
Abstract
Objective
The incidence rate of heat stroke (HS) has increased, with high morbidity and mortality rates, in recent years. Previous studies have suggested that vascular endothelial cell injury is one of the main pathological features of HS. Uncoupling protein 2 (UCP2) exhibits antioxidant activity under various stress conditions. This study aims to investigate the role of UCP2 in HS-induced vascular endothelial injury.
Method
To explore the mechanisms mediating vascular endothelial cell injury induced by HS, we established an HS model of HUVECs in vitro. The percentage of cell death and viability induced by HS were assessed using annexin V-FITC/PI staining and CCK8 assays. HS-induced mitochondrial membrane potential (ΔΨm) was detected by JC-1 staining. HS-induced mitochondrial superoxide was measured by MitoSOX staining, and analyzed by flow cytometry. UCP2, Drp1, phosphorylated Drp1, OPA1, and Mfn2 expression levels were measured by western blotting.
Results
HS triggered mitochondrial fragmentation and UCP2 upregulation in a time-dependent manner in HUVECs. As a specific Drp1 inhibitor, Mdivi‐1 pretreatment significantly promoted mitochondrial fission and apoptosis in HS-induced HUVECs. In addition, siRNA-mediated UCP2 knockdown further aggravated mitochondrial fragmentation and ΔΨm depolarization and increased mitochondrial ROS production and cell apoptosis in HS-induced HUVECs, which were abolished by Drp1 inhibition.
Conclusion
Our results indicate that UCP2 protects against HS-induced vascular endothelial damage and that it enhances mitochondrial function. These findings reveal that UCP2 can be a potential contributor to mechanism-based therapeutic strategies for HS.
Introduction
Heat stroke (HS) has proven to be a life-threatening condition, associated with high morbidity and mortality rates. It is characterized by a rapid increase in core body temperature to more than 40 °C, and is often associated with dry skin, hot, and central nervous system (CNS) dysfunction [Citation1]. Early diagnosis and intervention are necessary to limit its progression.
Vascular endothelial cells cover the luminal surface of the blood vessels and play a key role in maintaining the stability of microvascular permeability [Citation2]. It is hypothesized that vascular endothelial cell injury is one of the early pathological changes induced by HS, which includes vascular endothelial cell death and inflammation [Citation3]. Moreover, vascular endothelial dysfunction plays an essential role in the occurrence and development of HS [Citation4]. However, the mechanisms mediating vascular endothelial cell damage induced by HS remain inadequately understood.
Mitochondria are essential organelles in mammalian cells, and play a central role in metabolism, cell death, and cellular senescence. Mitochondrial dysfunction, in the form of permeabilization of the inner and/or outer membranes of organelles, can eventually lead to cell apoptosis or necrosis [Citation5,Citation6]. Growing evidence suggests that mitochondrial dysfunction induces the loss of cellular homeostasis, which contributes to cell death during HS [Citation7–9]. Mitochondrial function rests on the complex molecular machinery of mitochondrial dynamics-processes of fission and fusion [Citation10]. A precise balance in mitochondrial dynamics is closely related to the maintenance of mitochondrial functions and responses to external stress [Citation11]. Mitochondrial fission is driven by fission regulators, including mitochondrial fission factor (Mff), dynamin-related protein 1 (Drp1), and fission 1 (Fis1) [Citation12]. In particular, the phosphorylation of Drp1 regulates its translocation to the mitochondrial membrane to induce mitochondrial fission [Citation13,Citation14]. Mitochondrial fusion is mediated by Mitofusin 1/2 (Mfn1/2) and optic atrophy 1 (OPA1) anchor proteins that maintain the fusion of the mitochondrial outer and inner membranes [Citation15]. Therefore, researching the mechanism of mitochondrial homeostasis may provide an important breakthrough in HS therapy.
Mitochondrial uncoupling proteins (UCPs) are members of the anionic proton transporter family, which is mainly located in the inner membrane of mitochondria [Citation16]. Current available studies showed that the UCPs family could partially promote proton leakage across the inner membrane, dissipate the proton concentration gradient of the inner mitochondria membrane, and play critical roles in the regulation of respiratory activity and metabolic processes [Citation17,Citation18]. Among them, UCP1, UCP2, and UCP3 have been extensively studied owing to their biomedical importance [Citation19]. UCP3 is similar in tissue specificity to UCP1, which is exclusively expressed in brown adipose tissue and is a key molecule in thermogenesis [Citation20]. A previous report demonstrated that UCP1 plays a pivotal role in cardioprotective effects in myocardial I/R injury [Citation18,Citation21]. UCP2 is an essential member of the UCP family, which is distributed in various tissues, including the heart, kidney, spleen, and white adipose tissue, and plays an essential role in numerous biological processes, including basal metabolic rate regulation, oxidative stress response, and insulin resistance [Citation22,Citation23]. It has been reported that UCP2 deficiency exacerbated oxidative stress and aggravated the lipopolysaccharide (LPS)-induced renal injury, inflammation in vivo, whereas, UCP2 overexpression inhibited ROS production and protected HK-2 cells from LPS-induced injury in vitro [Citation24]. Besides, upregulation of UCP2 reduced mitochondrial ROS and improved endothelial function in endothelial cells [Citation25]. Therefore, further research is needed to determine whether UCP2 can be a master regulator of vascular endothelial injury and repair during HS.
Limited data is available on the role of UCP2 during HS. However, we found a significant elevation of UCP2 expression in HS-induced HUVECs, which was functionally related to mitochondrial dysfunction. The present study aimed to elucidate the mechanisms governing mitochondrial abnormalities and to clarify the role of UCP2 in this process. Specifically, we demonstrated that UCP2 may be a novel potential target for modulating the intensity of HS-mediated vascular endothelial damage.
Materials and methods
Cell culture and treatments
Primary human umbilical vein endothelial cells (HUVECs) were purchased from the Shanghai Institute of Cell Biology, Chinese Academy of Sciences. HUVECs were maintained in DMEM/F12 (FBS, Gibco BRL) supplemented with 10% (v/v) fetal bovine serum (FBS, Gibco BRL), 100 U/ml penicillin, and 100 µg/ml streptomycin at 37 °C in 5% CO2. For the heat stress treatment, the cell culture medium was replaced with a fresh medium, and then cells were placed in an incubator containing 5% CO2 and 95% air at 37 ± 0.5 °C (Cont) or 43 ± 0.5 °C (HS) for two hours. The cells were subsequently incubated at 37 °C, as indicated.
Mitochondria isolation and protein extraction
Mitochondria were isolated from HUVECs using the Cell Mitochondria Isolation Kit (Beyotime Biotechnology, China), according to the manufacturer’s protocols. Cells were grown on 100 mm plates until 80%–90% confluency and washed twice with pre-cooled phosphate-buffered saline (PBS). Cell pellets were resuspended in mitochondrial isolation buffer containing protease and phosphatase inhibitors (1 mM phenylmethanesulfonyl fluoride [PMSF], 2 mM phosphatase inhibitor cocktail A). The lysed cells were then resuspended with mitochondrial isolation reagent and centrifuged at 600 × g at 4 °C for 10 min. Supernatants were then discarded by centrifugation at 10,000 × g at 4 °C for 15 min to obtain a pellet containing mitochondria. Isolated mitochondria were added with mitochondria protein extraction reagent containing protease and phosphatase inhibitors for subsequent analysis.
MitoTracker red staining and immunofluorescent staining
Mitochondria in cells cultured on coverslips were labeled by incubating living cells with the MitoTracker Red fluorescent probe kit (250 nM for 30 min; Invitrogen, Waltham, USA). Cell culture media were removed and cells were washed three times with PBS, fixed in 4% paraformaldehyde for 15 min, and punched with 0.1% Triton X-100 for 10 min at room temperature. Cells were then incubated with the specific primary antibody, anti-Drp1 (8570, Cell Signaling), followed by staining with a fluorescent secondary antibody. Cells were subsequently counterstained with 4′ 6-diamidino-2-phenylindole (DAPI) to visualize the nuclei. Images were visualized using a confocal inverted laser microscope (Zeiss, Germany).
Western blot analysis
Total proteins were extracted from HUVECs using radioimmunoprecipitation assay (RIPA) lysis buffer supplemented with a 1× protease inhibitor cocktail. Protein concentrations were determined using an Enhanced BCA Protein Assay Kit (Beyotime Institute of Biotechnology, China). Proteins were separated using sodium dodecyl sulfate (SDS)-polyacrylamide gel, transferred onto polyvinylidene difluoride (PVDF) membranes, and blotted with 5% skimmed milk. The membranes were incubated overnight at 4 °C with primary antibodies and incubated with secondary antibodies for one hour at room temperature. These primary antibodies used were Drp1 (8570, Cell Signaling), p-Drp1-S616 (3455, Cell Signaling), OPA1 (ab42364, Abcam), Mfn2 (9482, Cell Signaling), Fis1 (10956-1-AP, Proteintech), UCP2 (89326, Cell Signaling), anti-GAPDH (AC002, ABclonal), and anti-VDAC1 (55259-1-AP, Proteintech). These secondary antibodies used were horseradish peroxidase-labeled goat anti-mouse (AS014, ABclonal) and anti-rabbit (AS003, ABclonal) antibodies. Quantification was performed by measuring band intensities using ImageJ software. Protein expression levels were normalized relative to the levels of VDAC1 or GAPDH.
Flow cytometry analysis
HUVECs were either exposed (experimental) or not exposed (control) to 43 °C for two hours, and then further incubated at 37 °C, as indicated. Cell apoptosis was investigated using the Annexin V-FITC Apoptosis Detection Kit (Invitrogen, USA). Cells (1 × 106) were collected, washed with ice-cold PBS, and resuspended in 400 μl of binding buffer containing Annexin V-FITC. The cells were incubated at room temperature in the dark for 10 min. Afterwards, the buffer was removed by centrifugation, and the cells were resuspended in a reaction buffer containing propidium iodide (PI) solution. After 10 min, the cells were immediately analyzed using a FACScalibur cytometer (Becton Dickinson, USA) to quantify cell apoptosis. Annexin V (+)/PI (-) cells in the lower right quadrant were considered to be early apoptotic cells.
CCK8 assay
Cell viability was measured using the Cell Counting Kit-8 (CCK8) assay by following the manufacturer’s instructions (Dojindo, Japan). HUVECs were seeded in 96-well plates at a density of 1 × 105/mL and treated accordingly. The medium was then replaced with 100 μl CCK-8 solution. After two hours, the absorbance at 450 nm was measured using a microplate reader (Multiskan MK3, Thermo Fisher Scientific).
Transfection of siRNA
HUVECs were transfected with target siRNA using Lipofectamine 2000, according to the manufacturer’s protocol. The sequences of oligonucleotides for control and UCP2 siRNA were synthesized by GenePharma (Shanghai, China). These siRNA targeting sequences were as follows: control siRNA, sense 5′-UUCUCCGAACGUGUCA CGUTT-3′ and antisense 5-ACGUGACACGUUCGGAGA ATT-3; UCP2 siRNA, sense 5′-GCCUGUAUGAUUCUGUCAATT −3′ and antisense 5′-UUGACAGAAUCAUACAGGCTT-3′. These transfected cells were collected and processed for subsequent experiments 72 h later.
Mitochondrial membrane potential (JC-1) assay
Mitochondrial membrane potential (ΔΨm) was quantified by measuring fluorescence intensities of red-shifted aggregates and green-shifted JC-1 monomers to evaluate mitochondrial viability using a JC-1 detection kit (Beyotime Biotechnology, China), according to the manufacturer’s instructions. JC-1 accumulates in functional mitochondria with high ΔΨm and forms red-shifted JC-1 aggregates, and damaged mitochondria with decreased ΔΨm and forms green-shifted JC-1 monomers. The green-shifted JC-1 monomers fluorescence is linearly correlated to a decrease in ΔΨm. We washed the cells with PBS buffer three times and prepared a JC-1 fluorescent probe. After staining at 37 °C for 30 min, red and green fluorescence was observed using a confocal inverted laser microscope (Zeiss, Germany).
Mitochondria ROS measurements
Mitochondrial ROS activity was detected using the fluorescent probe MitoSOX (Invitrogen, USA). Briefly, living HUVECs were incubated with 5 μM MitoSOX (red fluorescence) in HBSS and 5 μg/ml Hoechst (Beyotime Biotechnology, China) at 37 °C in the dark for 30 min. The fluorescence intensities of mitochondrial ROS probes were analyzed using a FACScalibur cytometer or a confocal inverted laser microscope (Zeiss, Germany).
Statistical analysis
All data are expressed as the mean ± standard error of the mean (SEM) from at least three independent experiments. One-way ANOVA was used to analyze the differences among three or more groups, followed by post hoc analysis. All statistical analyses were performed for statistical significance using the GraphPad software (version 6.0, GraphPad Software, CA, USA). Statistical significance was set at p < 0.05.
Results
Mitochondrial fragmentation and UCP2 upregulation in HS-induced HUVECs
Previous studies have indicated that HS-induced vascular endothelial cell death is associated with the accumulation of damaged mitochondria in cells [Citation26,Citation27]. To further study the effects of mitochondrial dysfunction on HUVECs, and then to treat HUVECs, we used JC-1 staining and MitoSOX fluorescent probe to measure ΔΨm and mitochondrial ROS production. As shown in , HS decreased ΔΨm, and significantly upregulated mitochondrial ROS in a time-dependent manner after HS. Given the necessary roles played by mitochondrial fragmentation, mitochondrial processes were measured. The expression of pro‐fission proteins in isolated mitochondria, such as p-Drp1S616, Drp1, and Fis1, was significantly upregulated after 0–6 h of incubation at 37 °C (). Additionally, the expression levels of pro‐fusion factors, such as OPA1 and Mfn2, were substantially downregulated during the early recovery periods after HS (). Therefore, our results showed that mitochondrial dysfunction was related to HS-induced endothelial cell death.
Figure 1. Mitochondrial dysregulation and UCP2 expression in HUVECs during the recovery period following HS. HUVECs were subjected to two hours of HS, and further incubation at 37 °C for zero, six, and twelve hours. (a) Mitochondrial membrane potential (ΔΨm) was detected by JC-1 staining in HUVECs induced by HS, Scale bar: 20 μm. Cells (in green) represent JC-1 monomers and (in red) JC-1 aggregates. (b) Effect of HS on protein expression of p-Drp1S616, Drp1, Fis1, OPA1, and Mfn2 in HUVECs induced by HS. VDAC1 was used as loading control. (c,d) Quantification of p-Drp1S616, Drp1, Fis1, OPA1, and Mfn2 expressions. (e) Effect of HS on protein expression of UCP2 in HUVECs induced by HS. n = 4, *p < 0.05 versus the Cont. group.
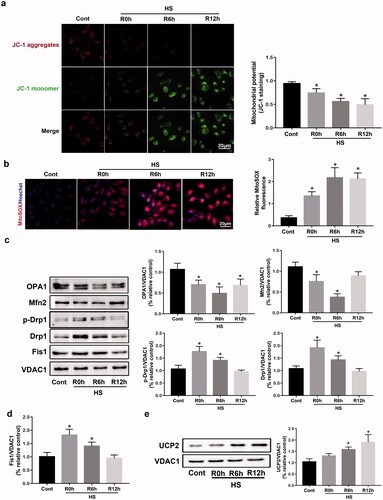
Subsequently, the protein expression of UCP2 was defined in an in vitro model of cultured HUVECs induced by HS. These results suggested that HS triggered a significant upregulation of UCP2 expression in a time-dependent manner between the control and HS groups (). The vital role and mechanism of UCP2 signaling in endothelial cell death induced by HS need further investigation.
UCP2‑knockdown exacerbation of HUVECs’ apoptosis induced by HS
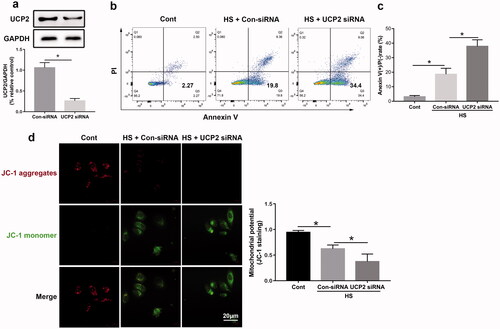
To further clarify the biological and clinical relevance of UCP2 in HS-mediated vascular endothelial damage, we next examined whether UCP2 siRNA transfection exacerbated HUVEC apoptosis induced by HS. It has been demonstrated that the number of Annexin V-FITC (+) and PI (-) cells in the lower right quadrant is indicative of early apoptosis. We found that UCP2 siRNA significantly reduced UCP2 protein expression (). Flow cytometry results revealed that UCP2 siRNA upregulated HS-induced apoptosis in HUVECs compared to HS groups (). UCP2-knockdown also caused a significant decrease in ΔΨm (). These findings indicated that UCP2 siRNA aggravated ΔΨm downregulation and increased mitochondrial ROS production and cell apoptosis in HUVECs induced by HS.
Figure 2. UCP2 deletion exacerbates cell death and mitochondrial dysregulation induced by HS. HUVECs were transfected with either control siRNA or UCP2 siRNA. Transfected HUVECs were subjected to two hours of HS, and further incubation at 37 °C for six hours. (a) Western blot and densitometric analyses of UCP2 protein expression. GAPDH was used as loading control. (b) The early apoptosis of HUVECs induced by HS was detected by Annexin V-FITC/PI staining. (c) Quantification of cell apoptosis in all groups. (d) The ΔΨm was detected by JC-1 staining in HUVECs induced by HS, Scale bar: 20 μm. n = 5, *p < 0.05 compared with the indicated groups.
UCP2 regulates HS-induced mitochondrial fission/fusion in HUVECs
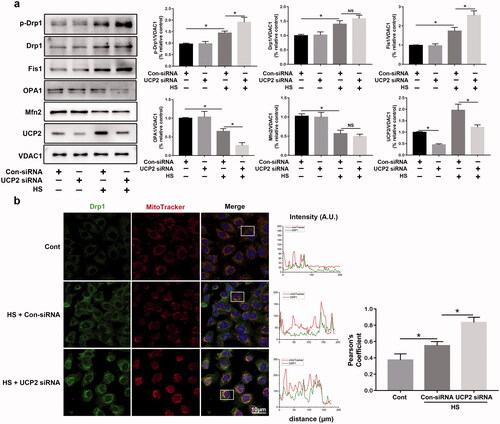
To further define the functional role of UCP2 in mitochondrial dynamics during HS, we demonstrated whether UCP2-knockdown regulated the expression of mitochondrial fission/fusion proteins during HS. To assess mitochondrial fission, we screened for Drp1, p-Drp1S616, and FIS1. Given the total DRP1 protein levels were upregulated by HS treatment (), we decided to normalize DRP1-S616 to VDAC1 to better indicate the underlying mechanism. As shown in , compared with control siRNA-treated cells, western blotting analysis results indicated that p-DRP1S616 phosphorylation was enhanced in UCP2-depleted HUVECs exposed to HS, while total DRP1 levels did not change. Interestingly, HS-induced downregulation of OPA1 protein levels in mitochondria was significantly reduced by UCP2 deletion but did not decrease total Mfn2 levels. Next, to observe alterations in mitochondrial morphology, immunofluorescence analysis was conducted; this indicated that HS-induced co-localization of the mitochondrial network and recruitment of Drp1 in HUVECs was upregulated by UCP2-knockdown (). Overall, these data confirmed the role of UCP2‐related mitochondrial fission/fusion machinery in HUVECs induced by HS.
Figure 3. UCP2 regulates mitochondrial fission in HUVECs induced by HS. HUVECs were transfected with either control siRNA or UCP2 siRNA. Transfected HUVECs were subjected to two hours of HS, and further incubation at 37 °C for six hours. (a) Western blot analysis for p-Drp1S616, Drp1, Fis1, OPA1, Mfn2, and UCP2 in mitochondria isolated from HUVECs induced by HS, and standardized to VDAC1. (b) Representative images showing mitochondrial morphology with MitoTracker Red (red), Drp1 (green), and DAPI (blue) staining in HUVECs induced by HS. The Pearson's coefficient indexes between Drp1 and MitoTracker Red fluorescence intensities were measured in 10 or more cells from three independent experiments. Scale bar: 10 μm. n = 4, *p < 0.05 compared with the indicated groups. NS = not significant.
Drp1 inhibition enhances HS-induced mitochondrial fission in HUVECs
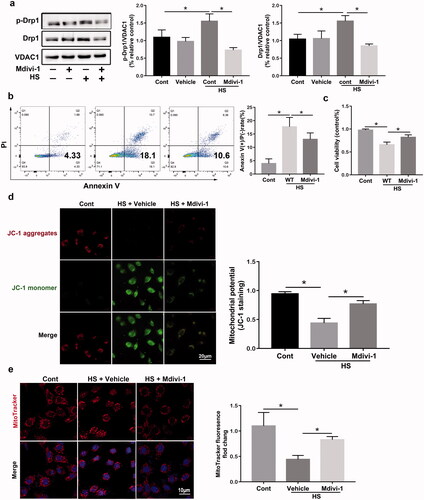
Mdivi‐1 has been identified as a selective Drp1 inhibitor that has widely been used extensively in vitro and vivo under different disease conditions[Citation28]. Mdivi-1 may act allosterically and prevent Drp1 oligomerization on the mitochondrial membrane, suggesting that inhibits Drp1 self-assembly into rings and its translocation into mitochondrial [Citation29]. To examine the regulation of Drp1 HS-mediated mitochondrial dysfunction in HUVECs, cells were pretreated with Mdivi-1(10 μM) for two hours and cultured under heatstroke conditions for two hours. Compared with the vehicle group, the western blotting assay indicated that Mdivi-1 treatment successfully decreased the p-Drp1S616 and Drp1 protein levels (). Flow cytometry results also revealed that HUVECs pretreated with Mdivi-1 reduced the number of Annexin V-FITC (+) and PI (-) cells compared to untreated cells (). Moreover, Mdivi-1 treatment significantly decreased the viability of HUVECs, as demonstrated by the CCK‑8 assays (). As shown in , HS caused ΔΨm downregulation to be significantly abolished by Mdivi‐1. To observe alterations in mitochondrial morphology, immunofluorescence analysis was conducted; this showed that Mdivi-1 treatment significantly promoted mitochondrial fragmentation induced by HS in HUVECs (). Collectively, these findings indicated that Drp1 inhibition by Mdivi-1 treatment markedly promoted mitochondrial fission and apoptosis.
Figure 4. Drp1 inhibition enhances HS-induced mitochondrial fission in HUVECs induced by HS. HUVECs were pretreated with Mdivi-1 (10 μM) for two hours and then subjected to two hours of HS, and further incubation at 37 °C for six hours. (a) The early apoptosis of HUVECs induced by HS was detected by Annexin V-FITC/PI staining. (b) Cell viability of HUVECs was determined using Cell Counting Kit-8 (CCK8) assays. (c) Representative images showing mitochondrial morphology with MitoTracker Red (red) staining in HUVECs induced by HS, Scale bar: 10 μm. (d) Representative images and quantification of ΔΨm were detected by JC-1 staining in HUVECs induced by HS, Scale bar: 20 μm. (e) Western blot analysis for p-Drp1S616 and Drp1 in mitochondria isolated from HUVECs induced by HS, and standardized to VDAC1. n = 4, *p < 0.05 compared with the indicated groups.
UCP2 regulates mitochondrial dysfunction via Drp1
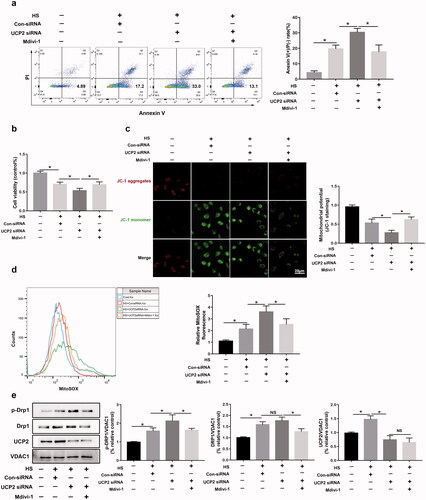
To examine UCP2 regulation of mitochondrial dysfunction and apoptosis, the Drp1 signaling pathway was investigated because previous studies have identified UCP2 as an upstream mediator of Drp1. HUVECs were pretreated with Mdivi-1(10 μM) before exposure to HS to inhibit Drp1 and to explore the relationship between UCP2 and the Drp1 signaling pathway. Flow cytometry results demonstrated that the addition of Mdivi‐1 before HS can inhibit cell apoptosis increased in UCP2 silencing HUVECs (). Furthermore, we found that Mdivi‐1 pro-treatment reversed the average viability of HUVECs in vitro (), indicating that UCP2-knockdown increased HS-induced HUVEC cell death, which was reduced by Drp1 inhibition. Consistently, the immunoblot analysis showed that the ΔΨm loss and mitochondrial ROS upregulated by silencing UCP2 were markedly abolished by Mdivi‐1 in HS HUVECs (). This finding was also supported via western blotting, which demonstrated that p-Drp1S616 phosphorylation was upregulated by UCP2 knock-down and was reversed markedly with Mdivi‐1 pro-treatment (). Taken together, silencing UCP2 sensitized HUVECs to HS-induced mitochondrial dysfunction, which was reduced by Drp1 inhibition.
Figure 5. UCP2 regulates mitochondrial fission and apoptosis via Drp1 in HUVECs induced by HS. HUVECs were transfected with either Con-siRNA or UCP2 siRNA. Transfected HUVECs were pretreated with Mdivi-1 (10 μM) for 2 h, and then subjected to two hours of HS, and further incubation at 37 °C for six hours. (a) The early apoptosis of HUVECs induced by HS was detected by Annexin V-FITC/PI staining. (b) The CCK8 assay assessed cell viability. (c) Mitochondrial membrane potential (ΔΨm) was detected by JC-1 staining in HUVECs induced by HS, Scale bar: 20 μm. (d) Representative images and quantification of mitochondrial O2. The formation was assessed by MitoSOX using flow cytometry. (e) Western blot analysis for p-Drp1S616, Drp1, and UCP2 in mitochondria isolated from HUVECs induced by HS, and standardized to VDAC1. n = 4, *p < 0.05 compared with the indicated groups. NS = not significant.
Discussion
HS incidents may continue to increase due to the proliferation of heatwaves caused by global warming over the past decades [Citation30,Citation31]. It is important to investigate the pathogenesis of HS and to develop effective prevention and treatment methods. Vascular endothelial injury can cause pathological changes in the cardiovascular system [Citation32]. Notably, vascular endothelium injury induced by HS is considered a key factor in multi-organ system dysfunction [Citation33,Citation34]. Thus, the molecular mechanisms underlying HS-induced vascular endothelial damage need to be further explored.
Mitochondrial fragmentation and swelling have been shown to be pathological features of cellular injury caused by HS [Citation35]. A recent study showed that preventing mitochondrial fragmentation can alleviate HS-induced brain injury, suggesting that targeting mitochondrial dynamics may be a therapeutic strategy for central nervous system damage caused by HS [Citation36]. In addition, the morphological appearance of mitochondria depends on the balance of mitochondrial dynamics, including mitochondrial fission and fusion [Citation37]. Drp1 is functionally regulated by post-translational modification of phosphorylation (S616) and then translocated to the mitochondrial membrane, leading to mitochondrial fragmentation, which is consistent with our results [Citation38,Citation39]. Here, p-Drp1S616, Drp1, and Fis1 were enhanced in HS-induced HUVECs, whereas OPA1 and Mfn2 were substantially downregulated in the acute phase of HS. Furthermore, HS decreased ΔΨM and significantly upregulated mitochondrial ROS in a time-dependent manner. These results suggest that mitochondrial dysfunction is closely related to HS-induced endothelial cell death.
Various studies have revealed the critical role of UCP2 in regulating mitochondrial function in many physiological processes [Citation40]. Some studies have demonstrated that enhancing UCP2 can improve mitochondrial dynamics in AKI [Citation23,Citation41]. Whether HS-induced UCP2 upregulation is related to mitochondrial dynamics is an important and interesting question. Here, we found that mitochondrial UCP2 increased in HS-induced HUVECs in a time-dependent manner. Interestingly, UCP2 depletion led to an elevated protein level of p-Drp1S616, which exacerbated mitochondrial fission in HUVECs exposed to HS. Meanwhile, HS-induced downregulation of OPA1 protein levels in mitochondria was also abolished by UCP2 deletion without changes in Mfn2 levels. These results indicated that UCP2 depletion increased the mitochondrial fission/fusion index and tuned the mitochondrial dynamics toward fission. These findings have substantiated the sufficiency of Drp1‐related mitochondrial fission in aggravating vascular endothelium damage; the mechanism of UCP2 renoprotection may depend on its capacity to limit mitochondrial fission and membrane depolarization via Drp1. In addition, UCP2 siRNA further upregulated HS-induced apoptosis of HUVECs compared to HS groups, suggesting that UCP2 can regulate apoptosis induced by HS. Therefore, targeting UCP2 could be a promising therapeutic strategy for the treatment of HS.
Mitochondria are the main source of intracellular ROS [Citation42]. The overproduction of ROS induces oxidative damage and disrupts cellular membrane structure and functions [Citation43]. Many mitochondrial processes are related to the electron transport chain and the resultant membrane gradient, ΔΨM [Citation43,Citation44]. Our results showed that silencing UCP2 aggravated HS-induced HUVECs’ ΔΨm loss, which was abolished by Drp1 inhibition. Consistent with the idea that UCP2 prevents mitochondrial ROS, we found that UCP2 knockdown aggravated HS-induced mitochondrial ROS production, suggesting that UCP2 importantly regulates mitochondrial ROS. All of these studies demonstrate that UCP2 carries out protective functions against oxidative stress under HS.
However, there are some limitations to the present study. First, the results of the present study are limited by the use of HUVECs in vitro, and further in vivo mechanisms need to be explored. Second, mitochondrial function was determined by two tightly linked elements: the ΔΨM and the ATP pool [Citation45]. Unfortunately, ATP levels have not yet been identified in HS-induced HUVECs. Finally, UCP2 knockdown had no effect on the expression of the mitochondrial fusion protein, Mfn2, after HS. We suspect that there may be other mechanisms involved in the influence of UCP2 on mitochondrial fusion. It is of considerable significance to detect whether UCP2 regulates mitochondrial fusion induced by HS.
Conclusion
This study provides strong evidence that HS triggers a significant upregulation of UCP2 expression in HUVECs. UCP2-knockdown sensitized HUVECs to HS-induced mitochondrial dysfunction and apoptosis, which were abolished by Drp1 inhibition. Therefore, we demonstrated that UCP2 can be used as a potential biomarker to diagnose HS. Our experiment also suggests a role for UCP2 as a potential target for mechanism-based therapeutic strategies for HS.
Author contributions
Wei Huang and Liangfeng Mao were responsible for primary data generation and the composition of this manuscript. Weidang Xie and Sumin Cai performed all the experiments. Qiaobing Huang and Zhongqing Chen acquired and interpreted the data. Yanan Liu and Chen confirmed the authenticity of the raw data. All authors analyzed the data, and reviewed and approved the final manuscript.
Disclosure statement
No potential conflict of interest is reported by the authors.
Additional information
Funding
Reference
- Bouchama A, Knochel JP. Heat stroke. N Engl J Med. 2002;346(25):1978–1988.
- Liu J, Zhu G, Xu S, et al. Analysis of miRNA expression profiling in human umbilical vein endothelial cells affected by heat stress. Int J Mol Med. 2017;40(6):1719–1730.
- Li L, Su Z, Zou Z, et al. Ser46 phosphorylation of p53 is an essential event in prolyl-isomerase Pin1-mediated p53-independent apoptosis in response to heat stress. Cell Death Dis. 2019;10(2):96.
- Huang W, Xie W, Gong J, et al. Heat stress induces RIP1/RIP3-dependent necroptosis through the MAPK, NF-κB, and c-Jun signaling pathways in pulmonary vascular endothelial cells. Biochem Biophys Res Commun. 2020;528(1):206–212.
- An H, Zhou B, Ji X. Mitochondrial quality control in acute ischemic stroke. J Cereb Blood Flow Metab. 2021;41(12):3157–3170.
- Tang C, Cai J, Yin X-M, et al. Mitochondrial quality control in kidney injury and repair. Nat Rev Nephrol. 2021;17(5):299–318.
- Chen Y, Leboutet R, Largeau C, et al. Autophagy facilitates mitochondrial rebuilding after acute heat stress via a DRP-1-dependent process. J Cell Biol. 2021;220(4):e201909139.
- Wang H-L, Xing G-D, Qian Y, et al. Dihydromyricetin attenuates heat stress-induced apoptosis in dairy cow mammary epithelial cells through suppressing mitochondrial dysfunction. Ecotoxicol Environ Saf. 2021;214:112078.
- Gu ZT, Li L, Wu F, et al. Heat stress induced apoptosis is triggered by transcription-independent p53, Ca(2+) dyshomeostasis and the subsequent bax mitochondrial translocation. Sci Rep. 2015;5:11497.
- Kraus F, Roy K, Pucadyil TJ, et al. Function and regulation of the divisome for mitochondrial fission. Nature. 2021;590(7844):57–66.
- Giacomello M, Pyakurel A, Glytsou C, et al. The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol. 2020;21(4):204–224.
- Xu T, Dong Q, Luo Y, et al. Porphyromonas gingivalis infection promotes mitochondrial dysfunction through Drp1-dependent mitochondrial fission in endothelial cells. Int J Oral Sci. 2021;13(1):28.
- Vongsfak J, Pratchayasakul W, Apaijai N, et al. The alterations in mitochondrial dynamics following cerebral ischemia/reperfusion injury. Antioxidants. 2021;10(9):1384.
- Zhang K, Guo M-Y, Li Q-G, et al. Drp1-dependent mitochondrial fission mediates corneal injury induced by alkali burn. Free Radic Biol Med. 2021;176:149–161.
- Heine KB, Hood WR. Mitochondrial behaviour, morphology, and animal performance. Biol Rev Camb Philos Soc. 2020;95(3):730–737.
- Pravednikova AE, Shevchenko SY, Kerchev VV, et al. Association of uncoupling protein (ucp) gene polymorphisms with cardiometabolic diseases. Mol Med. 2020;26(1):51.
- Ruiz-Ramírez A, López-Acosta O, Barrios-Maya MA, et al. Cell death and heart failure in obesity: Role of uncoupling proteins. Oxid Med Cell Longev. 2016;2016:9340654.
- Cadenas S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim Biophys Acta Bioenerg. 2018;1859(9):940–950.
- Robbins D, Zhao Y. New aspects of mitochondrial uncoupling proteins (UCPs) and their roles in tumorigenesis. Int J Mol Sci. 2011;12(8):5285–5293.
- Jia P, Wu X, Pan T, et al. Uncoupling protein 1 inhibits mitochondrial reactive oxygen species generation and alleviates acute kidney injury. EBioMedicine. 2019;49:331–340.
- Hou D, Fu H, Zheng Y, et al. Uncoupling protein 1 knockout aggravates isoproterenol-induced acute myocardial ischemia via AMPK/mTOR/PPARα pathways in rats. Transgenic Res. 2021.
- Vallejo FA, Vanni S, Graham RM. UCP2 as a potential biomarker for adjunctive metabolic therapies in tumor management. Front Oncol. 2021;11:640720.
- Qin N, Cai T, Ke Q, et al. UCP2-dependent improvement of mitochondrial dynamics protects against acute kidney injury. J Pathol. 2019;247(3):392–405.
- Ding Y, Zheng Y, Huang J, et al. UCP2 ameliorates mitochondrial dysfunction, inflammation, and oxidative stress in lipopolysaccharide-induced acute kidney injury. Int Immunopharmacol. 2019;71:336–349.
- Toral M, Romero M, Jiménez R, et al. Role of UCP2 in the protective effects of PPARβ/δ activation on lipopolysaccharide-induced endothelial dysfunction. Biochem Pharmacol. 2016;110-111:25–36.
- Liu J, Xu S, Liu S, et al. miR‑3613‑3p/MAP3K2/p38/caspase‑3 pathway regulates the heat‑stress‑induced apoptosis of endothelial cells. Mol Med Rep. 2021;24(3):633.
- Liu Y, Zhou G, Wang Z, et al. NF-κB signaling is essential for resistance to heat stress-induced early stage apoptosis in human umbilical vein endothelial cells. Sci Rep. 2015;5:13547.
- Chen Y, Zhang Z, Henson ES, et al. A DRP-1 dependent autophagy process facilitates rebuilding of the mitochondrial network and modulates adaptation capacity in response to acute heat stress during C. elegans development. Autophagy. 2021:1–2.
- Aishwarya R, Alam S, Abdullah CS, et al. Pleiotropic effects of mdivi-1 in altering mitochondrial dynamics, respiration, and autophagy in cardiomyocytes. Redox Biol. 2020;36:101660.
- Périard JD, Eijsvogels TMH, Daanen HAM. Exercise under heat stress: thermoregulation, hydration, performance implications, and mitigation strategies. Physiol Rev. 2021;101(4):1873–1979.
- Epstein Y, Yanovich R. Heatstroke. N Engl J Med. 2019;380(25):2449–2459.
- Zhou G, Chen Z, Li J, et al. Role of the receptor for advanced glycation end products in heat Stress-Induced endothelial hyperpermeability in acute lung injury. Front Physiol. 2020;11:1087.
- Zhang X, Chen B, Wu J, et al. Aspirin enhances the protection of Hsp90 from Heat-Stressed injury in cardiac microvascular endothelial cells through PI3K-Akt and PKM2 pathways. Cells. 2020;9(1):243.
- Gu ZT, Wang H, Li L, et al. Heat stress induces apoptosis through transcription-independent p53-mediated mitochondrial pathways in human umbilical vein endothelial cell. Sci Rep. 2014;4:4469.
- Chen K-L, Wang H-L, Jiang L-Z, et al. Heat stress induces apoptosis through disruption of dynamic mitochondrial networks in dairy cow mammary epithelial cells. In Vitro Cell Dev Biol Anim. 2020;56(4):322–331.
- Ni X-X, Nie J, Xie Q-Y, et al. Protective effects of hyperbaric oxygen therapy on brain injury by regulating the phosphorylation of Drp1 through ROS/PKC pathway in heatstroke rats. Cell Mol Neurobiol. 2020;40(8):1253–1269.
- Song J, Herrmann JM, Becker T. Quality control of the mitochondrial proteome. Nat Rev Mol Cell Biol. 2021;22(1):54–70.
- Geldon S, Fernández-Vizarra E, Tokatlidis K. Redox-Mediated regulation of mitochondrial biogenesis, dynamics, and respiratory chain assembly in yeast and human cells. Front Cell Dev Biol. 2021;9:720656.
- Mao X, Gu Y, Sui X, et al. Phosphorylation of Dynamin-Related protein 1 (DRP1) regulates mitochondrial dynamics and skeletal muscle wasting in cancer cachexia. Front Cell Dev Biol. 2021;9:673618.
- Jin X, Xiang Z, Chen Y-P, et al. Uncoupling protein and nonalcoholic fatty liver disease. Chin Med J. 2013;126(16):3151–3155.
- Zhou Y, Cai T, Xu J, et al. UCP2 attenuates apoptosis of tubular epithelial cells in renal ischemia-reperfusion injury. Am J Physiol Renal Physiol. 2017;313(4):F926–F37.
- Fontana F, Limonta P. The multifaceted roles of mitochondria at the crossroads of cell life and death in cancer. Free Radic Biol Med. 2021;176:203–221.
- Koch RE, Buchanan KL, Casagrande S, et al. Integrating mitochondrial aerobic metabolism into ecology and evolution. Trends Ecol Evol. 2021;36(4):321–332.
- Kanamori T, Miyazaki N, Aoki S, et al. Investigation of energy metabolic dynamism in hyperthermia-resistant ovarian and uterine cancer cells under heat stress. Sci Rep. 2021;11(1):14726.
- Kummer E, Ban N. Mechanisms and regulation of protein synthesis in mitochondria. Nat Rev Mol Cell Biol. 2021;22(5):307–325.