
Abstract
Background
Worldwide heat stroke incidence has increased in recent years and is associated with high morbidity and mortality. Therefore, it is critical to identify mechanisms that mediate heat stroke. Previous studies suggested that damage to the small intestine may be a major factor in heat stroke-related morbidity and mortality. However, the mechanism underlying heat stroke related small intestine injury remains unclear.
Methods
To explore how heat stroke promotes intestinal damage, we applied two well established models: mouse and IEC-6 cells heat stress (HS) to mimic heat stroke both in vivo and in vitro. The percentages of viability and cell death were assessed by WST-1 and LDH release assays. Induction of HS-induced cell death was analyzed by flow cytometry with Annexin V-FITC/PI staining. Flow cytometry was used to analyze HS-induced mitochondrial superoxide with MitoSOX staining. Malondialdehyde (MDA) levels and superoxide dismutase (SOD) levels were detected by ELISA. Flow cytometry was used to analyze HS-induced mitochondrial depolarization (low ΔΨm) with JC-1 staining. Histopathology changes in the ileum were detected by H&E staining.The ileum ultrastructure was observed by transmission electron microscopy (TEM). RIPK1, RIPK3, phosphorylated MLKL, and MLKL levels were detected by Western blot. RIPK1-RIPK3 complexes were measured by immunoprecipitation assay.
Results
HS increased both necrotic cell rate and RIPK1, RIPK3, and phosphorylated MLKL expression levels in IEC-6 cells. These increased expression levels promoted higher RIPK1-RIPK3 complex formation, leading to necrosome formation both in vivo and in vitro. Moreover, HS caused dyshomeostasis, an oxidative stress response, and mitochondrial damage, along with small intestinal tissue injury and cell death. However, IEC-6 cells or mice pretreated with the RIPK1 activity chemical inhibitor Nec-1 or RIPK3 activity chemical inhibitor GSK'872 significantly reversed these phenomena and promoted balance in oxidative stress response homeostasis. More importantly, the reactive oxygen species (ROS) scavenger N-acetyl-L-cysteine (NAC) pretreatment significantly inhibited HS-induced RIPK1/RIPK3-dependent necroptosis formation both in vivo and in vitro, suggesting that preventing necroptosis via scavenging ROS production might alleviate HS-induced small intestinal tissue injury and cell death.
Conclusion
This study provides strong evidence that HS causes damage to both the small intestine and intestinal epithelial cells, scavenging ROS production can significantly alleviate such RIPK1/RIPK3-dependent necroptosis, mediating HS-induced intestinal damage both in vitro and in vivo. These findings provide a clear target for future mechanism-based therapeutic strategies for patients diagnosed with heat stroke.
Keywords:
Background
Over the past few decades, record temperatures worldwide have resulted in increased intensity, frequency, and duration of heat waves. Given this dramatic climate change, the number of patients with heat-related illnesses will likely continue to rise [Citation1–3]. Yet despite several decades of research into the mechanism underlying heat stroke, it continues to have a high mortality rate and cause multiple organ dysfunction syndrome (MODS) in patients. Furthermore, research has only fostered a limited understanding of the mechanisms that MODS during heat stroke [Citation4,Citation5]. Therefore, it is important to investigate the pathogenesis of heat stroke and develop effective preventive measures. Previously, we demonstrated that pathological damage can occur to the small intestine and epithelial cells following heat stress (HS) [Citation6–10], including impaired integrity of the intestinal barrier and increased intestinal permeability [Citation8–10]. Interestingly, we found that the gastrointestinal tract is the primary organ affected by heat stroke [Citation11]. However, the molecular mechanisms underlying HS-induced intestinal injury have yet to be clarified.
One manner by which HS might promote intestinal injury is through oxidative stress [Citation11–13]. In the intestine, blood is redistributed away from splanchnic tissue toward peripheral tissue to maximize radiant heat dissipation when the body is in a high-temperature environment. Thus, due to in hyperthermia, hypoxia, or inflammation, epithelial cell death may occur throughout the gastrointestinal tract, which can trigger oxidative stress [Citation14,Citation15]. In animal models, HS-related oxidative stress has been shown to impair the integrity of the intestinal barrier, increase intestinal permeability, and induce epithelial cell apoptosis [Citation6,Citation9,Citation10,Citation12]. Therefore we hypothesize that alleviating oxidative stress may protect against HS-induced intestinal damage.
Since the effects of HS on cell health depend on the temperature and duration of heat exposure, HS-induced oxidative stress may lead to other types of cell death in the intestine that are still poorly understood. Recently, a new necroptosis pathway was discovered and characterized, expanding our understanding of the mechanisms leading to cell death [Citation16,Citation17]. It has only recently begun to emerge that there are different mechanisms underlying distinct types of necroptosis. Studies have indicated that necroptosis contributes to cell death under a variety of pathophysiological conditions, including infection, inflammation, acute organ injury, and ischemia/reperfusion disease [Citation17–20]. Under the pathophysiological condition of dysfunctional apoptotic signaling, receptor-interacting protein kinase 1 (RIPK1) and receptor-interacting protein kinase 3 (RIPK3) are known to auto- and trans-phosphorylate each other, forming RIPK1–RIPK3 complex, leading to further necrosome formation [Citation21,Citation22]. However, the precise molecular mechanism of how HS induces intestinal damage via reactive oxygen species (ROS) and subsequent necroptosis is still unknown.
In this study, we aimed to determine whether ROS and subsequent necroptosis are mediated by HS-induced damage in the small intestine and intestinal epithelial cells. We also examined whether ROS scavenger pretreatment could protect against HS-induced intestinal injury by alleviating oxidative stress.
Methods
Cell culture and treatments
Intestinal epithelial (IEC-6) cells were preserved at the Department of Pathophysiology, Southern Medical University. IEC-6 cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS). The establishment of a heat stressed-cell model was described in detail in our previous study [Citation23]. Cell culture dishes containing IEC-6 cells were sealed with Parafilm and immersed for 2 h in a circulating water bath that was thermo-regulated at either 37 ± 0.5 °C (control group) or 43 °C (HS group), and cells were further incubated at 37 °C for 24 h. IEC-6 cells were pretreated with or without 20 μM Nec-1 or 10 μM GSK’ 872 or 2 mM N-acetyl-L-cysteine (NAC) before 43 °C of HS [Citation6,Citation24].
Cell viability assays
IEC-6 cells were either exposed (experimental condition) or not exposed (control condition) to 43 °C for 2 h, then further incubated at 37°C for 24 h. Afterward, cell proliferation was tested using the Premixed WST-1 Cell Proliferation Reagent manual (Clontech Laboratories Inc., USA). The enzymatic activity of lactate dehydrogenase (LDH) was assayed using a commercially available kit manual (JianChen Co, China).
Flow cytometry analysis
IEC-6 cells were either exposed (experimental) or not exposed (control) to 43 °C for 2 h, then further incubated at 37°C for 24 h. Cell death was detected according to the Annexin V-FITC apoptosis detection kit manual (Invitrogen). Approximately 1 × 106 cells were collected, washed with ice-cold phosphate-buffed saline (PBS), and re-suspended in a binding buffer containing Annexin V-FITC. The cells were incubated at room temperature in the dark for 10 min, then the buffer was removed by centrifugation. Cells were re-suspended in a reaction buffer containing propidium iodide (PI), and then immediately analyzed by flow cytometry to quantify cell apoptosis and death.
ROS measurements
IEC-6 cells were exposed (experimental condition) or not exposed (control condition) to 43 °C for 2 h, then further incubated for 24 h. Mitochondrial ROS (O2-.) was detected using a fluorescent probe MitoSOX (Molecular Probes). Cells or tissue were incubated with 4 µM MitoSOX (red fluorescence) at 37 °C in the dark for 30 min. The fluorescence intensities of ROS (O2-.) probes were analyzed by flow cytometry.
Mitochondrial membrane potential assay
Mitochondrial membrane potential (MMP) was detected using a fluorescent probe JC-1 (Invitrogen, CA, USA). In normal membrane potentials in mitochondria, JC-1 forms aggregates that fluoresce red; however, in damaged and/or depolarized mitochondria, JC-1 forms monomers that fluoresce green. Cells were exposed or not exposed to 43 °C for 2 h and further incubated at 37°C for 24 h, then incubated in DMEM containing 5 µmol/L JC-1 at 37 °C for 15 min. Images were captured via inverted laser scanning confocal microscopy (Model 510; Carl Zeiss, Jena, Germany). Relative fluorescence was subsequently tested by flow cytometry (BD FACSVerseTM; BD, USA). Data were analyzed using BD FAC Suite software.
Animals
A total of 104 specific pathogen-free 6- to 8-week-old male C57BL/6 mice were purchased from the Experimental Animal Center of Southern Medical University in Guangzhou, China [certification number: SYXK (Yue) 2016-0167]. The mice were housed individually in controlled environmental conditions with a 12-h light/dark cycle and unrestricted access to pellet food and water throughout the study. All surgical interventions were performed under anesthesia, using a mixture of 13.3% urethane and 0.5% chloralose (0.65 ml/100 g body weight, intraperitoneally). The experimental protocols were approved by the Animal Care and Use Committee of Southern Medical University, Guangzhou, China. The care of the animals was in accordance with the National Institutes of Health Guidelines, as well as with those of Chinese National.
Induction of HS
The mice were pretreated either with (experimental condition) or without (control condition) 1.8 mg/kg Nec−1 or 1.9 mmol/kg GSK' 872, or 100 mg/kg NAC (intraperitoneal) 1 h before HS [Citation25–27]. The establishment of heat stressed-animal model was described in detail in our previous study [Citation7,Citation23]. The mice in the HS group were placed in a pre-warmed incubator, maintained at 35.5 ± 0.5 °C with relative humidity of 60 ± 5%, without food and water. Rectal core temperature (Tc) was continuously monitored with a rectal thermometer until the Tc reached 42 °C. The mice in the control group were sham-heated at a temperature of 25 ± 0.5 °C and humidity of 35 ± 5% for a time comparable to that of the HS group. Mice were anesthetized by intraperitoneal injection with a mixture of 13.3% urethane and 0.5% chloralose (0.65 ml/100 g body weight, intraperitoneally), then sacrificed 20 h after HS exposure, and the small intestine (ileum) was isolated.
Histopathological analysis
Small intestine (ileum) samples were quickly excised, sliced into transverse or longitudinal sections, and fixed in 10% neutral-buffered formalin. The tissues were then embedded in paraffin blocks, and serial sections were stained with hematoxylin and eosin for microscopic evaluation at 200× magnification. Morphological changes were blind assessed and graded by two certified veterinary pathologists using the intestinal injury score developed by Chiu et al [Citation28].
Morphological observation
The morphological changes in the small intestine (ileum) and mitochondria were observed by transmission electron microscopy (TEM). The small intestine (ileum) was fixed with 2.5% glutaraldehyde and stained with cacodylate-buffered osmium tetroxide (OsO4). Sections were prepared and examined under an electron microscope (Philips CM10; Philips, Eindhoven, the Netherlands).
Western blot analysis
Proteins were extracted from cells or small intestine (ileum) using a Total Protein Extraction Kit (Beyotime Institute of Biotechnology, China) following the manufacturer’s protocols. The protein concentrations of the extracts were determined using an Enhanced BCA Protein Assay Kit (Beyotime Institute of Biotechnology, China). Western blot analysis was performed using RIPK1 and RIPK3 antibodies (1:1000; Abcam or Cell Signaling Technology). A horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG antibody was used as the secondary antibody (1:2500, Zhongshan Inc, China) and the signal was visualized using enhanced chemiluminescence (Pierce, Rockford, IL, USA).
Co-immunoprecipitation
After exposure to HS, cells or tissue were harvested for lysates preparation using RIPA buffer (Sigma). Cells or tissue lysates were incubated on ice for 20 min, vortexed, and centrifuged at 6600 g for 10 min to remove cellular debris. Protein concentrations were determined with Bradford Reagent (Sigma). Three milligram of cells or tissue lysates was incubated with 3 mg of anti-RIPK1 (Cell Signaling Technology) overnight at 4 °C. The immunocomplexes were collected by incubating the sample with a mix of Protein A-Agarose and Protein G-Sepharose (Sigma) overnight at 4 °C. The beads were washed once with RIPA buffer once, and twice with PBS, then resuspended in 2× Lysis buffer, loaded directly onto a 10% SDS-polyacrylamide gel and then subjected to Western blot with the indicated antibodies (RIP3, 1:1000). IgG served as the negative control.
Statistical analysis
All data were analyzed for statistical significance using IBM SPSS Statistics, version 20.0. Data were from at least three independent experiments performed in triplicate, expressed as mean ± SD. Statistical comparisons of the results were performed using one-way analysis of variance (ANOVA). p < 0.05 was considered statistically significant.
Results
HS-induced RIPK1/RIPK3-dependent necroptosis activation both in vivo and in vitro
The thermal response of the mice to HS was described in detail in our previous studyCitation7. Briefly, Tc quickly increased above 39˚C during the first 15 min of incubation, followed by a decrease to ∼38 °C. After a slow increase over ∼3 h, Tc rose above 42 °C, and the mice with a Tc above 42 °C succumbed to heat stroke (). As shown in , after exposure of IEC-6 cells to HS at 37 °C (the control group), 39 °C, 41 °C, 43 °C, or 45 °C for 2 h, followed by further incubation at 37 °C for 24 h, cell viability declined drastically after IEC-6 cells were cultured at elevated temperatures. Then, after exposure of IEC-6 cells to HS at 43 °C for 2 h, and further incubation at 37 °C for different times increments (1 h, 6 h, 12 h, 24 h and 48 h), cell viability was shown to decline in a time-dependent manner.
Figure 1. HS-induced RIPK1/RIPK3-dependent necroptosis activation in vivo. (A) The temperature rise curve of mice subjected to HS. Mice were exposed to a temperature controlled chamber (ambient temperature 35.5 ± 0.5 °C and 60 ± 5% relative humidity) until rectal core temperature (Tc) reached 42 °C. (B) Mice were exposed to a temperature controlled chamber (ambient temperature 35.5 ± 0.5 °C and 60 ± 5% relative humidity) until Tc reached 42 °C. The mice were sacrificed at 20 h after HS exposure and the small intestine (ileum) was isolated. RIPK1, RIPK3, phosphorylated MLKL, and MLKL levels were detected by Western blot. (C) Mice were exposed to a temperature controlled chamber (ambient temperature 35.5 ± 0.5 °C and 60 ± 5% relative humidity) until rectal core temperature (Tc) reached 42 °C. The mice were sacrificed at 20 h after HS exposure and the small intestine (ileum) was isolated. RIPK1-RIPK3 complexes were measured by immunoprecipitation assay with IgG serving as the negative control. (D) Mice were pretreated with or without 1.8 mg/kg Nec-1 or 1.9 mmol/kg GSK’ 872 (intraperitoneal) at 1 h, followed by exposure to a temperature-controlled chamber (ambient temperature 35.5 ± 0.5 °C and 60 ± 5% relative humidity) until Tc reached 42 °C. The mice were sacrificed at 20 h after HS exposure and the small intestine (ileum) was isolated. RIPK1, RIPK3, phosphorylated MLKL, and MLKL levels were detected by Western blot, (n = 6).
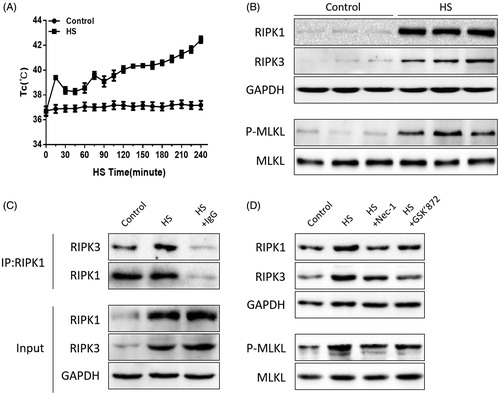
Figure 2. HS-induced RIPK1/RIPK3-dependent necroptosis activation in IEC-6 cells. (A) IEC-6 cells underwent HS (37 °C, 39 °C, 41 °C, 43 °C, or 45 °C) for 2 h and were then further incubated at 37 °C for 24 h; IEC-6 cells were subsequently exposed to HS at 43 °C for 2 h and were further incubated at 37 °C for different time increments (1 h, 6 h, 12 h, 24 h and 48 h). Cell viability percentage was measured by WST-1. (B) IEC-6 cells underwent HS (43 °C) for 2 h and were further incubated at 37 °C for 24 h. RIPK1, RIPK3, phosphorylated MLKL and MLKL levels were detected by Western blot. (C) IEC-6 cells underwent HS (43 °C) for 2 h and were further incubated at 37 °C for 24 h. RIPK1-RIPK3 complexes were measured by immunoprecipitation assay with IgG serving as the negative control. (D) IEC-6 cells were pretreated with or without 20 μM Nec-1 or 10 μM GSK’ 872, then underwent HS (43 °C) for 2 h and were further incubated at 37 °C for 24 h. RIPK1, RIPK3, phosphorylated MLKL, and MLKL levels were detected by Western blot. The data shown represent the mean ± SD from at least three independent experiments, performed in triplicate.
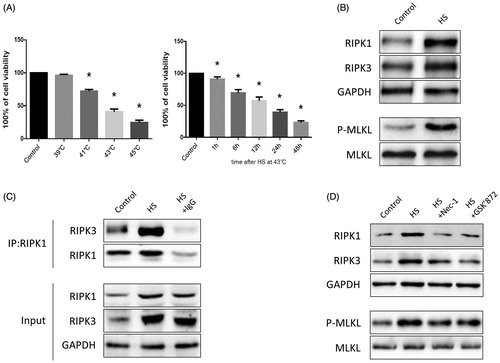
RIP1K, RIPK3, and phosphorylated MLKL were positively correlated with necrosome formation and activation, which plays a crucial role in the necroptotic signaling pathway [Citation24,Citation25]. Thus, we explored the relative expression of RIPK1, RIPK3, and phosphorylated MLKL in both the heat-stressed IEC-6 cells and animal small intestine (ileum). Western blotting assays indicated that HS treatment increased RIPK1, RIPK3, and phosphorylated MLKL expression compared with the non-treated group both in vitro and in vivo (, Citation2(B)). Furthermore, immunoprecipitation analysis showed that HS treatment increased RIPK3 expression in RIPK1-containing complexes both in vitro and in vivo (, Citation2(C)). Next, we pretreated the IEC-6 cells or mice with RIPK1 activity chemical inhibitor Nec-1 or RIPK3 activity chemical inhibitor GSK’872, and found that inhibition of RIPK1 or RIPK3 activity significantly decreased RIPK1, RIPK3, and phosphorylated MLKL expression compared to the non-treated group both in vivo and in vitro (, Citation2(D)).
Nec-1 or GSK’872 pretreatment reduced cell injury and death induced by HS
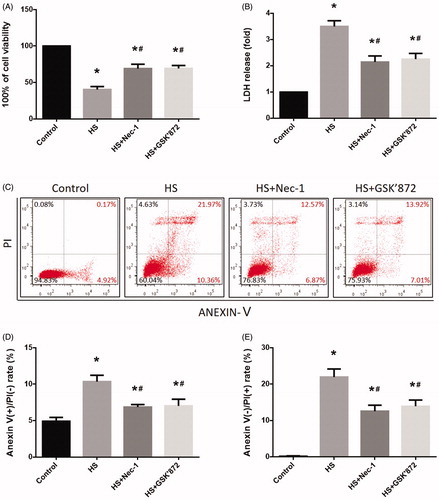
To determine whether Nec-1 or GSK’872 could reduce HS-induced IEC-6 cell injury, we examined cell viability and LDH release. Compared to control cells, IEC-6 cells demonstrated a 60% reduction in viability and released a three-fold increase of LDH after HS (). However, cells that were pretreated with Nec-1 or GSK’872 were relatively resistant to the toxic effect of HS (). To further investigate the effects of Nec-1 or GSK’872 on HS-induced IEC-6 cell death, we treated the cells that were defined by flow cytometry as having double-positive staining for Annexin V and PI. As shown in , HS resulted in a 21.97% and 10.36% increase of necrotic cells and apoptotic cells, respectively, while pretreatment with Nec-1 or GSK’872 markedly reduced HS-induced IEC-6 cell death ().
Figure 3. Nec-1 or GSK’872 pretreatment reduced cell injury and death induced by HS. IEC-6 cells were pretreated with or without 20 μM Nec-1 or 10 μM GSK’ 872 then underwent HS (43 °C) for 2 h and were further incubated at 37 °C for 24 h. (A) Cell viability percentage was measured by WST-1. (B) The folds of cell death were detected by LDH release assays. (C) Induction of HS-induced cell death was analyzed by flow cytometry with Annexin V-FITC/PI staining. (D) Quantification of the apoptotic cell percentages induced by HS analyzed by flow cytometry with Annexin V-FITC/PI staining. (E) Quantification of the necrotic cell percentages induced by HS analyzed by flow cytometry with Annexin V-FITC/PI staining. The data represent the mean ± SD of at least three independent experiments performed in triplicate. *p < 0.05 compared to the control (37 °C) group; #p < 0.05 compared to the HS (43 °C) group.
Nec-1 or GSK’872 pretreatment reduced HS-induced mitochondrial ROS production and promoted MMP recovery
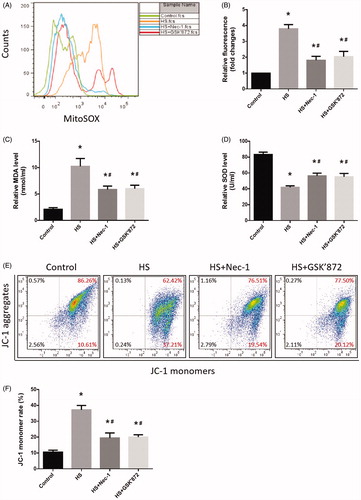
Samples subjected to the HS assay demonstrated a significant increase in levels of mitochondrial ROS in IEC-6 cells (). Notably, pretreatment with Nec-1 or GSK’872 showed a significant inhibitory effect on HS-induced mitochondrial ROS overproduction (). In addition, HS resulted in a significant increase in malondialdehyde (MDA) concentration (), and superoxide dismutase SOD activities were reduced in IEC-6 cells (). Pretreatment with Nec-1 or GSK’872 reversed these phenomena and promoted the balance of oxidative stress response homeostasis in IEC-6 cells (). Next, we determined the amount of mitochondrial depolarization (low MMP) by quantifying the change in JC-1 fluorescence from red to green. We found that the proportion of IEC-6 cells with low MMP increased from 10.61% in untreated cells to 37.21% in HS cells, while pretreatment with Nec-1 or GSK’872 notably decreased the HS-mediated mitochondrial depolarization, promoting IEC-6 cell MMP recovery ().
Figure 4. Nec-1 or GSK’872 pretreatment reduced HS-induced mitochondrial ROS production and promoted MMP recovery. IEC-6 cells were pretreated with or without 20 μM Nec-1 or 10 μM GSK’ 872, then underwent HS (43 °C) for 2 h and were further incubated at 37 °C for 24 h. (A) Cells were treated with 4 µM MitoSOX for mitochondrial superoxide detection. Flow cytometry was used to analyze HS-induced mitochondrial superoxide. (B) Quantification of the mitochondrial superoxide induced by HS. (C) Quantification of the malondialdehyde (MDA) levels induced by HS. (D) Quantification of the superoxide dismutase (SOD) levels induced by HS. (E) Cells were labeled with 5 µmol/L JC-1. Flow cytometry was used to analyze HS-induced mitochondrial depolarization (low ΔΨm). (F) Quantification of the low ΔΨm percentage induced by HS analyzed by flow cytometry. The data shown represent the mean ± SD from at least three independent experiments performed in triplicate. *p < 0.05, compared to the control (37 °C) group; #p < 0.05, compared to the HS (43 °C) group.
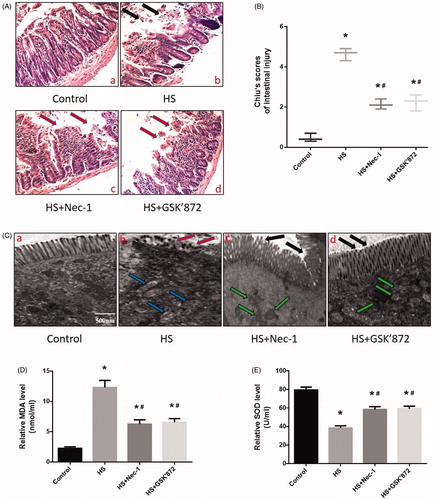
Nec-1 or GSK’872 pretreatment reduced HS-induced intestinal injury in vivo
To further explore the effects of Nec-1 or GSK’872 pretreatment on HS-induced intestinal damage, adult male mice were pretreated with Nec-1 or GSK’872 and histopathologic examination was performed on the ileum. The control group that did not undergo HS showed no marked damage to the small intestinal mucosa (). Conversely, animals exposed to HS demonstrated profound damage to the epithelium of the small intestine, manifested as villous stroma broadening, focal necrosis, and some epithelial cell detachment accompanied by marked edema and congestion (), black arrows). Chiu score was increased in HS animals (). Notably, HS-induced intestinal damage was prevented in mice that were pretreated with Nec-1 or GSK’872 (). In addition, we observed sloughing of the epithelium off the basement membrane at the villus tips in the tissue of the animals subjected to HS, when compared with the control tissue, as obtained from TEM (), red arrows). Again, we observed that pretreatment with Nec-1 or GSK’872 significantly reversed or prevented HS-induced intestinal injuries (, black arrows). Further examination revealed HS animals displayed a greater number of swollen mitochondria with shortened internal cristae compared to non-stressed mice (, blue arrows) and mice that received prior Nec-1 or GSK’872 treatment (, green arrows). Further in vivo experiments revealed that HS resulted in a significant increase in MDA concentration (), and SOD activities were reduced in the small intestine (ileum) (). Pretreatment with Nec-1 or GSK’872 reversed these phenomena and promoted the balance of oxidative stress response homeostasis in vivo ().
Figure 5. Nec-1 or GSK’872 pretreatment reduced HS-induced intestinal injury in vivo. Mice were pretreated with or without 1.8 mg/kg Nec-1 or 1.9 mmol/kg GSK’ 872 (intraperitoneal) at 1 h followed by exposure to a temperature controlled chamber (ambient temperature 35.5 ± 0.5 °C and 60 ± 5% relative humidity) until rectal core temperature (Tc) reached 42 °C. The mice were sacrificed at 20 h after HS exposure and the small intestine (ileum) was isolated. (A) Histopathology changes in the ileum were detected by H&E staining (magnification, ×200). HS resulted in profound damage to the small intestine epithelium, manifested as villous stroma broadening, focal necrosis, and some epithelial cell detachment accompanied by marked edema and congestion (A-b, black arrows). (B) Intestinal injury was evaluated by Chiu scores in mice. (C) The ileum ultrastructure was observed by TEM. Changes in microvillus height (C-b, red arrows), and mitochondria (C-b, blue arrows) were apparent in the HS group ileum. (D) Quantification of the malondialdehyde (MDA) levels induced by HS. E. Quantification of the superoxide dismutase (SOD) levels induced by HS. *p < 0.05, compared to the control (37 °C) group; #p < 0.05, compared to the HS group, (n = 6).
Scavenging ROS production inhibited HS-induced RIPK1/RIPK3-dependent necroptosis activation both in vivo and in vitro
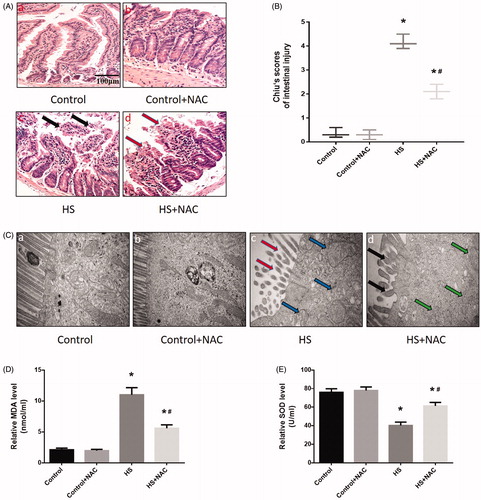
The above results indicated that HS-induced RIPK1/RIPK3-dependent necrosome formation and activation and inhibition of RIPK1 or RIPK3 activity not only reduced RIPK1, RIPK3, and phosphorylated MLKL expression, but also reduced IEC-6 cell death and small intestine injury. Previous studies indicated that oxidative stress might be a crucial factor in HS-induced intestinal injury. To investigate whether ROS was involved in HS-induced necrosome formation and activation, we treated the ROS scavenger NAC in IEC-6 cells and mice before HS. As expected, both IEC-6 cells and mice pretreated with the ROS scavenger NAC showed dramatically reduced levels of RIPK1, RIPK3, and phosphorylated MLKL (, . Further, immunoprecipitation analysis confirmed that NAC reduced RIPK1-RIPK3 complex formation both in vitro and in vivo (, . Moreover, the overall survival rate in the NAC pretreated group was significantly higher than in the HS group (). Furthermore, IEC-6 cells pretreated with NAC were relatively resistant to HS-induced toxic effects and cell death, manifested as NAC promoted HS-induced cell viability recovery, reduced LDH release, and necrotic cells and apoptotic (). In addition, pretreatment with NAC promoted the balance of oxidative stress response homeostasis in IEC-6 cells, including inhibited HS-induced mitochondrial ROS overproduction and MDA concentration, and increased SOD activities (). NAC pretreatment notably decreased HS-mediated mitochondrial depolarization, promoting ICE-6 cell MMP recovery (). Further in vivo experiments revealed that NAC pretreatment significantly reversed or prevented HS-induced intestinal tissue and mitochondrial injuries (). Pretreatment with NAC also inhibited MDA concentration and increased SOD activities and promoted the balance of oxidative stress response homeostasis in vivo ().
Figure 6. Scavenging ROS production relieved HS-induced RIPK1/RIPK3-dependent IEC-6 cells injury and death. IEC-6 cells were pretreated with or without 2 mM NAC, then underwent HS (43 °C) for 2 h and were further incubated at 37 °C for 24 h. (A) RIPK1, RIPK3, phosphorylated MLKL, and MLKL levels were detected by Western blot. (B) RIPK1-RIPK3 complexes were measured by immunoprecipitation assay with IgG serving as the negative control. (C) Cell viability percentages were measured by WST-1. (D) The folds of cell death were detected by LDH release assays. (E) Induction of HS-induced cell death was analyzed by flow cytometry with Annexin V-FITC/PI staining. (F) Quantification of the apoptotic cells percentage induced by HS analyzed by flow cytometry with Annexin V-FITC/PI staining. (G) Quantification of the necrotic cells percentage induced by HS analyzed by flow cytometry with Annexin V-FITC/PI staining. The data shown represent the mean ± SD from at least three independent experiments performed in triplicate. *p < 0.05, compared to the control (37 °C) group; #p < 0.05, compared to the HS (43 °C) group.
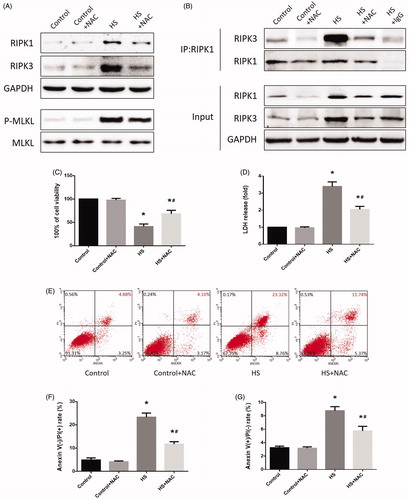
Figure 7. Scavenging ROS production reduced HS-induced RIPK1/RIPK3-dependent mitochondrial ROS production and promoted MMP recovery. IEC-6 cells were pretreated with or without 2 mM NAC, then underwent HS (43°C) for 2h and were further incubated at 37°C for 24h. A. Cells were treated with 4μM MitoSOX for mitochondrial superoxide detection. Flow cytometry was used to analyze HS-induced mitochondrial superoxide. B. Quantification of the mitochondrial superoxide induced by HS. C. Quantification of the malondialdehyde (MDA) levels induced by HS. D. Quantification of the superoxide dismutase (SOD) levels induced by HS. E. Cells were labeled with 5 μmol/L JC-1. Flow cytometry was used to analyze HS-induced mitochondrial depolarization (low ΔΨm). F. Quantification of the low ΔΨm percentage induced by HS analyzed by flow cytometry. The data shown represent the mean ±SD from at least three independent experiments performed in triplicate. *P < 0.05, compared to the control (37°C) group; #P < 0.05, compared to the HS (43°C) group.
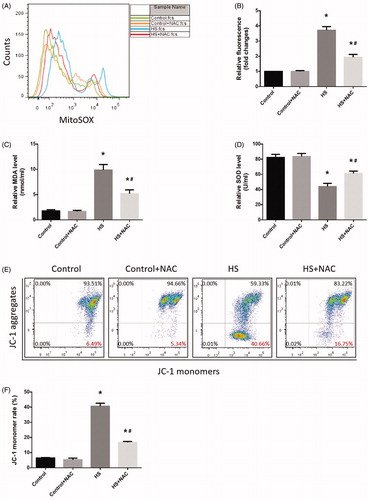
Figure 8. Scavenging ROS production improved HS-induced RIPK1/RIPK3-dependent mice survival. Mice were pretreated with or without 100mg/kg NAC (intraperitoneal) at 1h followed by exposure to a temperature controlled chamber (ambient temperature 35.5±0.5°C and 60±5% relative humidity) until rectal core temperature (Tc) reached 42°C. The mice were sacrificed at 20h after HS exposure and the small intestine (ileum) was isolated. RIPK1, RIPK3, phosphorylated MLKL, and MLKL levels were detected by Western blot (n=8). B. RIPK1-RIPK3 complexes were detected by immunoprecipitation assay with IgG serving as the negative control (n=8). C. Kaplan-Meier results showing mice survival duration in each group (n=8). *P < 0.05, compared to the control (37°C) group; #P < 0.05, compared to the HS group.
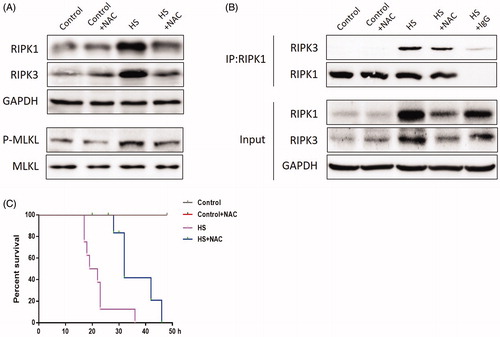
Figure 9. Scavenging ROS production reduced HS-induced intestinal damage in vivo. Mice were pretreated with or without 100 mg/kg NAC (intraperitoneal) at 1 h followed by exposure to a temperature controlled chamber (ambient temperature 35.5 ± 0.5 °C and 60 ± 5% relative humidity) until rectal core temperature (Tc) reached 42 °C. The mice were sacrificed at 20 h after HS exposure and the small intestine (ileum) was isolated. (A) Histopathology changes in the ileum were detected by H&E staining (magnification ×200). HS resulted in profound damage to the small intestine epithelium, manifested as villous stroma broadening, focal necrosis, and some epithelial cell detachment accompanied by marked edema and congestion (A-c, black arrows). (B) Intestinal injury was evaluated by Chiu scores in mice. (C) The ileum ultrastructure was observed by TEM. Changes in microvillus height (C-c, red arrows), and mitochondria (C-c, blue arrows) were apparent in HS group ileum. (D) Quantification of the malondialdehyde (MDA) levels induced by HS. (E) Quantification of the superoxide dismutase (SOD) levels induced by HS. *p < 0.05, compared to the control (37 °C) group; #p < 0.05, compared to the HS group, (n = 6).
Discussion
Heat stroke is a serious condition, with tissue damage and multi-organ dysfunction being the primary symptoms that can lead to death [Citation4,Citation5]. The gastrointestinal tract has long been considered the “motor” of multiple organ failure, with intestinal damage being widely accepted as a key factor in various critical illnesses, including heat stroke [Citation7,Citation29]. Heat stroke can damage the intestinal villi and alter the integrity of the intestinal membrane [Citation8–10]. Following heat stroke there is reduced blood flow to the small intestine to preserve flow to vital organs, such as the brain and heart [Citation30,Citation31]. Consequently, the small intestinal epithelial tissue experiences ischemia under hypoxic conditions, which may lead to permanent tissue damage [Citation30,Citation31]. This process can greatly impair the small intestinal villus epithelial cells, induce excessive epithelial cell death, and increase mortality risk [Citation30,Citation31].
Necroptosis is a newly identified form of programed cell death and displays unique morphological features that are distinct from apoptosis or necrosis [Citation16]. Necroptosis is initiated by necrosome formation, the main component of which is the RIPK1-RIPK3 complex [Citation21,Citation22]. Activated RIPK3 can also phosphorylate the pseudokinase MLKL, causing it to translocate to the plasma membrane and induce cell permeabilization, eventually leading to necroptosis [Citation32]. Necroptosis and necroptosis relevant proteins have also been shown to be involved in the etiology of various nonmalignant disorders characterized by unwarranted cell loss and/or a prominent inflammatory components [Citation17–20]. Despite this, necroptosis in HS-induced intestinal injury has not previously been experimentally evaluated or verified. To our knowledge this study is the first to confirm necroptosis in HS-induced intestinal injury both in vitro and in vivo. With HS treatment, the expression levels of RIPK1, RIPK3, and phosphorylated MLKL were significantly increased as were the levels of RIPK1-RIPK3 necrosome in both IEC-6 cells and the small intestine of mice after HS. However, pretreatment of mice with the chemical inhibitors Nec-1 or GSK’872 significantly inhibited decreased RIPK1, RIPK3, phosphorylated MLKL expression, and RIPK1-RIPK3 necrosome. Thus, these results indicate that necroptosis appears to be a mechanism of HS-induced intestinal injury both in vitro and in vivo.
Extreme heat has long been considered a major extracellular stimulus from which an organism must protect itself [Citation33,Citation34]. Intense HS can have devastating effects on an organism, primarily caused by direct toxicity to cells. Previous studies have found that, within a few hours, temperatures greater than 42 °C, a critical human body temperature point, will result in cell death, from apoptosis to necrosis; Furthermore, temperatures greater than 45 °C can promote cell death from necrosis within a few minutes [Citation33–35], indicating that the effect of HS on cells depends on both temperature and duration. In the present study, after exposure of IEC-6 cells to HS at 37 °C (the control group), 39 °C, 41 °C, 43 °C, or 45 °C for 2 h, followed by further incubation at 37 °C for 24 h, cell viability drastically declined after IEC-6 cells were cultured at elevated temperatures. Further, after IEC-6 cells were exposed to HS at 43˚C for 2 h, followed by further incubation at 37 °C at different times increments (1 h, 6 h, 12 h, 24 h, and 48 h), cell viability was shown to decline in a time-dependent manner.
Additionally, we found that under HS conditions in which IEC-6 cells were incubated at 43 °C for 2 h and then further incubated at 37 °C for 24 h, there was a 21.97% increase in necrotic cells and a 10.36% increase in apoptotic cells in IEC-6 samples. Importantly, pretreatment of IEC-6 cells with the chemical inhibitors Nec-1 or GSK’872, which interfere with RIPK1 and RIPK3 activity, respectively, markedly reduced the HS-induced IEC-6 cell death rate. While previous studies have shown that HS induces a large number of apoptotic cells in IEC-6 cells [Citation30,Citation31,Citation36], in this study, we focused on what happens in IEC-6 cells during the recovery time after HS. We found that without timely intervention after HS, necrosis would occur. This may explain why patients who suffer from heat stroke also experience systemic inflammatory response syndrome and multi-organ dysfunction, even when removed from extreme heat conditions. In addition, pretreatment of mice with the chemical inhibitors Nec-1 or GSK’872 significantly reversed or prevented HS-induced intestinal tissue and mitochondrial injuries. Therefore, it is plausible that RIPK1/RIPK3-dependent necroptosis mediates HS-induced intestinal tissue injury and cell death.
Recently, there have been reports illustrating that oxidative stress is related to intestinal epithelial barrier dysfunction induced by HS [Citation11,Citation12,Citation37] and that antioxidants have the ability to counteract HS effects such as increased intestinal permeability and intestinal epithelial damage [Citation37,Citation38]. In the current study, we found that HS caused mitochondrial depolarization that led to the accumulation of large amounts of ROS in the mitochondria of IEC-6 cells. In vivo experiments demonstrated that HS induces morphological injuries in the small intestine of mice, including villous stroma broadening, focal necrosis, swollen mitochondria, epithelial cell edema, and congestion. We also found that HS resulted in dyshomeostasis from an oxidative stress response in the small intestine. Remarkably, both animals and cells that were pretreated with the well-established ROS scavenger NAC showed a dramatic reversal of HS-induced oxidative stress injury. NAC pretreatment significantly inhibited HS-induced necrosome formation both in vitro and in vivo, including decreased expression levels of RIPK1, RIPK3, and phosphorylated MLKL, as well as RIPK1-RIPK3 necrosome levels. Although we did not explore the intermediate mechanisms of HS-induced intestinal necroptosis, these results suggest that prevention of HS-induced necroptosis of the small intestine may be accomplished via scavenging ROS production. In addition, through scavenging ROS production the IEC-6 cells were relatively resistant to HS-induced toxic effects, cell death, and mitochondrial depolarization. In vivo experiments also revealed that pretreatment with NAC could significantly reverse or prevent HS-induced intestinal tissue and mitochondria injuries. These findings indicated that oxidative stress is the main cause of RIPK1/RIPK3-dependent necroptosis mediated HS-induced intestinal injury, and via scavenging ROS production we can alleviate such intestinal damage both in vivo and in vitro.
Conclusion
This study provides strong evidence that HS causes damage to both the small intestine and intestinal epithelial cells. This intestinal injury is mediated through dyshomeostasis from an oxidative stress response, mitochondrial damage, and RIPK1-RIPK3-dependent necrosome formation. Scavenging ROS production can significantly alleviate such RIPK1/RIPK3-dependent necroptosis, mediating HS-induced intestinal damage both in vitro and in vivo. These findings provide a clear target for future mechanism-based therapeutic strategies for patients diagnosed with heat stroke.
Author contributions
Li Li and Hongping Tan were responsible for primary data generation and composed this manuscript. Zhimin Zou, Junjie Zhou, Jian Gong, and Na Peng performed all experiments. Zhengtao Gu and Marc Maegele acquired and interpreted the data. Daozhang Cai and Lei Su conceived and designed the research and interpreted the data. All authors read and approved the final manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Reference
- Wang Y, Bobb JF, Papi B, et al. Heat stroke admissions during heat waves in 1,916 US counties for the period from 1999 to 2010 and their effect modifiers. Environ Health. 2016;15(1):83.
- Pryor RR, Roth RN, Suyama J, Hostler D. Exertional heat illness: emerging concepts and advances in prehospital care. Prehosp Disaster Med. 2015;30(3):297–305.
- Gu S, Huang C, Bai L, et al. Heat-related illness in China, summer of 2013. Int J Biometeorol. 2016;60(1):131–137.
- Leon LR, Bouchama A. Heat stroke. Compr Physiol. 2015;5(2):611–647.
- Leon LR, Helwig BG. Heat stroke: role of the systemic inflammatory response. J Appl Physiol. 2010;109(6):1980–1988.
- Yi G, Li L, Luo M, et al. Heat stress induces intestinal injury through lysosome- and mitochondria-dependent pathway in vivo and in vitro. Oncotarget. 2017;8(25):40741–40755.
- Liu Z, Sun X, Tang J, et al. Intestinal inflammation and tissue injury in response to heat stress and cooling treatment in mice. Mol Med Rep. 2011;4(3):437–443.
- Xiao G, Tang L, Yuan F, et al. Eicosapentaenoic acid enhances heat stress-impaired intestinal epithelial barrier function in Caco-2 cells. PLoS One. 2013;8(9):e73571.
- Xu QL, Guo XH, Liu JX, et al. Blockage of protease-activated receptor 1 ameliorates heat-stress induced intestinal high permeability and bacterial translocation. Cell Biol Int. 2015;39(4):411–417.
- Xiao G, Yuan F, Geng Y, et al. Eicosapentaenoic acid enhances heatstroke-impaired intestinal epithelial barrier function in rats. Shock. 2015;44(4):348–356.
- Pearce SC, Mani V, Boddicker RL, et al. Heat stress reduces intestinal barrier integrity and favors intestinal glucose transport in growing pigs. PLoS One. 2013;8(8):e70215.
- Liu F, Cottrell JJ, Furness JB, et al. Selenium and vitamin E together improve intestinal epithelial barrier function and alleviate oxidative stress in heat-stressed pigs. Exp Physiol. 2016;101(7):801–810.
- Cui Y, Gu X. Proteomic changes of the porcine small intestine in response to chronic heat stress. J Mol Endocrinol. 2015;55(3):277–293.
- Lambert GP. Stress-induced gastrointestinal barrier dysfunction and its inflammatory effects. J Anim Sci. 2009;87(suppl_14):E101–108.
- Zuhl M, Schneider S, Lanphere K, et al. Exercise regulation of intestinal tight junction proteins. Br J Sports Med. 2014;48(12):980–986.
- Vandenabeele P, Galluzzi L, Vanden Berghe T, et al. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11(10):700–714.
- Karunakaran D, Geoffrion M, Wei L, et al. Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci Adv. 2016;2(7):e1600224.
- Galluzzi L, Kepp O, Chan FK, et al. Necroptosis: mechanisms and relevance to disease. Annu Rev Pathol Mech Dis. 2017;12(1):103–130.
- Rogers C, Fernandes-Alnemri T, Mayes L, et al. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017;8(1):14128.
- Qing DY, Conegliano D, Shashaty MG, Seo J, et al. Red blood cells induce necroptosis of lung endothelial cells and increase susceptibility to lung inflammation. Am J Respir Crit Care Med. 2014;190(11):1243–1254.
- Cho YS, Challa S, Moquin D, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137(6):1112–1123.
- Tian F, Yao J, Yan M, et al. 5-Aminolevulinic acid-mediated sonodynamic therapy inhibits RIPK1/RIPK3-dependent necroptosis in THP-1-derived foam cells. Sci Rep. 2016;6(1):21992.
- Li L, Su Z, Zou Z, et al. Ser46 phosphorylation of p53 is an essential event in prolyl-isomerase Pin1-mediated p53-independent apoptosis in response to heat stress. Cell Death Dis. 2019;10(2):96.
- Chen S, Lv X, Hu B, et al. RIPK1/RIPK3/MLKL-mediated necroptosis contributes to compression-induced rat nucleus pulposus cells death. Apoptosis: An International Journal on Programmed Cell Death. 2017;22(5):626–638.
- Yang XS, Yi TL, Zhang S, et al. Hypoxia-inducible factor-1 alpha is involved in RIP-induced necroptosis caused by in vitro and in vivo ischemic brain injury. Sci Rep. 2017;7(1):5818.
- Jiang L, Yan Q, Liu RH, et al. Preventive and therapeutic effect of N-Acetyl-l-cysteine on infection-associated preterm labor in mice. Asian Pac J Trop Med. 2016;9(2):197–200.
- Zhou Y, Dai W, Lin C, et al. Protective effects of necrostatin-1 against concanavalin A-induced acute hepatic injury in mice. Mediators Inflammation. 2013;2013:1–15.
- Chiu CJ, McArdle AH, Brown R, et al. Intestinal mucosal lesion in low-flow states. I. A morphological, hemodynamic, and metabolic reappraisal. Arch Surg. 1970;101(4):478–483.
- Bouchama A, Roberts G, Al Mohanna F, et al. Inflammatory, hemostatic, and clinical changes in a baboon experimental model for heatstroke. J Appl Physiol. 2005;98(2):697–705.
- Gao Z, Liu F, Yin P, et al. Inhibition of heat-induced apoptosis in rat small intestine and IEC-6 cells through the AKT signaling pathway. BMC Vet Res. 2013;9(1):241.
- Mei C, He SS, Yin P, et al. Magnolol pretreatment attenuates heat stress-induced IEC-6 cell injury. J Zhejiang Univ Sci B. 2016;17(6):413–424.
- Jouan-Lanhouet S, Riquet F, Duprez L, et al. Necroptosis, in vivo detection in experimental disease models. Semin Cell Dev Biol . 2014;35:2–13.
- Gu ZT, Wang H, Li L, et al. Heat stress induces apoptosis through transcription-independent p53-mediated mitochondrial pathways in human umbilical vein endothelial cell. Sci Rep. 2015;4(1):4469.
- Li L, Tan H, Yang H, et al. Reactive oxygen species mediate heat stress-induced apoptosis via ERK dephosphorylation and Bcl-2 ubiquitination in human umbilical vein endothelial cells. Oncotarget. 2017;8(8):12902–12916.
- Buckley IK. A light and electron microscopic study of thermally injured cultured cells. Lab. Invest. 1972;26(2):201–209.
- Zhang Y, Zhao H, Liu T, et al. Activation of transcription factor AP-1 in response to thermal injury in rat small intestine and IEC-6 cells. BMC Gastroenterol. 2015;15(1):83.
- He S, Guo Y, Zhao J, et al. Ferulic acid protects against heat stress-induced intestinal epithelial barrier dysfunction in IEC-6 cells via the PI3K/Akt-mediated Nrf2/HO-1 signaling pathway. Int J Hyperther. 2018;35(1):112–121.
- Varasteh S, Fink-Gremmels J, Garssen J, et al. alpha-Lipoic acid prevents the intestinal epithelial monolayer damage under heat stress conditions: model experiments in Caco-2 cells. Eur J Nutr. 2018;57(4):1577–1589.