
Abstract
Background
Radiofrequency ablation (RFA) is a first-line treatment for early-stage hepatocellular carcinoma (HCC). However, the recurrence after RFA remains an urgent challenge. Current studies have shown that residual tumor after RFA is an important cause of recurrence.
Objective
We hypothesized that the products of dead tumor cells after RFA have direct effects on the development of residual tumors. Further, we investigated the underlying mechanisms.
Methods
The proliferation and invasion ability of HepG2 and Huh7 cells were assessed using CCK-8, colony formation, EdU, transwell invasion and migration assay. Immunofluorescence and western blotting were used to show HMGB1 released from dead tumor cells. The levels of MMP2, MMP9, CyclinE1 and pERK1/2 were determined using western blotting. Finally, in vivo validation was performed in BALB/c nude mice xenograft tumor models.
Results
The products of dead tumor cells after thermal treatment can promote the proliferation and invasion of residual HCC cells. Dead tumor cells could release high-mobility group box 1 (HMGB1) after thermal treatment. Similar to the products of dead tumor cells, the recombinant protein of HMGB1 can promote the proliferation and invasion of residual HCC cells. Moreover, HMGB1 could bind to receptor of advanced glycation end-products. Then, it activated the ERK1/2 pathway and significantly upregulated the expressions of MMP2, MMP9, and CyclinE1.
Conclusion
Our study reveals that HMGB1 released by dead tumor cells after thermal treatment can promote the proliferation and invasion of residual HCC cells. Hence, the HMGB1/RAGE/ERK1/2 pathway is a potential target for improving the prognosis of HCC after radiofrequency ablation.
Introduction
Hepatocellular carcinoma (HCC) is one of the most common malignant tumors worldwide and is an important cause of cancer-related mortality [Citation1]. Currently, its main treatments include surgical resection, liver transplantation, local ablation, and radiotherapy [Citation2–4]. Radiofrequency ablation (RFA) is a first-line treatment for early-stage HCC. However, HCC recurrence after RFA remains an urgent challenge. Current studies showed that insufficient radiofrequency ablation (IRFA) resulted in the development of tumor residual disease and the progression of residual cancer.
Previous studies have shown that heat stimulation and hypoxic microenvironment after IRFA directly stimulated not only residual HCC cells but also other non-immune cells, such as endothelial and hepatic stellate cells [Citation5–8]. Finally, it promoted epithelial-mesenchymal transformation, autophagy, and cancer stem cells, which ultimately promoted the progression of residual HCC [Citation9–11]. In addition, dead tumor cells after IRFA released a large volume of cell debris and tumor-associated antigens, thereby recruiting and activating immune cells and causing complex immune responses [Citation12–14]. The components of tumor necrosis products after IRFA are complex. In addition to the recruitment of immune cells, their direct effects on residual hepatocellular carcinoma cells have not been evaluated.
If tumor cells are exposed to external stimuli, they may be damaged or can even die. Damaged or dead tumor cells then release or secrete damage-associated molecular patterns (DAMPs), which activate immune cells and cause a series of immune responses [Citation15–17]. High mobility group box 1 (HMGB1), a typical DAMP, has been reported to promote the proliferation of a variety of tumors [Citation18,Citation19]. HMGB1 is mainly located in the nucleus and is a highly conserved chromosomal protein that binds to DNA and plays an important role in maintaining the stability of chromosomes [Citation20]. If cells become damaged or if they die, HMGB1 is released outside of the cell. Some studies have revealed that HMGB1 released by dead colon and cervical cancer cells during radiotherapy stimulated the proliferation of residual tumor cells [Citation21]. However, the release of HMGB1 caused by dead tumor cells after IRFA and its effect on residual HCC cells have not been investigated.
Therefore, this study aimed to assess the proliferation and invasion of residual cancer cells after the release of HMGB1 by dead tumor cells after IRFA. Further, the associated mechanisms were investigated.
Material and methods
Cell culture and heat treatment
The human HCC cell lines Huh7 and HepG2 were cultivated in Dulbecco’s Modified Eagle Medium (Biological Industries, Israel) supplemented with 10% fetal bovine serum (Biological Industries, Israel) and 1% penicillin-streptomycin. Next, they were maintained in a humidified atmosphere with 5% CO2 at 37 °C. The thermal treatment in vitro was performed as follows: The cells were subjected to water baths at different temperatures for 10 min and were returned to the incubator for further use.
Conditioned medium preparation
The serum-free medium was replaced when the cell density reached 80%–90% and subjected to a water bath at 60 °C for 10 min. After thermal treatment, the cell culture supernatant was collected at different time intervals, centrifuged at 3000 rpm for 10 min, filtered with a 0.22-um filter, and stored at −80 °C for further use.
Drug treatment
Recombinant human HMGB1 (R&D Systems, the USA), ethyl pyruvate (Sigma, the USA), SCH772984 (GLPBIO, the USA), C29 (GLPBIO, the USA), FPS-ZM1 (APExBIO, the USA), and TAK-242 (APExBIO, the USA) were used.
Proliferation assay
The proliferation of HepG2 and Huh7 cells was assessed using CCK-8, colony formation, and EdU assays. CCK-8 and EdU assays were performed according to the manufacturer’s instructions to detect the proliferation ability of cells after different treatments. In the clone formation experiment, after different treatments, the cells were digested and re-seeded into plates (1000 cells per well), and the medium was changed every 3 days. Next, the cells were fixed and stained after 7–10 days.
Cell migration and invasion assay
The invasion and metastasis ability of HepG2 and Huh7 cells were evaluated using transwell invasion and migration assay. After different treatments, the cells were digested and resuspended with serum-free medium. Then, 5*104 cells were seeded into the transwell chambers. After 24 and 48 h, the chambers were fixed, stained, and finally photographed.
Immunofluorescence analyses
An immunofluorescence assay was used to label the distribution of HMGB1. The cells treated at 37 °C and 60 °C were fixed, perforated, and blocked using conventional methods. Then, the cells were incubated with HMGB1 antibodies (Servicebio, 1:500) overnight, followed by fluorescent antibody AF488 (Abcam, 1:2000) the next day. Finally, the cells were stained with 4′, 6-diamidino-2′-phenylindole (DAPI) and photographed.
Western blot analysis
Western blot analysis was performed using conventional methods. MMP2 (Proteintech, 1:1000), MMP9 (ImmunoWay, 1:1000), cyclin D1 (Signalway Antibody, 1:1000), cyclin E1 (Signalway Antibody, 1:1000), ERK (ImmunoWay, 1:1000), p-ERK (ImmunoWay, 1:1000), Akt (ImmunoWay, 1:1000), p-Akt (ImmunoWay, 1:1000), GAPDH (Proteintech, 1:5000) were used.
Xenograft tumor models
The animal procedures were approved by the Animal Care and Use Committee of Sun Yat-sen University (No. SYSU-IACUC-2022-B0896). BALB/c nude mice aged 4–5 weeks received 5 × 106 HepG2 cells in the armpit. Based on previous experiments [Citation22], When the volume of tumor xenograft reached 500 mm3, the mice were anesthetized with isoflurane (RWD Life Science Co., China). The radiofrequency needle was pierced into a non-central location of the tumor at 3 Watts for 30 s (90 Joules) to simulate clinical IRFA. FPS-ZM1 was administered on the day and after the first day of ablation and every other day for 2 weeks. The size of the tumor and the weight of the mice were measured during each drug treatment.
Statistical analysis
Statistical data were expressed as mean ± standard deviation and were analyzed using the Statistical Package for the Social Sciences software version 25.0. The t-test was used to compare between-group differences. A p-value of < 0.05 was considered statistically significant. GraphPad Prism v8.0 was used to draw the charts.
Results
Products of dead tumor cells after IRFA promoted the proliferation and invasion of residual HCC cells
To validate the simulation conditions of complete radiofrequency ablation, CCK-8 assay was used to assess the relative cell viability of tumor cells 24 h after thermal treatment in water baths at different temperatures (37 °C, 40 °C, 42 °C, 45 °C, 48 °C, 51 °C, and 60 °C) for 10 min. Results showed that the survival rate of tumor cells gradually decreased with increased temperature (Figure S1(A,B)). According to previous studies [Citation7,Citation23], thermal treatment with a 48 °C water bath for 10 min was selected for insufficient ablation to mimic residual tumor cells and with a 60 °C water bath for 10 min for complete ablation to simulate the complete ablation area in the ablation center.
To determine the effect of products from dead tumor cells on the proliferation of residual HCC cells after IRFA, HepG2 and Huh7 cells after thermal treatment at 48 °C were cultured with control medium (CM) and supernatant of dead cells (SDC), respectively. Cell proliferation ability was evaluated using CCK-8, clonal formation, and EdU assay. The OD values (p < 0.01; p < 0.01), colony-forming efficiency (p < 0.01; p < 0.05), and cell replication activity (p < 0.01; p < 0.01) of HepG2 and Huh7 cells in the SDC group were significantly higher than those in the CM group (). Hence, tumor cell necrosis products significantly enhanced the proliferation of residual HCC cells.
Figure 1. Products of dead tumor cells after IRFA promoted the proliferation and invasion of residual HCC cells. (A–C) Proliferation of HepG2 and Huh7 cells cultured with control medium (CM) or supernatant of dead cells (SDCs) were analyzed using the CCK-8 assay (A), clonal formation assay (B), and EdU assay (C), Scale bars, 100 µm. (D,E) Migration and invasion capability of HepG2 (D) and Huh7 (E) cells cultured with CM or SDC were analyzed using transwell migration and invasion assay. Scale bars, 50 µm, *p < 0.05, **p < 0.01, ***p < 0.001.
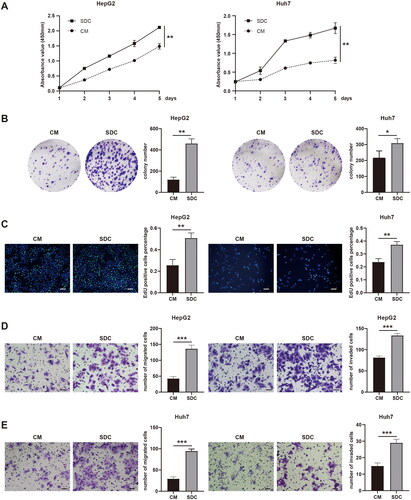
In addition, the effect of products from dead tumor cells on the invasion ability of residual HCC cells was continually explored. After the cells were cultured in the control medium and supernatant of dead cells, a transwell assay was performed to evaluate the migration and invasion abilities of HepG2 and Huh7 cells. Quantitative analysis of images showed that the migration and invasion rate of HepG2 and Huh7 cells with necrotic supernatant was significantly higher than that with control medium (p < 0.001, p < 0.001, p < 0.001, and p < 0.01) (). The abovementioned experimental results showed that tumor cell necrosis products could significantly enhance the proliferation and invasion ability of residual HCC cells.
HMGB1 was released from dead tumor cells after thermal treatment
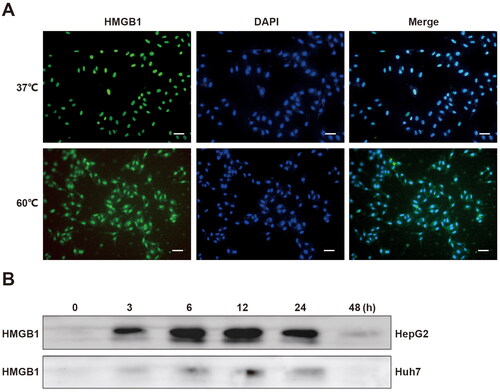
If tumor cells are exposed to certain stimuli, they become damaged or even die and subsequently release or secrete DAMPs [Citation17]. Previous studies have shown that DAMPs activated immune cells, thereby leading to a series of immune reactions [Citation15,Citation16]. However, recent studies have revealed that dead tumor cells during radiotherapy released HMGB1, a typical DAMPs, and promoted the proliferation of residual tumor cells [Citation21]. Therefore, the necrotic tumor cells after thermal treatment might also release HMGB1. Then, immunofluorescence experiments were performed on normal cultured and thermal-treated tumor cells to assess whether there were changes in the distribution of HMGB1. We found that in normal tumor cells, HMGB1 was mainly distributed in the nucleus. However, after thermal treatment, some HMGB1 translocated from the nucleus to the cytoplasm (). Subsequently, we collected the supernatant of necrotic cells at different times after thermal treatment. Via western blot analysis, we found that HMGB1 existed in the supernatant of necrotic tumor cells after thermal treatment (). Therefore, HMGB1 could be released from dead tumor cells after thermal treatment.
Figure 2. HMGB1 was released from dead tumor cells after thermal treatment. (A) Immunofluorescence of HMGB1 was performed on HepG2 cells treated in a water bath at 60 °C or 37 °C, Scale bars, 50 µm. (B) Western blot analyses were performed on HMGB1 in the supernatant of HepG2 and Huh7 cells at 0, 3, 6, 12, 24, and 48 h after thermal treatment at 60 °C.
HMGB1 from dead tumor cells promoted the proliferation and invasion of residual tumor cells
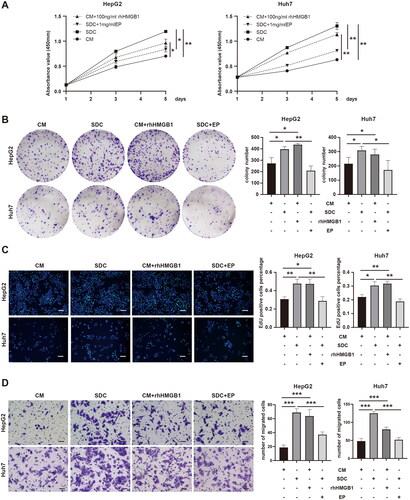
After confirming the release of HMGB1 from dead tumor cells after thermal treatment, whether HMGB1 derived from dead tumor cells could enhance the proliferation and invasion ability of residual tumor cells were continually assessed using recombinant human HMGB1 protein (rhHMGB1) and its inhibitor ethyl pyruvate (EP). After determining the appropriate drug concentration (Figure S2(A,D)), rhHMGB1 was added into the control medium (the CM + rhHMGB1 group), and ethyl pyruvate was added to the supernatant (the SDC + EP group). Then, CCK-8, clonal formation, EdU, transwell migration, and invasion assays were used to evaluate cell proliferation and invasion ability. The OD values (p < 0.05, p < 0.001), colony-forming efficiency (p < 0.05, p < 0.05), and cell replication activity (p < 0.05, p < 0.01) of HepG2 and Huh7 cells in the CM + rhHMGB1 group were significantly higher than those in the CM group (). The OD values (p < 0.05, p < 0.01), colony-forming efficiency (p < 0.01, p < 0.05), and cell replication activity (p < 0.01, p < 0.01) of HepG2 and Huh7 cells in the SDC + EP group were significantly lower than those in the SDC group (). Similar results were observed in transwell migration and invasion experiments ( and S2(E)). Based on the abovementioned experimental results, HMGB1 derived from dead tumor cells after thermal treatment can promote the proliferation and invasion of residual tumor cells.
Figure 3. HMGB1 from dead tumor cells promoted the proliferation and invasion of residual tumor cells. (A–C) The proliferation of HepG2 and Huh7 cells cultured with CM, SDC, CM + 100 ng/mL rhHMGB1, or SDC + 1 mg/mL EP was analyzed with CCK-8 assay (A), clonal formation assay (B), and EdU assay (C), Scale bars, 100 µm. (D) The migration capability of HepG2 and Huh7 cells cultured with CM, SDC, CM + 100 ng/mL rhHMGB1, or SDC + 1 mg/mL EP was analyzed using transwell migration and invasion assay, Scale bars, 50 µm, *p < 0.05, **p < 0.01, ***p < 0.001.
HMGB1 promoted the proliferation and invasion of residual tumor cells by activating the ERK1/2 pathway
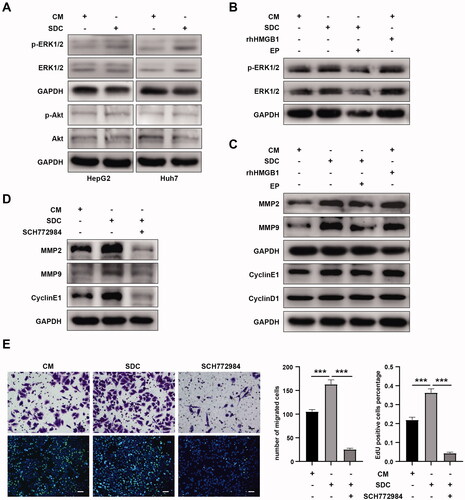
We further explored how HMGB1 promoted the proliferation and invasion of residual tumor cells. Results showed that the supernatant of dead tumor cells activated the ERK1/2 signaling pathway, but not the Akt signaling pathway (). Similar to the supernatant of dead tumor cells, rhHMGB1 could also activate the ERK1/2 signaling pathway in residual HCC cells. Meanwhile, EP inhibited the activation of the ERK1/2 signaling pathway by the supernatant of dead tumor cells (). Based on the abovementioned experimental results, HMGB1 from dead tumor cells after IRFA activated the ERK1/2 signaling pathway in residual HCC cells. In addition, the supernatant of dead cells promoted the expression of MMP2, MMP9, and cyclin E1 in residual HCC cells (). Analogously, rhHMGB1 also upregulated the expression of MMP2, MMP9, and CyclinE1. Meanwhile, EP inhibited the expression of MMP2, MMP9, and cyclin E1 in residual HCC cells upregulated by the supernatant of dead tumor cells (). Therefore, HMGB1 in the supernatant of dead tumor cells after IRFA upregulated the expression of MMP2, MMP9, and cyclin E1 in residual HCC cells. To validate the association between the activation of the ERK1/2 pathway and the upregulation of MMP2, MMP9, and cyclin E1 expression, residual HCC cells were treated with p-ERK1/2 inhibitor SCH772984. We found that if ERK1/2 phosphorylation was inhibited (Figure S1(C)), the supernatant of dead tumor cells could not upregulate the expression of MMP2, MMP9, and cyclin E1 in residual HCC cells () nor could it enhance the proliferation and migration ability of residual HCC cells (). Based on the abovementioned experimental results, HMGB1 from dead tumor cells after IRFA upregulated the expression of MMP2, MMP9, and cyclin E1 by activating the ERK1/2 signaling pathway and promoted the proliferation and migration of residual HCC cells.
Figure 4. HMGB1 promoted the proliferation and invasion of residual tumor cells by activating the ERK1/2 pathway. (A) Western blot analyses of p-ERK1/2, ERK1/2, p-Akt, and Akt in HepG2 and Huh7 cells cultured with CM or SDC were performed. (B) Western blot analyses of p-ERK1/2 and ERK1/2 in HepG2 cells cultured with CM, SDC, CM + 100 ng/mL rhHMGB1, or SDC + 1 mg/mL EP were performed. (C) Western blot analyses of MMP2, MMP9, cyclin E1, and cyclin D1 in HepG2 cells cultured with CM, SDC, CM + 100 ng/mL rhHMGB1, or SDC + 1 mg/mL EP were performed. (D) Western blot analyses of MMP2, MMP9, and cyclin E1 in HepG2 cells cultured with CM, SDC, or SDC + SCH772984 (inhibitors of p-ERK) were performed. (E) Migration capability and proliferation of HepG2 cells cultured with CM, SDC, and SDC + SCH772984 (inhibitors of p-ERK) were analyzed using EdU and transwell migration assay. *p < 0.05, **p < 0.01, ***p < 0.001.
HMGB1 activated the ERK1/2 pathway via the receptor for advanced glycation end products
According to previous studies, the common receptors of HMGB1 include the receptor for advanced glycation end product (RAGE), TLR2, and TLR4. Therefore, we continually explored the mechanism through which receptor HMGB1 activated the ERK1/2 signaling pathway to promote the proliferation and invasion of residual HCC cells. The FPS-ZM1 (RGAE inhibitor), C29 (TLR2 inhibitor), and TAK-242 (TLR4 inhibitor) were used to initially screen the receptors of HMGB1 using EdU and transwell migration assay at different concentrations. With increased concentration of FPS-ZM1, the ability of the supernatant to promote the proliferation and invasion of residual HCC cells gradually decreased (p < 0.01, p < 0.001, p < 0.001, and p < 0.001), but not C29 and TAK-242 (). Therefore, RAGE was the most likely receptor for HMGB1. To further validate this finding, we supplemented the EdU, CCK-8, transwell migration, and invasion assay. All the abovementioned experiments had similar results. The FPS-ZM1 inhibited the ability of supernatant to promote the proliferation and invasion of residual HCC cells (, p < 0.001, p < 0.01, p < 0.001, and p < 0.001). Moreover, FPS-ZM1 inhibited the ERK1/2 signaling pathway activated by supernatant in residual HCC cells (Figure S1(D)). Therefore, HMGB1 from the dead tumor cells after IRFA promoted the proliferation and migration of residual HCC cells by activating the ERK1/2 signaling pathway via RAGE.
Figure 5. HMGB1 activated the ERK1/2 pathway via the receptor for advanced glycation end products (RAGE). (A) Proliferation of HepG2 cells cultured with SDC, SDC + FPS-ZM1 (1 µM, 5 µM, 10 µM; inhibitors of RAGE), SDC + C29 (10 µM, 2 µM, 50 µM; inhibitors of TLR2), or SDC + TAK-242 (1 µM, 5 µM; inhibitors of TLR4E) were analyzed using EdU assay, Scale bars, 100 µm. (B) Migration ability of HepG2 cells cultured with SDC, SDC + FPS-ZM1 (1 µM, 5 µM, 10 µM; inhibitors of RAGE), SDC + c29 (10 µM, 2 µM, 50 µM; inhibitors of TLR2), or SDC + TAK-242 (1 µM, 5 µM; inhibitors of TLR4E) was analyzed using transwell migration assay, Scale bars, 50 µm. (C) Proliferation of HepG2 cells cultured with SDC or SDC + 5 µM FPS-ZM1 was analyzed using EdU assay, Scale bars, 100 µm. (D) Proliferation of HepG2 cells cultured with SDC or SDC + 5 µM FPS-ZM1 was analyzed using CCK-8 assay. (E) Migration capability of HepG2 cells cultured with SDC or SDC + 5 µM FPS-ZM1 was analyzed using a transwell migration assay, Scale bars, 50 µm. (F) Invasion ability of HepG2 cells cultured with SDC or SDC + 5 µM FPS-ZM1 was analyzed using a transwell invasion assay, Scale bars, 50 µm. *p < 0.05, **p < 0.01, ***p < 0.001, n.s.: not significant.
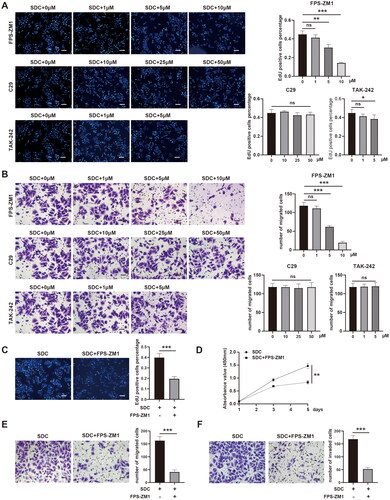
FPS-ZM1 treatment after IRFA inhibited the progression of residual tumor
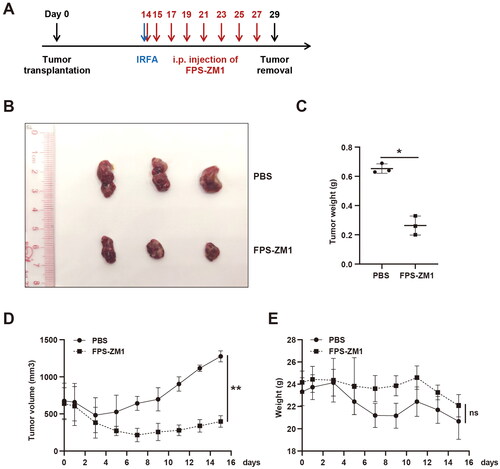
HMGB1 from dead tumor cells promoted the proliferation of residual HCC cells by binding to RAGE. Hence, we explored if interfering with HMGB1/RAGE could slow the progression of residual cancer after IRFA. Therefore, a subcutaneous xenograft tumor mice model was established, and insufficient radiofrequency ablation was performed after the tumor reached 500 mm3. FPS-ZM1 was administered on the day and one day after ablation and every other day for 2 weeks (). The size of the tumor and the weight of the mice were evaluated during each drug treatment. Finally, the tumor was resected and weighed. Results showed that FPS-ZM1 treatment significantly slowed the growth rate of residual cancer after IRFA, and it had minimal effect on the body weight of mice ().
Figure 6. FPS-ZM1 treatment after IRFA inhibited the progression of residual cancer. (A) Design of animal experiments. The blue arrow indicates IRFA, and the red arrows represent the time of FPS-ZM1 treatments. (B) Representative tumor image of the PBS and FPS-ZM1 groups, n = 3. (C). Tumor weight of the PBS and FPS-ZM1 group, n = 3. (D). Tumor growth curve of the PBS and FPS-ZM1 group, n = 3. (E). Body weight of BALB/c nude mice treated with PBS or FPS-ZM1, n = 3; *p < 0.05, **p < 0.01, ***p < 0.001, n.s.: not significant.
Discussion
RFA is a first-line treatment for early- and very-early-stage HCC. However, the recurrence rate of HCC after RFA is a clinical challenge that cannot be ignored. Previous studies have shown that the 5-year local tumor recurrence rate of HCC after RFA is as high as 20.4% [Citation24]. And the residual tumor is the most important risk factor for HCC recurrence after IRFA.
Dead tumor cells after IRFA produced a large volume of cell debris and tumor-associated antigens. Current studies have focused on the effects of dead tumor cell products on immune cells after IRFA. The products of dead tumor cells recruited and activated immune cells, which caused complex immune responses [Citation12–14,Citation25,Citation26]. However, whether the products of dead tumor cells have a direct effect on residual HCC cells after IRFA has not been investigated yet. Recent studies have shown that products released by dead pancreatic cancer cells after radiotherapy promoted the invasion of residual pancreatic cancer cells [Citation27]. Hence, they may affect residual HCC cells, thereby promoting residual tumor progression.
The current study found that the supernatant of dead tumor cells could significantly enhance the proliferation and invasion abilities of residual HCC cells. Subsequently, we found that HMGB1 was released from dead tumor cells after thermal treatment and that HMGB1 from dead tumor cells could promote the proliferation and invasion abilities of residual HCC cells. Furthermore, HMGB1 derived from dead tumor cells activated the ERK1/2 signaling pathway via RAGE, upregulated the expression of MMP2, MMP9, and CyclinE1, and promoted the proliferation and migration of residual HCC cells.
HMGB1, a DNA chaperone, is mainly located in the nucleus where it binds to DNA and plays an important role in maintaining the stability of the chromosome nucleus [Citation7,Citation20,Citation23]. Previous studies have shown that HMGB1 can be released extracellularly from injured or necrotic cells or actively secreted by immune cells to mediate immune responses during injury [Citation18,Citation28–30]. In addition, exocrine HMGB1 can bind to the surface receptors of tumor cells and promote cell proliferation and invasion. Previous studies have shown that HMGB1 released from dead colon and cervical cancer cells during radiotherapy could stimulate the proliferation of residual tumor cells [Citation21]. This is consistent with the findings of our study. Moreover, other studies have shown that HMGB1 can translocate from the nucleus to the cytoplasm and bind to mitochondrial DNA in the cytoplasm to promote the proliferation of tumor cells [Citation31]. Therefore, whether there is any change in the distribution of HMGB1 in residual HCC cells after IRFA and what factors cause the ectopic HMGB1 must be further investigated. HMGB1 may affect residual cancer progression in several ways after IRFA.
RAGE and TLRs are classical receptors of HMGB1, which are widely expressed in the cell membrane. They can bind to extracellular HMGB1 and mediate immune responses [Citation27,Citation30,Citation32,Citation33]. Previous studies have shown that HMGB1 binding to RAGE in the mitochondria of pancreatic cancer cells enhances the activity of mitochondrial complex I via the MEK/ERK signaling pathway, increase ATP production, and finally promote the growth of pancreatic tumor [Citation34]. This study showed that HMGB1 derived from dead tumor cells after IRFA promoted the proliferation and invasion of residual cancer cells via RAGE, but not TLRs. Moreover, animal experiments have revealed that the use of RAGE inhibitors after IRFA significantly inhibited the progression of residual tumors. This finding suggests that a novel combination therapy strategy can be used.
The MEK/ERK signaling pathway is a classical pathway correlated with tumor proliferation and invasion. Based on HCC studies, the MEK/ERK signaling pathway was correlated with stemness characteristics, EMT changes, and neovascularization of HCC cells, and it promoted tumor growth, invasion, and metastasis in HCC [Citation35–38]. In addition, in a study on the mechanism of HCC progression after IRFA, the activation of the MEK/ERK signaling pathway was closely correlated with the progression of residual HCC [Citation39–42]. In this research, dead tumor cell-derived HMGB1 significantly activated ERK1/2 signaling via RAGE, and ERK1/2 activation was closely correlated with residual cancer progression. This finding was consistent with previous findings, and it further confirmed the upstream mechanism of the ERK1/2 signaling pathway.
The current study had several limitations. That is, only the effect of HMGB1 in the supernatant of dead tumor cells was validated. The components in the supernatant of dead tumor cells after IRFA are complex. In addition to the fact that HMGB1 promotes the progression of residual cancer, other components can also highly likely to promote the progression of residual cancer, which must be further evaluated. In addition, only cell and animal experiments were performed in this study. It was challenging to obtain clinical samples in the short term after ablation. Hence, clinical sample validation was not performed.
In conclusion, HMGB1 was released from dead tumor cells after IRFA, and HMGB1 activated the ERK1/2 signaling pathway via RAGE, upregulated the expression of MMP2, MMP9, and CyclinE1, and ultimately promoted the progression of residual cancer. Therefore, targeting the HMGB1/RAGE/ERK signaling pathway might be a novel therapeutic strategy after IRFA.
Supplemental Material
Download PDF (2.6 MB)Acknowledgments
The authors acknowledged the Sun Yat-sen Memorial Hospital of Sun Yat-sen University for technical assistance.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249.
- Chen LT, Martinelli E, Cheng AL, et al. Pan-Asian adapted ESMO clinical practice guidelines for the management of patients with intermediate and advanced/relapsed hepatocellular carcinoma: a TOS-ESMO initiative endorsed by CSCO, ISMPO, JSMO, KSMO, MOS and SSO. Ann Oncol. 2020;31(3):334–351.
- Vogel A, Cervantes A, Chau I, et al. Hepatocellular carcinoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2019;30(5):871–873.
- EASL clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2018;69(1):182–236.
- Kong J, Yao C, Dong S, et al. ICAM-1 activates platelets and promotes endothelial permeability through VE-Cadherin after insufficient radiofrequency ablation. Adv Sci. 2021;8(4):2002228.
- Zhang R, Lin XH, Ma M, et al. Periostin involved in the activated hepatic stellate cells-induced progression of residual hepatocellular carcinoma after sublethal heat treatment: its role and potential for therapeutic inhibition. J Transl Med. 2018;16(1):302.
- Su T, Liao J, Dai Z, et al. Stress-induced phosphoprotein 1 mediates hepatocellular carcinoma metastasis after insufficient radiofrequency ablation. Oncogene. 2018;37(26):3514–3527.
- Tong Y, Yang H, Xu X, et al. Effect of a hypoxic microenvironment after radiofrequency ablation on residual hepatocellular cell migration and invasion. Cancer Sci. 2017;108(4):753–762.
- Zhao Z, Wu J, Liu X, et al. Insufficient radiofrequency ablation promotes proliferation of residual hepatocellular carcinoma via autophagy. Cancer Lett. 2018;421:73–81.
- Iwahashi S, Shimada M, Utsunomiya T, et al. Epithelial-mesenchymal transition-related genes are linked to aggressive local recurrence of hepatocellular carcinoma after radiofrequency ablation. Cancer Lett. 2016;375(1):47–50.
- Chen Y, Bei J, Liu M, et al. Sublethal heat stress-induced O-GlcNAcylation coordinates the warburg effect to promote hepatocellular carcinoma recurrence and metastasis after thermal ablation. Cancer Lett. 2021;518:23–34.
- Mizukoshi E, Yamashita T, Arai K, et al. Enhancement of tumor-associated antigen-specific T cell responses by radiofrequency ablation of hepatocellular carcinoma. Hepatology. 2013;57(4):1448–1457.
- Zerbini A, Pilli M, Laccabue D, et al. Radiofrequency thermal ablation for hepatocellular carcinoma stimulates autologous NK-cell response. Gastroenterology. 2010;138(5):1931–1942.
- Hiroishi K, Eguchi J, Baba T, et al. Strong CD8(+) T-cell responses against tumor-associated antigens prolong the recurrence-free interval after tumor treatment in patients with hepatocellular carcinoma. J Gastroenterol. 2010;45(4):451–458.
- Krombach J, Hennel R, Brix N, et al. Priming anti-tumor immunity by radiotherapy: dying tumor cell-derived DAMPs trigger endothelial cell activation and recruitment of myeloid cells. Oncoimmunology. 2019;8(1):e1523097.
- Garg AD, Galluzzi L, Apetoh L, et al. Molecular and translational classifications of DAMPs in immunogenic cell death. Front Immunol. 2015;6:588.
- Krysko DV, Garg AD, Kaczmarek A, et al. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12(12):860–875.
- Wang S, Zhang Y. HMGB1 in inflammation and cancer. J Hematol Oncol. 2020;13(1):116.
- Pu Z, Duda DG, Zhu Y, et al. VCP interaction with HMGB1 promotes hepatocellular carcinoma progression by activating the PI3K/AKT/mTOR pathway. J Transl Med. 2022;20(1):212.
- Kang R, Zhang Q, Zeh HJ, et al. HMGB1 in cancer: good, bad, or both? Clin Cancer Res. 2013;19(15):4046–4057.
- He S, Cheng J, Sun L, et al. HMGB1 released by irradiated tumor cells promotes living tumor cell proliferation via paracrine effect. Cell Death Dis. 2018;9(6):648.
- Liu X, Zhang W, Xu Y, et al. Targeting PI3Kγ/AKT pathway remodels LC3-Associated phagocytosis induced immunosuppression after radiofrequency ablation. Adv Sci. 2022;9(7):e2102182.
- Tan L, Chen S, Wei G, et al. Sublethal heat treatment of hepatocellular carcinoma promotes intrahepatic metastasis and stemness in a VEGFR1-dependent manner. Cancer Lett. 2019;460:29–40.
- Lee MW, Kang D, Lim HK, et al. Updated 10-year outcomes of percutaneous radiofrequency ablation as first-line therapy for single hepatocellular carcinoma < 3 cm: emphasis on association of local tumor progression and overall survival. Eur Radiol. 2020;30(4):2391–2400.
- Shi L, Wang J, Ding N, et al. Inflammation induced by incomplete radiofrequency ablation accelerates tumor progression and hinders PD-1 immunotherapy. Nat Commun. 2019;10(1):5421.
- Shi L, Chen L, Wu C, et al. PD-1 blockade boosts radiofrequency ablation-elicited adaptive immune responses against tumor. Clin Cancer Res. 2016;22(5):1173–1184.
- Chen X, Zhang L, Jiang Y, et al. Radiotherapy-induced cell death activates paracrine HMGB1-TLR2 signaling and accelerates pancreatic carcinoma metastasis. J Exp Clin Cancer Res. 2018;37(1):77.
- Rivera Vargas T, Apetoh L. Danger signals: chemotherapy enhancers? Immunol Rev. 2017;280(1):175–193.
- Pistoia V, Pezzolo A. Involvement of HMGB1 in resistance to tumor vessel-targeted, monoclonal antibody-based immunotherapy. J Immunol Res. 2016;2016:3142365.
- Sims GP, Rowe DC, Rietdijk ST, et al. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367–388.
- Liu Y, Yan W, Tohme S, et al. Hypoxia induced HMGB1 and mitochondrial DNA interactions mediate tumor growth in hepatocellular carcinoma through toll-like receptor 9. J Hepatol. 2015;63(1):114–121.
- Liu Y, Yin H, Zhao M, et al. TLR2 and TLR4 in autoimmune diseases: a comprehensive review. Clin Rev Allergy Immunol. 2014;47(2):136–147.
- Hudson BI, Lippman ME. Targeting RAGE signaling in inflammatory disease. Annu Rev Med. 2018;69:349–364.
- Kang R, Tang D, Schapiro NE, et al. The HMGB1/RAGE inflammatory pathway promotes pancreatic tumor growth by regulating mitochondrial bioenergetics. Oncogene. 2014;33(5):567–577.
- Chen J, Ding C, Chen Y, et al. ACSL4 promotes hepatocellular carcinoma progression via c-Myc stability mediated by ERK/FBW7/c-Myc axis. Oncogenesis. 2020;9(4):42.
- Chen X, Zhang S, Wang Z, et al. Supervillin promotes epithelial-mesenchymal transition and metastasis of hepatocellular carcinoma in hypoxia via activation of the RhoA/ROCK-ERK/p38 pathway. J Exp Clin Cancer Res. 2018;37(1):128.
- Chan LH, Zhou L, Ng KY, et al. PRMT6 regulates RAS/RAF binding and MEK/ERK-mediated cancer stemness activities in hepatocellular carcinoma through CRAF methylation. Cell Rep. 2018;25(3):690–701.e698.
- Liu L, Cao Y, Chen C, et al. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006;66(24):11851–11858.
- Liu Z, Dai H, Jia G, et al. Insufficient radiofrequency ablation promotes human hepatoma SMMC7721 cell proliferation by stimulating vascular endothelial growth factor overexpression. Oncol Lett. 2015;9(4):1893–1896.
- Dong S, Kong J, Kong F, et al. Insufficient radiofrequency ablation promotes epithelial-mesenchymal transition of hepatocellular carcinoma cells through akt and ERK signaling pathways. J Transl Med. 2013;11:273.
- Kong J, Kong L, Kong J, et al. After insufficient radiofrequency ablation, tumor-associated endothelial cells exhibit enhanced angiogenesis and promote invasiveness of residual hepatocellular carcinoma. J Transl Med. 2012;10(1):230.
- Yoshida S, Kornek M, Ikenaga N, et al. Sublethal heat treatment promotes epithelial-mesenchymal transition and enhances the malignant potential of hepatocellular carcinoma. Hepatology. 2013;58(5):1667–1680.