimicking the hypovascular microenvironment following RFA could further enhance autophagy and increase the proportion of G0/G1 of cell cycles in heat-treated residual HCC cells. This result is consistent with the finding from a recent study that transarterial embolization-like ischemia inducing cell cycle arrest dependent on autophagy of surviving HCC cells [Citation32]. More importantly, HGF at a low concentration promoted autophagic survival of heat-treated residual HCC cells while high concentration of HGF promoted cell proliferation. Dual inhibition targeting autophagy and HGF/c-Met signaling could potentiate heat-induced apoptosis and reduced residual tumor growth.
HCC often arises from liver cirrhosis and activated HSCs are the main contributor to fibrotic microenvironment [Citation12,Citation33,Citation34]. Activated HSCs were accumulated at the border of ablative zone after RFA. In our previous studies, we showed that activated HSCs promoted progression of heat-treated residual HCC. Autophagy is a fundamentally protective mechanism of cell survival against various stresses [Citation35,Citation36]. Recently, two studies have shown that activation of autophagy is induced by incomplete RFA [Citation18,Citation37]. In this study, we add to the growing body of knowledge by focusing on activated HSCs in tumor microenvironment that modulates autophagic survival and proliferation of heat-treated residual HCC cells, leading to residual HCC recurrence and progression. Furthermore, we dissected a dynamic process that HCC cells survived sublethal heat treatment and regained cell proliferation to progression via HSC-triggered HGF/c-Met signaling pathway, revealing a close cooperation between activated HSCs and post-heat residual surviving HCC cells. In other words, activated HSCs facilitated progression of heat-treated residual HCC via the increased autophagy and the decreased apoptosis to enhance autophagic survival and proliferation. The shift from autophagic survival to proliferation of heat-treated surviving HCC cells was modulated by paracrine effects of HGF from activated HSCs.
HGF is highly expressed by activated HSCs and involved in promoting HCC proliferation by binding to its receptor c-Met and activating its downstream ERK1/2 signaling [Citation38–42]. In addition, activation of c-Met/ERK pathway can modulate autophagy processes [Citation43,Citation44]. ERK2 expression was also found to be closely correlated with cyclinD1 expression in TCGA-HCC data. In the present study, we found that HGF at low concentration could enhance the autophagy to sustain autophagic survival of heat-treated residual HCC cells via activating c-Met/ERK signaling and autophagy (Atg5, Beclin1, P62), suggesting HGF with low concentration sustains residual HCC cells to survive in a dormant state via autophagy. This may provide an explanation why some recurrence derived from residual HCC cells occurs after a long period of disease resolution following a “successful” RFA.
In view of the effects of HGF on modulating autophagy and proliferation of heat-treated residual HCC cells, targeting autophagy and c-Met signaling were proposed as an effective approach to suppress tumor progression of residual HCC after RFA. Autophagy inhibitor could decrease the autophagic survival and increase the apoptosis of heat-treated residual HCC cells. The c-Met inhibition reduced HGF-mediated promotion of proliferation. In this study, we showed that combined treatment of inhibiting autophagy and c-Met synergistically thwarted tumor progression of residual HCC after incomplete RFA via the suppressed autophagy, the decreased proliferation and the increased apoptosis.
In conclusion (), we demonstrate that activated HSCs promote post-thermal residual HCC progression via modulating biological statues from autophagic survival to proliferation by HGF/c-Met signaling. As a potential new treatment, targeting autophagy and HGF/c-Met signaling could suppress progression of post-heat residual HCC to improve therapeutic effects after incomplete RFA.
Doc S1, Tables S1-S3 and Figure S1
Download PDF (724.5 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Bruix J, Sherman M. American Association for the Study of Liver Diseases. Management of hepatocellular carcinoma: an update. Hepatology. 2011;53:1020–1022.
- Llovet JM, Brú C, Bruix J. Prognosis of hepatocellular carcinoma: the BCLC staging classification. Semin Liver Dis. 1999;19:329–338.
- Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379:1245–1255.
- European Association For The Study Of The Liver. European Organisation for Research and Treatment Of Cancer EASL-EORTC clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2012;56:908–943.
- Lee JM, Park JW, Choi BI. KLCSG-NCC Korea Practice Guidelines for the management of hepatocellular carcinoma: HCC diagnostic algorithm. Dig Dis. 2014;32:764–777.
- Obara K, Matsumoto N, Okamoto M, et al. Insufficient radiofrequency ablation therapy may induce further malignant transformation of hepatocellular carcinoma. Hepatol Int. 2008;2:116–123.
- Curley SA, Izzo F, Delrio P, et al. Radiofrequency ablation of unresectable primary and metastatic hepatic malignancies: results in 123 patients. Ann Surg. 1999;230:1–8.
- Huang J, Yan L, Cheng Z, et al. A randomized trial comparing radiofrequency ablation and surgical resection for HCC conforming to the Milan criteria. Ann Surg. 2010;252:903–912.
- Feng K, Yan J, Li X, et al. A randomized controlled trial of radiofrequency ablation and surgical resection in the treatment of small hepatocellular carcinoma. J Hepatol. 2012;57:794–802.
- Rozenblum N, Zeira E, Bulvik B, et al. Radiofrequency ablation: inflammatory changes in the periablative zone can induce global organ effects, including liver regeneration. Radiology. 2015;276:416–425.
- Tompson AI, Conroy KP, Henderson NC. Hepatic stellate cells: central modulators of hepatic carcinogenesis. BMC Gastroenterology. 2015;15:63.
- Coulouarn C, Corlu A, Glaise D, et al. Hepatocyte-stellate cell cross-talk in the liver engenders a permissive inflammatory microenvironment that drives progression in hepatocellular carcinoma. Cancer Res. 2012;72:2533–2542.
- Coulouarn C, Clément B. Stellate cells and the development of liver cancer: therapeutic potential of targeting the stroma. J Hepatol. 2014;60:1306–1309.
- Kimmelman AC, White E. Autophagy and tumor metabolism. Cell Metab. 2017;25:1037–1043.
- Kondo Y, Kanzawa T, Sawaya R, et al. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5:726–734.
- Ding ZB, Hui B, Shi YH, et al. Autophagy activation in hepatocellular carcinoma contributes to the tolerance of oxaliplatin via reactive oxygen species modulation. Clin Cancer Res. 2011;17:6229–6238.
- Peng WX, Wan YY, Gong AH, et al. Egr-1 regulates irradiation-induced autophagy through Atg4B to promote radioresistance in hepatocellular carcinoma cells. Oncogenesis. 2017;6:e292.
- Zhao Z, Wu J, Liu X, et al. Insufficient radiofrequency ablation promotes proliferation of residual hepatocellular carcinoma via autophagy. Cancer Lett. 2018;421:73–81.
- Zhang R, Yao RR, Li JH, et al. Activated hepatic stellate cells secrete periostin to induce stem cell-like phenotype of residual hepatocellular carcinoma cells after heat treatment. Sci Rep. 2017;7:2164.
- Zhang R, Ma M, Dong G, et al. Increased matrix stiffness promotes tumor progression of residual hepatocellular carcinoma after insufficient heat treatment. Cancer Sci. 2017;108:1778–1786.
- Lee DH, Lee JM, Yoon JH, et al. Thermal injury-induced hepatic parenchymal hypoperfusion: risk of hepatocellular carcinoma recurrence after radiofrequency ablation. Radiology. 2017;282:880–891.
- Wang Y, Liu J, Tao Z, et al. Exogenous HGF prevents cardiomyocytes from apoptosis after hypoxia via up-regulating cell autophagy. Cell Physiol Biochem. 2016;38:2401–2413.
- Lau EY, Lo J, Cheng BY, et al. Cancer-associated fibroblasts regulate tumor-initiating cell plasticity in hepatocellular carcinoma through c-Met/FRA1/HEY1 signaling. Cell Rep. 2016;15:1175–1189.
- Kanaji N, Yokohira M, Nakano-Narusawa Y, et al. Hepatocyte growth factor produced in lung fibroblasts enhances non-small cell lung cancer cell survival and tumor progression. Respir Res. 2017;18:118.
- Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7:504–516.
- Arnold L, Enders J, Thomas SM. Activated HGF-c-met axis in head and neck cancer. Cancers (Basel). 2017;9:169.
- Tsukada Y, Miyazawa K, Kitamura N. High intensity ERK signal mediates hepatocyte growth factor-induced proliferation inhibition of the human hepatocellular carcinoma cell line HepG2. J Biol Chem. 2001;276:40968–40976.
- Snoeren N, Huiskens J, Rijken AM, et al. Viable tumor tissue adherent to needle applicators after local ablation: a risk factor for local tumor progression. Ann Surg Oncol. 2011;18:3702–3710.
- Yoshida S, Kornek M, Ikenaga N, et al. Sublethal heat treatment promotes epithelial-mesenchymal transition and enhances the malignant potential of hepatocellular carcinoma. Hepatology. 2013;58:1667–1680.
- Thompson SM, Callstrom MR, Butters KA, et al. Role for putative hepatocellular carcinoma stem cell subpopulations in biological response to incomplete thermal ablation: in vitro and in vivo pilot study. Cardiovasc Intervent Radiol. 2014;37:1343–1351.
- Koda M, Maeda Y, Matsunaga Y, et al. Hepatocellular carcinoma with sarcomatous change arising after radiofrequency ablation for well-differentiated hepatocellular carcinoma. Hepatol Res. 2003;27:163–167.
- Gade TPF, Tucker E, Nakazawa MS, et al. Ischemia induces quiescence and autophagy dependence in hepatocellular carcinoma. Radiology. 2017;283:702–710.
- Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–172.
- Amann T, Bataille F, Spruss T, et al. Activated hepatic stellate cells promote tumorigenicity of hepatocellular carcinoma. Cancer Sci. 2009;100:646–653.
- Song YJ, Zhang SS, Guo XL, et al. Autophagy contributes to the survival of CD133+ liver cancer stem cells in the hypoxic and nutrient-deprived tumor microenvironment. Cancer Lett. 2013;339:70–81.
- Yu T, Guo F, Yu Y, et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell. 2017;170:548–563.
- Wang X, Deng Q, Feng K, et al. Insufficient radiofrequency ablation promotes hepatocellular carcinoma cell progression via autophagy and the CD133 feedback loop. Oncol Rep. 2018;40:241–251.
- Guirouilh J, Castroviejo M, Balabaud C, et al. Desmoulière A, Rosenbaum J. Hepatocarcinoma cells stimulate hepatocyte growth factor secretion in human liver myofibroblasts. Int J Oncol. 2000;17:777–781.
- Guirouilh J, Le Bail B, Boussarie L, et al. Expression of hepatocyte growth factor in human hepatocellular carcinoma. J Hepatol. 2001;34:78–83.
- Efimova EA, Glanemann M, Liu L, et al. Effects of human hepatocyte growth factor on the proliferation of human hepatocytes and hepatocellular carcinoma cell lines. Eur Surg Res. 2004;36:300–307.
- Giordano S, Columbano A. Met as a therapeutic target in HCC: facts and hopes. J Hepatol. 2014;60:442–452.
- Rhee H, Kim HY, Choi JH, et al. Keratin 19 expression in hepatocellular carcinoma is regulated by fibroblast-derived HGF via a MET-ERK1/2-AP1 and SP1 axis. Cancer Res. 2018;78:1619–1631.
- Park EJ, Umh HN, Kim SW, et al. ERK pathway is activated in bare-FeNPs-induced autophagy. Arch Toxicol. 2014;88:323–336.
- Barrow-McGee R, Kishi N, Joffre C, et al. Beta 1-integrin-c-Met cooperation reveals an inside-in survival signalling on autophagy-related endomembranes. Nat Commun. 2016;7:11942.