
Abstract
Objective: Heat stroke (HS) elicits the systemic inflammatory responses that result in multiple organ dysfunction (MOD). Heat shock response and autophagy are activated during heat stress for removal of damaged organelles and proteins, emerging as a major regulator of cellular homeostasis. Ethyl pyruvate (EP) is a derivative of pyruvic acid and possesses antioxidant and anti-inflammatory effects. This study aims to investigate the effects of EP on MOD in HS rats and explore the possible mechanisms.
Method: Anesthetized rats were placed in a heating chamber (42 °C) to elevate the core body temperature attaining to 42.9 °C. Rats were then moved to room temperature and monitored for 6 h. EP (60 mg/kg, i.v.) was administered 30 min prior to heat exposure.
Results: Results showed that EP significantly reduced HS-induced increases in plasma levels of LDH, CPK, GPT and CK-MB, reversed the decrease of platelet counts, and alleviated intestinal mucosal and pulmonary damage. Moreover, EP reduced pro-inflammatory protein, including TNF-α, IL-6, IL-1β, HMGB1 and iNOS, and induced stress proteins, heme oxygenase-1 (HO-1), heat shock protein (HSP) 70 and HSP90 in the liver of HS rats. The levels of HS-activated autophagy-regulatory proteins were affected by EP, in which the phosphorylated mTOR and AKT were reduced, and the phosphorylated AMPK increased, accompanied with upregulation in ULK1, Atg7, Atg12 and LC3II, and downregulation of p62.
Conclusion: In conclusion, EP ameliorated HS-induced inflammatory responses and MOD, and the underlying mechanism is associated with the induction of the stress proteins HO-1 and HSP70 as well as restorage of autophagy.
Introduction
Climate change and the rise in global temperature increase the frequency of extreme heat events, or heat waves, leading to the elevated risk of heat-related illnesses and deaths. Heat-related illnesses include clinical conditions ranging from heat cramps and syncope to heat exhaustion and heat stroke (HS) [Citation1]. Heat stress initiates thermoregulatory responses, including sweating, increased cardiac output and redistribution of blood flow to reduce body temperature [Citation2]. When the ability to physiologically adjust to heat stress has become compromised, cardiac output is insufficient to deal with the high thermoregulatory demand, leading to rapid elevation of core body temperature, which consequently causes direct cell damage, a systemic inflammatory response, and multiple organ dysfunction (MOD) [Citation3].
Heat stress causes drastic disturbances of cellular homeostasis, critically shifting the conformational equilibrium of a subset of proteins to the unfolded states. Under these stress conditions, stress proteins, e.g., heat shock protein 90 (HSP90) and HSP70 are transcriptionally upregulated to participate in the restoration of proteostasis [Citation4]. HSPs prevent protein aggregation by shielding the hydrophobic surfaces exposed by partially unfolded proteins and facilitate refolding [Citation5]. Additionally, for clearance of the misfolded proteins, these chaperones cooperate with various proteolytic systems, primarily autophagy and ubiquitin–proteasome system [Citation6]. Autophagy is a highly conserved catabolic process in all eukaryotic organisms and transports cytoplasmic target materials to lysosomes for degradation. It is induced by various conditions of cellular stress to exert cytoprotective functions for maintenance of cellular homeostasis, resulting in the prevention of cell damage and promotion of cell survival in different forms of stress, including nutrient deprivation, growth factor depletion, infection and hypoxia [Citation7]. Autophagy is deregulated in various human pathogenesis, including aging, cancer, neurodegenerative disorders, infectious disease and immunity [Citation8]. It has been shown to confer a protective effect in heat stress [Citation9]. Our previous study has indicated that impaired autophagy function is involved in the process of HS-induced myocardial injuries, and restoration of autophagy prevents the deleterious effects [Citation10]. Another stress protein heme oxygenase-1 (HO-1) is the inducible HO isozyme and transcriptionally upregulated in response to heat stress. HO-1 induction is a widely accepted strategy used by cells to neutralize a variety of stress conditions via its antioxidant and anti-inflammatory effects [Citation11]. Accumulating evidence has prompted that regulation of autophagy pathway is the novel anti-inflammatory target of HO-1induction [Citation12].
Ethyl pyruvate (EP), a derivative of pyruvic acid, has been shown to alleviate MOD and increase survival rate in many animal disease models, including sepsis, hemorrhagic shock and ischemia–reperfusion injury via exerting as a reactive oxygen species (ROS) scavenger and anti-inflammatory capacity [Citation13,Citation14]. Anti-inflammatory effects of EP are evidenced by the inhibition of NF-κB activation, resulting in the reduction of cytokine release, e.g., TNF-α, IL-1, IL-6 and high-mobility group box (HMGB) 1, as well as iNOS expression [Citation15]. We have previously shown that EP possessed anticoagulant effects in lipopolysaccharide (LPS)-induced disseminated intravascular coagulation (DIC) rat model and improvement of organ functions by reduction of IL-6 release and suppression of tissue factor expression. The protective effect of EP is associated with the induction of HO-1 expression [Citation16]. Furthermore, EP attenuated liver injuries in several experimental animal models, including acute pancreatitis [Citation17], alcohol, obstructive jaundice [Citation18], ischemia–reperfusion [Citation19], high-fat diet [Citation20], as well as reduced acute fatal liver injury attributed from concanavalin A, d-galactosamine and acetaminophen [Citation15]. However, the beneficial effect of EP on HS has not been investigated yet. In the present study, we explored the protective effect of EP on the MOD caused by HS in rats, and further investigated the possible mechanisms, including anti-inflammatory response, stress protein induction and activation of autophagy.
Materials and methods
Experimental animals and groups
Male Wistar rats (8 weeks old, 290–320 g) were obtained from BioLASCO Co., Ltd. (Taipei, Taiwan). This study was approved by the Institutional Animal Care and Use Committee of the National Defense Medical Center, Taiwan (number of permission: IACUC-15-207). All procedures for animal handling were in accordance with the Guideline for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication no. 85-23, revised in 1996).
Under anesthesia (urethane 1.2 g/kg, i.p.), the left femoral artery of rats was cannulated with a polyethylene-50 (PE-50) catheter and connected to a pressure transducer (MLT844, ADInstruments Pty Ltd, Castle Hill, Australia) for the measurement of mean arterial blood pressure (MAP) and heart rate (HR) displayed on a polygraph recorder (ML 785, PowerLab, ADInstruments Pty Ltd, Castle Hill, Australia). The left femoral vein was cannulated for the administration of normal saline and drugs. After the surgical procedure was complete, rats were allowed to stabilize all vital signs for 30 min. The MAP, HR and core temperature (Tco) were continuously monitored throughout the experimental period in all groups.
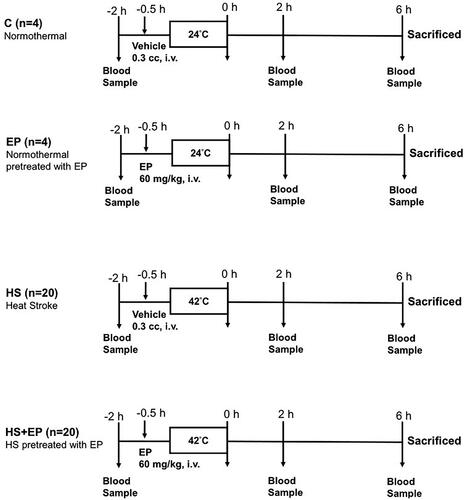
The rats were divided into four groups as indicated in : (1) normothermal control group (C): 30 min after receiving the vehicle of EP, 0.3 ml of Ringer’s solution (i.v. bolus), rats were arranged in a heating chamber at room temperature (24 °C) (N = 4); (2) EP alone group (EP): 30 min after intravenously given EP 60 mg/kg (Sigma-Aldrich, St. Louis, MO), rats were arranged in a heating chamber at room temperature (24 °C) for 45 min (N = 4); (3) HS group: 30 min after receiving the vehicle of EP, 0.3 ml of Ringer’s solution (i.v.), rats were placed in a heating chamber (42 °C) until the rectal Tco elevated to 42.9 °C to induce HS (N = 16); (4) HS + EP group: HS rats were pretreated with EP 60 mg/kg (i.v.) at 30 min prior to heat stress (N = 16). The average time of the rats stayed in the heating chamber top reach the peak temperature was 45 min. After removing out the heating chamber, rats were placed at room temperature (24 °C) throughout the experimental period. The survival rate was monitored for 6 h and rats were then sacrificed by the injection of pentobarbital (50 mg/kg, i.v.). Blood and organs, livers, lungs and ileums were collected immediately at 6 h for further analysis.
Figure 1. Experimental protocol. C: control group; Ringer’s solution 0.3 cm3, the vehicle of ethyl pyruvate, was administered intravenously, and no heat stress was given (normothermal); EP group: ethyl pyruvate (60 mg/kg, i.v.) was administered, and no heat stress was given (normothermal); HS group: Ringer’s solution 0.3 cm3 was administered 30 min before heat stress; HS + EP group: ethyl pyruvate (60 mg/kg, i.v.) was administered 30 min before heat stress. After HS induction, rats were placed at room temperature (24 °C) and monitored for 6 h.
Biochemical assays and platelet counts
Plasma levels of lactate dehydrogenase (LDH) were determined as the cell toxicity indicator. Hepatic function indicator glutamic pyruvic transaminase (GPT), renal function indicator creatinine (CRE) and rhabdomyolysis marker creatine phosphokinase (CPK) were measured at 1 h before, and 0, 2 and 6 h after HS. Myocardia injury indicator creatine kinase-MB (CK-MB) was measured at 6 h after heat stress, using a Fuji DRI-CHEM 3030 analyzer (Fuji Photo Film, Tokyo, Japan). Platelet counts were performed in a complete blood count (CBC) measurement with Sysmex XN-450 automatic hematology analyzer (Sysmex, Kobe, Japan).
Western blot analysis
The livers and lungs of rats were isolated 6 h after HS challenge and immediately frozen in liquid nitrogen and stored at −80 °C until processed. Total proteins of the livers were isolated by 1× RIPA lysis buffer. Nuclear extracts from tissues were obtained using a nuclear extraction kit (Merck Millipore, Burlington, MA). The protein concentration of supernatant of cell lysate and the nuclear extract was measured by the BCA kit (Thermo Fisher, Rockford, IL). Samples containing equal amounts of protein were separated on 10% sodium dodecyl sulfate-polyacrylamide gels and transferred to a nitrocellulose membrane. After blocking in 5% bovine serum albumin for 1 h, blots were then incubated overnight at 4 °C with the following primary antibodies: anti-TNF-α (GeneTex, Irvine, CA), anti-IL-6 (Novus Biologicals, Centennial, CO), anti-IL-1β (Abcam, Cambridge, MA), anti-HMGB1 (Cell Signaling, Beverly, MA), anti-iNOS (BD Biosciences, Heidelberg, Germany), anti-NF-κB phosphorylated (p)-p65 (GeneTex, Irvine, CA), anti-p65 (Abcam, Cambridge, MA), anti-p-IκB, anti-LC3B, anti-Atg7, anti-Atg5–12, anti-p62/SQSTM1 (p62), anti-phosphorylated mammalian target of rapamycin (p-mTOR), anti-mTOR, anti-AMP-activated protein kinase (AMPK), anti-p-AMPK, anti-p-AKT, anti-AKT, anti-ULK1 (Cell Signaling, Danvers, MA), anti-HSP70, HSP90, HO-1 (Enzo Life Sciences, Farmingdale, NY), anti-β-actin (GeneTex, Irvine, CA), nuclear HSF1 (Cell Signaling, Danvers, MA) and lamin B1 (Abcam, Cambridge, MA) for 16 h. The membranes were washed and then probed with horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG (Cell Signaling, Danvers, MA). Proteins were visualized using enhanced chemiluminescence reagents (Bio-Rad, Hercules, CA) and quantified by densitometry of the blots using ImageJ software (Bethesda, MD).
Histopathological analysis
The lung and ileum were fixed in 10% neutral-buffered formalin, and embedded in paraffin blocks, and sliced into transverse sections. Serial sections were dyed with hematoxylin and eosin stains for microscopic examination (×200 magnification). Morphological changes, including the extent and range of mucosal epithelial cell degeneration and necrosis, villus height, and structural damage were assessed and graded by certificated pathomorphological changes were assessed and graded by certified pathologists using the intestinal injury score.
Statistical analysis
The data are expressed as mean ± SEM. Statistical evaluation was performed with one-way ANOVA followed by the Newman–Keuls test. A two-way ANOVA and LSD post hoc comparison test was used to compare the means of arterial blood pressure (MAP) or Tco between the groups and time. Chi-square test followed by Fisher’s exact test was used for comparison of the survival distributions between groups of rats. The Bonferroni test was used to correct multiple comparison of survival distribution. Kaplan–Meier’s method was used to calculate the survival rate, and the log-rank test was used to test differences between groups. p<.05 was accepted as indicating statistical significance.
Results
Effects of EP on MAP and Tco in rats with HS
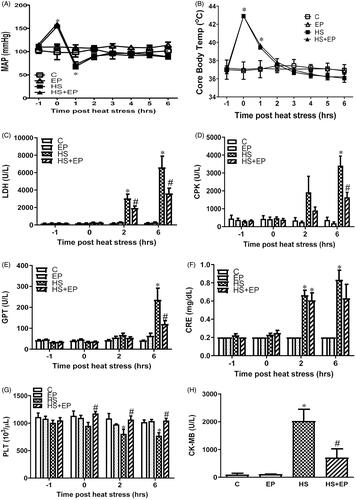
Upon removing out of the heating chamber, the MAP of rats in both HS and HS + EP groups was dramatically increased 1.58-fold (up to 154.1 ± 1.4 mmHg) and 1.63-fold (up to 158.9 ± 2.4 mmHg), respectively, which were significantly higher than that of C group (97.3 ± 10.9 mmHg) (p<.05). One hour later, the rats in both HS-treated groups appeared hypotension, followed by the gradual recovery of MAP. As shown in , there were no significant differences of MAP among groups at 6 h. shows that the Tco of rats exposed to HS was highest at 0 h and then gradually lowered to the normal range of body temperature. Six hours after HS, the Tco in both HS-treated groups was not significantly different among groups; meanwhile, they were not significantly lower than that of C group (p>.05).
Figure 2. Effects of pretreatment with ethyl pyruvate (EP) on (A) mean arterial blood pressure (MAP), (B) core body temperature, (C) plasma levels of lactate dehydrogenase (LDH), (D) creatine phosphokinase (CPK), (E) glutamic pyruvate transaminase (GPT), (F) creatinine (CRE), (G) platelet counts (PLT) and (H) creatine kinase-MB (CK-MB) in HS rats. Data are expressed as mean ± SEM. *p< .05 versus C group; #p< .05 versus HS group.
Effects of EP on HS-induced organs damage in rats
The baseline levels of LDH, CPK, GPT and CRE in the plasma were not significantly different among groups (). After HS challenge, these biomarkers were significantly increased as compared to C group at 6 h in HS group (p < .05) (HS group: LDH: 31.0-fold, CPK: 9.0-fold, GPT:5.8-fold and CRE: 3.1-fold increases of C group). Pretreatment with EP significantly reduced plasma levels of LDH, CPK and GPT when compared with HS group at 6 h (p < .05) (HS + EP group: LDH: 45.2%, CPK: 51.7% and GPT: 49.6% decreases of HS group). In addition, platelet counts were not significantly different among groups before HS exposure. However, EP pretreatment also prevented the HS-induced decreases of platelet counts at 0, 2 and 6 h (p < .05) (HS group: 35% decrease of C group; EP + HS group: 31% increases of HS group) ().
In addition, we measured plasma levels of CK-MB, the principal indicator in the early stage of myocardia injury. The result showed that plasma levels of CK-MB were significantly higher in HS group than those of C group (p<.05) (), an effect that was drastically reversed by EP pretreatment (p<.05).
Effects of EP on inflammation-related proteins expression in the livers of HS rats
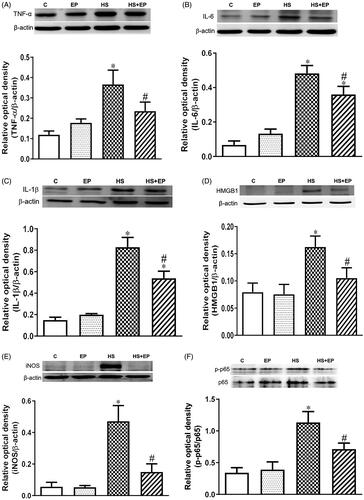
shows that the HS challenge caused increases in protein expression of TNF-α, IL-6, IL-1β, HMGB1 and iNOS in the livers at 6 h, which were significantly reduced in EP + HS group (p<.05). In addition, activation of NF-κB plays a key role in mediating the inflammatory responses [Citation21], and its activity was determined by the phosphorylation of p65 (p-p65) subunit. As shown in , NF-κB p-p65/p65 ratio was significantly increased in the HS group, which was significantly attenuated by EP pretreatment (p<.05).
Figure 3. Effects of pretreatment with ethyl pyruvate (EP) on pro-inflammatory proteins (A) TNF-α, (B) IL-6, (C) IL-1β, (D) HMGB1, (E) iNOS and (F) phosphorylated NF-κB p65 (p-p65) in the livers of HS rats. Data are expressed as mean ± SEM (n = 4-11). *p< .05 versus C group; #p< .05 versus HS group.
Effects of EP on stress protein expression in livers of HS rats
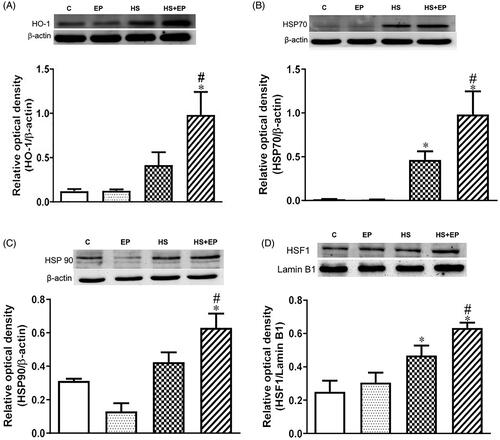
shows that protein expression of HO-1, HSP70 and HSP90 in the livers was slightly induced in the livers of HS group. Intriguingly, EP treatment significantly enhanced the HS-induced HO-1, HSP70 and 90 protein expression. Furthermore, the nuclear heat shock factor (HSF)-1 protein, the main transcription factor for HSP70 and HSP90 production, was drastically enhanced by EP () (p<.05).
Figure 4. Effects of pretreatment with ethyl pyruvate (EP) on stress proteins, including (A) HO-1, (B) HSP70 and (C) HSP90, and nuclear HSF1 protein levels in the livers of HS rats. Data are expressed as mean ± SEM (n = 4–11). *p< .05 versus C group; #p< .05 versus HS group.
Effects of EP on the expression of autophagy marker proteins in livers of HS rats
The protein levels of Atg7, Atg5–12, LC3B, p62/SQSTM1(p62) and p-mTOR have been recognized as autophagy-associated markers [Citation22], and were detected to explore the relationship between the protective effect of EP and autophagy. Six hours after HS, the levels of Atg7, and Atg5–12 complex, and the ratio of LC3B-II/LC3B-I levels were significantly increased in EP + HS group as compared with the HS group (). In addition, it has been reported that adaptor protein p62 is responsible for carrying the target substrate into the autophagosome for degradation, and its amount correlates with autophagic degradation in an inverse manner [Citation23]. As shown in , protein expression of p62 in HS group was significantly higher than that of HS + EP group, indicating that EP may prevent HS-induced dysfunction of autophagosome degradation.
Figure 5. Effects of pretreatment with ethyl pyruvate (EP) on autophagy-related proteins (A) Atg7, (B) Atg5–12, (C) LC3B, (D) p62, (E) mTOR, (F) AMPK, (G) AKT and (H) ULK1 protein expression in the livers of HS rats. Data are expressed as mean ± SEM (n = 4–11). *p< .05 versus C group; #p< .05 versus HS group.
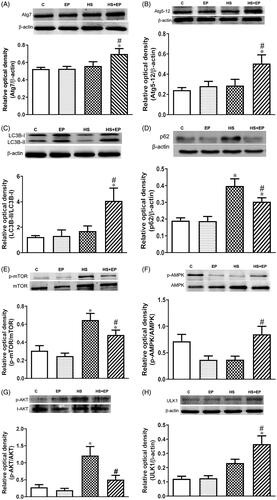
The activation of mTOR is involved in the suppression of autophagy [Citation24]. The result in showed that HS markedly increased the phosphorylation of mTOR (p-mTOR; at the serine 2448 site) and pretreatment with EP significantly attenuated the increase of p-mTOR expression at 6 h. In addition, AMPK and AKT have been reported to control autophagy. Autophagy is induced by AMPK and inhibited by AKT [Citation25]. Phosphorylated AMPK levels in the livers of rats were decreased after HS. Pretreatment of EP with HS rats significantly increased the ratio of hepatic p-AMPK/AMPK levels, and decreased p-AKT/t-AKT ratio, when compared with HS group (). AMPK-mediated induction of autophagy acts by directly inducing phosphorylation of Unc-51-like kinase 1 (ULK1) [Citation25]. In consistent with previous observation, we found that pretreatment of EP in HS rats (EP + HS group) significantly increased the levels of ULK1 when compared with HS group ().
EP attenuated heat stroke-induced intestinal and lung injuries
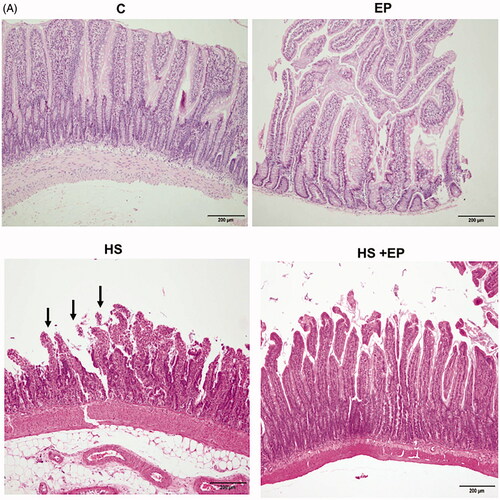
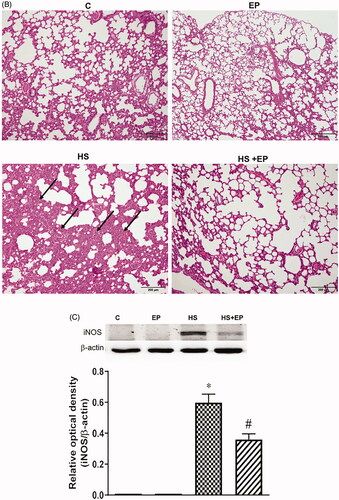
The histological changes of intestine and lung were obtained by H&E stain. As shown in , no obvious damage was observed in the mucosa of ileum of C and EP groups. In HS group, villous tip mucosal disintegration of the epithelium and necrosis with inflammatory cells infiltration were observed (arrow), indicating the intestinal mucosa was damaged by heat stress. This effect was attenuated by EP pretreatment. There was no significant damage of lung tissue in C and EP groups (). The HS group displayed marked alveolar structure damage with patchy inflammatory infiltrate (arrow) as well as septal widening. Pretreatment with EP improved the deleterious effects caused by HS. Moreover, the iNOS protein expression in the lung of HS rats was significantly induced and EP pretreatment markedly reversed the increase of iNOS protein ().
Figure 6. Effects of pretreatment with ethyl pyruvate (EP) on pathological changes of the (A) ileum and (B) lung and (C) protein expression of iNOS in the lung of HS rats. Sections were stained with hematoxylin and eosin, each ×400 (original magnification). Scale bar = 200 μm.
Effects of EP on survival rate in rats with heat stress
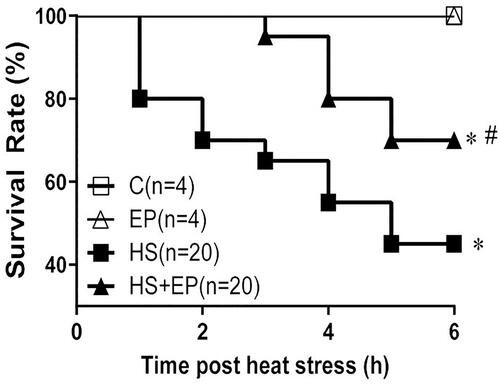
Six hours after HS, the survival rate in HS group was 45% (9/20), which was significantly lower than that of C group (100%, 4/4 animals) (p<.05). The survival rate of HS + EP group was 70% (14/20 animals), which was significantly higher than that of HS group (p> .05) ().
Figure 7. Effects of pretreatment with ethyl pyruvate (EP) on survival rate of HS rats. The survival rate was calculated per hour and observed during 6 h after HS induction. *p< .05 versus C group, #p < .05 versus HS group.
Discussion
In the present study, we demonstrated the protective effect of EP on MOD caused by heat stress in vivo. EP alleviated coagulopathy and the injuries of livers, hearts, lungs, skeletal muscles and intestinal epithelium caused by heat stress. The anti-inflammatory effects of EP were evidenced by attenuation of protein expression of cytokines (TNF-α, IL-6, IL-1β and HMGB1), iNOS and NF-κB p-p65, which may contribute to the protective effects. The underlying mechanism of action is associated with the induction of HSP70, HSP90 and HO-1, as well as the preservation of autophagy, thus contributing to the improvement of survival rates in HS rats ().
Figure 8. Schematic depiction of the protective mechanisms of EP in heat stroke. EP augments stress proteins HSP70, HSP90 and HO-1 expression and activates autophagy to reduce heat stroke-induced inflammatory responses, contributing to the improvement of multiple organ dysfunction.
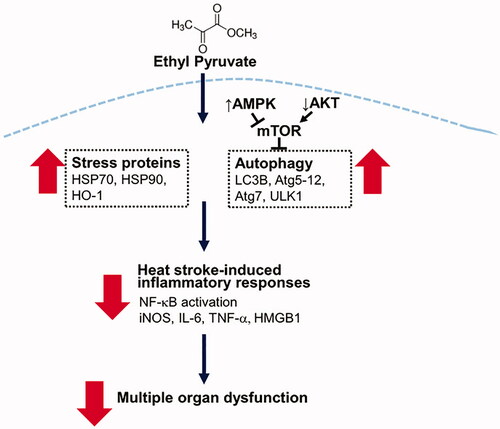
EP has been shown to alleviate outcomes from MOD in experimental animal models of inflammation, such as sepsis, acute pancreatitis, acute respiratory distress syndrome (ARDS), alcoholic liver injury, acute viral myocarditis and acute kidney injury [Citation15,Citation17]. Here, we further demonstrated the beneficial effect of EP on heat stress-induced MOD. Current hypothesis proposes that heat stress evokes the systemic inflammatory response and thermoregulatory response is initiated to increase cardiac output, and skin blood flow, and reduce visceral blood flow for compensation of the core body temperature elevation [Citation3]. This compensatory response results in hypoxia and oxidative stress of the gut and increase of gut epithelial permeability, leading to the leakage of endotoxin from the intestine into circulation and eventually elicits systemic inflammatory responses and MOD. Here, we showed that EP prevented MOD caused by HS, which is closely associated with the anti-inflammatory effect.
We showed that intracellular expression of HSP70 was induced by EP pretreatment in HS rats, which was associated with its anti-inflammatory response. HSPs are synthesized by cells in response to heat stress. Activation of heat shock transcription factor 1 (HSF1) is central to the transcriptional induction of the stress response in cells. Under normal physiological conditions, HSF1 mostly bound to HSP90 in the cytoplasm. During heat stress, it could be displaced from HSP90 by stress-denatured proteins and phosphorylated to shift the monomeric HSF1 to an active trimeric form. This trimeric form of HSF1 enters the nucleus and binds at the heat shock element (HSE) to promote the transactivation function of HSF1, resulting in the translation of HSPs, including HSP90 and HSP70 [Citation6]. HSP70 detects the denatured or incorrectly folded proteins and forms a complex with these proteins, leading to correct folding, compartmentalization in organelles, or to proteolytic degradation. The cytoprotective effects of HSPs are partially mediated via their ability to stabilize intracellular protein structures. It has been shown that HSP70 plays a critical role in mediating the anti-inflammatory effects on macrophage cytokine expression. Intracellular HSP70 can interact with IKK to prevent IκB phosphorylation, resulting in the suppression of NF-κB activation [Citation26]. In addition, HSP70 inhibits LPS-induced NF-κB activation by binding with TRAF6, the essential adaptor protein downstream of TLR4 and reduce the downstream inflammatory cytokine production in macrophages [Citation27]. Furthermore, in animal models of severe sepsis and HS, preemptive induction of HSP70 improves MOD [Citation9,Citation28–30]. Induction of HSP70 via activating HSF-1 interferes the binding of NF-κB to the TNF-α transcription element, resulting in inhibition of the TNF-α transcription, which results in the amelioration of hepatic dysfunction caused by LPS [Citation28]. Additionally, HSP70 can be induced by oxidative stress and prevent NF-κB-induced iNOS expression, leading to limiting the generation of both ROS and peroxynitrite [Citation31]. In the present study, EP pretreatment significantly enhanced heat stress-induced HSF1 translocation into nucleus, as well as HSP70 expression, suggesting that induction of HSPs plays a key role in the protective effect.
HO-1 exerts its antioxidant capacity mediated by the degradation of pro-oxidant free heme and the generation of biliverdin and bilirubin, the major components for antioxidant action [Citation11]. We have previously shown that EP induced HO-1 expression to alleviate lung injury in endotoxemic rats [Citation16]. Similar report also indicated that EP inhibited LPS-stimulated macrophages from releasing iNOS and HMGB1 [Citation32]. HMGB1 is a ubiquitous nuclear protein and late inflammatory mediator in sepsis that can be secreted by immune cells, including macrophages and neutrophils. Emerging evidence supports that EP significantly inhibits the release of HMGB1 from stimulated macrophages [Citation33] and acts an ROS scavenger [Citation13,Citation14]. In this study, EP may exert its antioxidant action by induction of HSP70 and HO-1, and by scavenging free radicals, leading to the attenuation of oxidative stress and inflammatory responses in HS rats.
Activation of autophagy is an adaptive process in response to various conditions of stress, such as starvation, infection, hypoxia, oxidative stress, protein aggregation and endoplasmic reticulum stress. The major function of autophagy is to provide nutrients for the maintenance of cellular functions during different forms of stress. Autophagy has been shown to act as a major cytoprotective system via selectively eliminating unwanted and harmful cytosolic materials, such as protein aggregates and damaged mitochondria. Furthermore, autophagy regulates the homeostasis of immune cells, including neutrophils, macrophages and lymphocytes, which play an important role in inflammatory responses [Citation34]. Under nutrient-rich conditions, mTORC1 associates with the ULK1 complex. Upon starvation, mTORC1 is dephosphorylated and results in dissociation of ULK1 complex from mTORC1. ULK1 initiates autophagy by phosphorylating components of the autophagy machinery to promote phagophore nucleation. Several autophagy-related proteins (ATGs), such as ATG4, ATG 7, ATG 3, ATG12–ATG5–ATG16L, PE-conjugated ATG8s (LC3II) and ATG9 participate in phagophore expansion. Conversely, one kind of autophagy receptor, p62, can help cargo sequestration. p62 carries the target substrate for degradation into the autophagosome, and its amount inversely correlates with autophagic degradation [Citation23]. As the phagophore expands, the membrane bends to ultimately generate a spherical autophagosome, which traffics to and fuses with an endosome and/or lysosome, becoming an autolysosome [Citation7,Citation35]. In addition to the mTORC1 pathway, AMPK and AKT have been reported to regulate autophagy. Reduced glucose or energy levels increase the AMP/ATP ratio and result in the activation of AMPK and TSC (tuberous sclerosis complex), which consists of the TSC1–TSC2 tumor suppressor heterodimer. This activation of AMPK and TSC results in inhibition of the mTOR pathway and inhibition of cell growth and activation of autophagy. In contrast, activated AKT directly phosphorylates TSC2, leading to increases in a small protein, the RAS-like GTPase RHEB levels, which results in mTOR activation and autophagy inhibition [Citation25]. It has been shown that activation of autophagy is one of the protective mechanisms against neuronal and myocardial damages in HS [Citation10,Citation36]. In the present study, pretreatment of EP demonstrated enhancement of autophagy activation during heat stress, evidenced by attenuation of phosphorylated mTOR (p-mTOR) and p-AKT, and increases in p-AMPK, ULK1, ATG7 and ATG12 levels, suggesting that the beneficial effect of EP is mediated via preserving the adequate function of autophagy to maintain cellular homeostasis during HS. However, EP has been reported to inhibit autophagy and attenuate to hepatic ischemia/reperfusion injury [Citation37], LPS-induced acute lung injury [Citation38] and CaCl2-induced tubular epithelial cell injury [Citation39]. The controversial results may be attributed to the different forms of stress. Heat stress caused the intense and immediate insult to cells; therefore, autophagy needs to be activated in a short time to maintain cellular function. Despite the fact that heat stress can activate cellular autophagy process to provide cytoprotection, it did not last to 6 h after HS. Pretreatment with EP maintained the activation of autophagy to 6 h post HS, accompanied by amelioration of HS-induced organ dysfunction. Therefore, we suggest that preservation of autophagy function during HS may contribute to the protective effect of EP.
HSP70 has been reported to assist autophagy. It interacts with the co-chaperone Bag3, and the small heat shock protein (HSP), HspB8, forming a ternary complex, which links to the ubiquitin ligase CHIP and ubiquitinated client protein. The preloading complex is recruited by the ubiquitin-binding protein p62/SQSTM1, which binds simultaneously to ubiquitinated client proteins and LC3II, targeting for autophagic clearance [Citation40]. In addition, localization of HSP70 to the lysosomes stabilizes the lysosomal membrane to promote autophagy [Citation41]. Induction of HSP70 has been reported to protect against HS-induced organ damage, which is associated with the activation of the autophagic process [Citation9,Citation30]. On the other hand, recent studies have revealed that regulation of the autophagy pathway is novel anti-inflammatory targets of HO-1 [Citation12,Citation42]. By induction of autophagy, HO-1 protects against hepatocyte cell death and liver injury from infection/sepsis in mice [Citation43]. In LPS-activated macrophages, HO-1 triggers the activation of autophagy to exert anti-inflammatory effects [Citation44]. HO-1 dependent induction of autophagy reduces hepatocellular injury by liver ischemia–reperfusion [Citation45]. Therefore, enhancement of heat stress-induced HSP70 and HO-1 expression by EP may exert the anti-inflammatory effect via sustaining autophagic function in HS.
Our study provided preventive potential of EP in HS-induced systemic inflammatory responses. However, HS is mainly characterized by the leakage of LPS from the intestine into the systemic circulation, which leads to MOD and mimics the pathophysiological features of sepsis [Citation46]. It should be stressed that this pretreatment study does not allow any conclusions that may offer clinical strategies. Nonetheless, several studies also indicated that post-treatment therapy failed to improve organ function and survival [Citation47]. Thus, a growing number of studies investigated the prophylactic strategies for systemic inflammatory responses during heat stress [Citation48–50]. It is still somewhat challenging to conceive whether a pretreatment therapy would have practical application except for the extreme and risky circumstances. However, the practical timing of this supplement would be difficult to conduct clinically because one would have to predict that unexpected circumstances are imminent. One implication of our study is that EP supplement provides prophylactic values in protecting the integrity of the gut barrier to limit HS-induced endotoxemia as well as the anti-inflammatory capacities in mitigating the risks of HS. In addition, EP is relatively safe for human consumption as a food additive and could be an attractive candidate as a preventive medication of unexpected HS events.
In conclusion, EP alleviated heat stress-induced MOD via its anti-inflammatory effect. Induction of HSP70 and preservation of autophagy may contribute to the beneficial effects of EP on heat stress-related illness.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Gauer R, Meyers BK. Heat-related illnesses. Am Fam Physician. 2019;99:482–489.
- Argaud L, Ferry T, Le QH, et al. Short- and long-term outcomes of heatstroke following the 2003 heat wave in Lyon, France. Arch Intern Med. 2007;167:2177–2183.
- Epstein Y, Yanovich R. Heatstroke. N Engl J Med. 2019;380:2449–2459.
- Soti C, Nagy E, Giricz Z, et al. Heat shock proteins as emerging therapeutic targets. Br J Pharmacol. 2005;146:769–780.
- Yenari MA, Liu J, Zheng Z, et al. Antiapoptotic and anti-inflammatory mechanisms of heat-shock protein protection. Ann N Y Acad Sci. 2005;1053:74–83.
- Vabulas RM, Raychaudhuri S, Hayer-Hartl M, et al. Protein folding in the cytoplasm and the heat shock response. Cold Spring Harb Perspect Biol. 2010;2:a004390.
- Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19:349–364.
- Saha S, Panigrahi DP, Patil S, et al. Autophagy in health and disease: a comprehensive review. Biomed Pharmacother. 2018;104:485–495.
- Tsai YC, Lam KK, Peng YJ, et al. Heat shock protein 70 and AMP-activated protein kinase contribute to 17-DMAG-dependent protection against heat stroke. J Cell Mol Med. 2016;20:1889–1897.
- Shen HH, Tseng YS, Kuo NC, et al. Alpha-lipoic acid protects cardiomyocytes against heat stroke-induced apoptosis and inflammatory responses associated with the induction of Hsp70 and activation of autophagy. Mediators Inflamm. 2019;2019:8187529.
- Motterlini R, Foresti R. Heme oxygenase-1 as a target for drug discovery. Antioxid Redox Signal. 2014;20:1810–1826.
- Ryter SW. Heme oxygenase-1/carbon monoxide as modulators of autophagy and inflammation. Arch Biochem Biophys. 2019;678:108186.
- Fink MP. Ethyl pyruvate. Curr Opin Anaesthesiol. 2008;21:160–167.
- Kao KK, Fink MP. The biochemical basis for the anti-inflammatory and cytoprotective actions of ethyl pyruvate and related compounds. Biochem Pharmacol. 2010;80:151–159.
- Yang R, Zhu S, Tonnessen TI. Ethyl pyruvate is a novel anti-inflammatory agent to treat multiple inflammatory organ injuries. J Inflamm (Lond). 2016;13:37.
- Kung CW, Lee YM, Cheng PY, et al. Ethyl pyruvate reduces acute lung injury via regulation of iNOS and HO-1 expression in endotoxemic rats. J Surg Res. 2011;167:e323–e331.
- Yao L, Cheng C, Yang X, et al. Ethyl pyruvate and analogs as potential treatments for acute pancreatitis: a review of in vitro and in vivo studies. Pancreatology. 2019;19:209–216.
- Wang P, Gong G, Wei Z, et al. Ethyl pyruvate prevents intestinal inflammatory response and oxidative stress in a rat model of extrahepatic cholestasis. J Surg Res. 2010;160(2):228–235.
- Soh S, Jun JH, Song JW, et al. Ethyl pyruvate attenuates myocardial ischemia–reperfusion injury exacerbated by hyperglycemia via retained inhibitory effect on HMGB1. Int J Cardiol. 2018;252:156–162.
- Olek RA, Ziolkowski W, Wierzba TH, et al. Effect of ethyl pyruvate on skeletal muscle metabolism in rats fed on a high fat diet. Nutrients. 2013;5:2372–2383.
- Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-kappa B. Annu Rev Cell Biol. 1994;10:405–455.
- Glick D, Barth S, MacLeod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12.
- Katsuragi Y, Ichimura Y, Komatsu M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015;282:4672–4678.
- Gurusamy N, Das DK. Is autophagy a double-edged sword for the heart? Acta Physiol Hung. 2009;96:267–276.
- Dokladny K, Myers OB, Moseley PL. Heat shock response and autophagy – cooperation and control. Autophagy. 2015;11(2):200–213.
- Ran R, Lu A, Zhang L, et al. Hsp70 promotes TNF-mediated apoptosis by binding IKK gamma and impairing NF-kappa B survival signaling. Genes Dev. 2004;18:1466–1481.
- Chen H, Wu Y, Zhang Y, et al. Hsp70 inhibits lipopolysaccharide-induced NF-kappaB activation by interacting with TRAF6 and inhibiting its ubiquitination. FEBS Lett. 2006;580:3145–3152.
- Ambade A, Mandrekar P. Oxidative stress and inflammation: essential partners in alcoholic liver disease. Int J Hepatol. 2012;2012:853175.
- Bruemmer-Smith S, Stüber F, Schroeder S. Protective functions of intracellular heat-shock protein (HSP) 70-expression in patients with severe sepsis. Intensive Care Med. 2001;27:1835–1841.
- Wang JL, Ke DS, Lin MT. Heat shock pretreatment may protect against heatstroke-induced circulatory shock and cerebral ischemia by reducing oxidative stress and energy depletion. Shock. 2005;23:161–167.
- Radons J. The human HSP70 family of chaperones: where do we stand? Cell Stress Chaperones. 2016;21:379–404.
- Jang HJ, Kim YM, Tsoyi K, et al. Ethyl pyruvate induces heme oxygenase-1 through p38 mitogen-activated protein kinase activation by depletion of glutathione in RAW 264.7 cells and improves survival in septic animals. Antioxid Redox Signal. 2012;17:878–889.
- Ulloa L, Ochani M, Yang H, et al. Ethyl pyruvate prevents lethality in mice with established lethal sepsis and systemic inflammation. Proc Natl Acad Sci U S A. 2002;99(19):12351–12356.
- Qian M, Fang X, Wang X. Autophagy and inflammation. Clin Transl Med. 2017;6:24.
- Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. 2014;20:460–473.
- Liu TT, Hu CH, Tsai CD, et al. Heat stroke induces autophagy as a protection mechanism against neurodegeneration in the brain. Shock. 2010;34:643–648.
- Shen M, Lu J, Dai W, et al. Ethyl pyruvate ameliorates hepatic ischemia–reperfusion injury by inhibiting intrinsic pathway of apoptosis and autophagy. Mediators Inflamm. 2013;2013:1–12.
- Zhu Q, Wang H, Wang H, et al. Protective effects of ethyl pyruvate on lipopolysaccharide‑induced acute lung injury through inhibition of autophagy in neutrophils. Mol Med Rep. 2017;15:1272–1278.
- Zhao J, Cheng J, Li C, et al. Ethyl pyruvate attenuates CaCl2-induced tubular epithelial cell injury by inhibiting autophagy and inflammatory responses. Kidney Blood Press Res. 2018;43:1585–1595.
- Fernández-Fernández MR, Valpuesta JM. Hsp70 chaperone: a master player in protein homeostasis. F1000Res. 2018;7:1497.
- Chatterjee S, Burns TF. Targeting heat shock proteins in cancer: a promising therapeutic approach. Int J Mol Sci. 2017;18:1978.
- Kuo NC, Huang SY, Yang CY, et al. Involvement of HO-1 and autophagy in the protective effect of magnolol in hepatic steatosis-induced NLRP3 inflammasome activation in vivo and in vitro. Antioxidants (Basel). 2020;9(10):924.
- Carchman EH, Rao J, Loughran PA, et al. Heme oxygenase-1-mediated autophagy protects against hepatocyte cell death and hepatic injury from infection/sepsis in mice. Hepatology. 2011;53:2053–2062.
- Waltz P, Carchman EH, Young AC, et al. Lipopolysaccharide induces autophagic signaling in macrophages via a TLR4, heme oxygenase-1 dependent pathway. Autophagy. 2011;7:315–320.
- Yun N, Cho HI, Lee SM. Impaired autophagy contributes to hepatocellular damage during ischemia/reperfusion: heme oxygenase-1 as a possible regulator. Free Radic Biol Med. 2014;68:168–177.
- Lim CL. Heat sepsis precedes heat toxicity in the pathophysiology of heat stroke-a new paradigm on an ancient disease. Antioxidants (Basel). 2018;7(11):149.
- Bhargava R, Altmann CJ, Andres-Hernando A, et al. Acute lung injury and acute kidney injury are established by four hours in experimental sepsis and are improved with pre, but not post, sepsis administration of TNF-α antibodies. PLoS One. 2013;8:e79037.
- Lam KK, Cheng PY, Lee YM, et al. The role of heat shock protein 70 in the protective effect of YC-1 on heat stroke rats. Eur J Pharmacol. 2013;699:67–73.
- Lin X, Zhao T, Lin CH, et al. Melatonin provides protection against heat stroke-induced myocardial injury in male rats. J Pharm Pharmacol. 2018;70:760–767.
- Phillips NA, Welc SS, Wallet SM, et al. Protection of intestinal injury during heat stroke in mice by interleukin-6 pretreatment. J Physiol. 2015;593:739–752.