
Abstract
Purpose
c-Jun N-terminal kinases (JNKs) comprise a subfamily of mitogen-activated protein kinases (MAPKs). The JNK group is known to be activated by a variety of stimuli. However, the molecular mechanism underlying heat-induced JNK activation is largely unknown. The aim of this study was to clarify how JNK activity is stimulated by heat.
Methods and materials
The expression levels of various MAPK members in HeLa cells, with or without hyperthermia treatment, were evaluated via western blotting. The kinase activity of MAPK members was assessed through in vitro kinase assays. Cell death was assessed in the absence or presence of siRNAs targeting MAPK-related members.
Results
Hyperthermia decreased the levels of MAP3Ks, such as ASK1 and MLK3 which are JNK kinase kinase members, but not those of the downstream MAP2K/SEK1 and MAPK/JNK. Despite the reduced or transient phosphorylation of ASK1, MLK3, or SEK1, downstream JNK was phosphorylated in a temperature-dependent manner. In vitro kinase assays demonstrated that heat did not directly stimulate SEK1 or JNK. However, the expression levels of DUSP16, a JNK phosphatase, were decreased upon hyperthermia treatment. DUSP16 knockdown enhanced the heat-induced activation of ASK1–SEK1–JNK pathway and apoptosis.
Conclusion
JNK was activated in a temperature-dependent manner despite reduced or transient phosphorylation of the upstream MAP3K and MAP2K. Hyperthermia-induced degradation of DUSP16 may induce activation of the ASK1–SEK1–JNK pathway and subsequent apoptosis.
Introduction
Hyperthermia is a well-known method for cancer treatment. Hyperthermia increases the cell temperature and induces many biochemical changes, such as the generation of reactive oxygen species, an increased intracellular calcium ion concentration [Citation1,Citation2]. Heat-induced protein denaturation and aggregation are the key events in the disruption of cellular homeostasis [Citation3,Citation4]. Intracellular protein degradation is regulated by multiple proteolytic pathways, including lysosome-, calcium-, and proteasome-dependent mechanisms [Citation5,Citation6]. Hyperthermia induces the proteasomal degradation of cellular signaling proteins, anti-apoptotic regulators and DNA repair proteins [Citation7,Citation8]. However, the molecular mechanisms underlying thermal cellular signaling and their roles in thermal killing are largely unknown.
Mitogen-activated protein kinase (MAPK) signaling pathways mediate cellular responses to various extracellular stimuli, including growth factors and environment stresses [Citation9]. The MAPK pathway features a three-kinase cascade, including MAPKs (e.g., ERK, p38, and JNK), MAPK kinases (MAP2Ks; e.g., MEK and MKK), and MAPKK kinases (MAP3Ks; e.g., ASK1, MLK3, and TAK1). MAP3Ks phosphorylate and activate MAP2Ks, which in turn phosphorylate MAPKs. Increasing evidence from biochemical and genetic analyses suggests that MAP3Ks link various extracellular stimuli to cytoplasmic and nuclear effectors by activating downstream MAPK pathways [Citation10]. Twenty-one MAP3Ks have been shown to activate the known MAP2Ks. The ERK cascade is mostly activated by agonists of tyrosine kinase-encoded receptors and G protein-coupled receptors that induce mitogenesis or differentiation [Citation10,Citation11], whereas the JNK and p38 cascades are strongly activated by proinflammatory cytokines or extracellular stresses [Citation12,Citation13].
The MAP3Ks of the JNK pathway include Ste20/Ste11/Ste7 (STE) family kinases such as Apoptosis Signal-regulating Kinase 1 (ASK1), and members of the tyrosine kinase-like (TLK) family such as Mixed Lineage Kinase (MLKs), and TGFβ-Activated Kinase-1 (TAK1) [Citation14]. ASK-1, functions, as its name indicates, for regulating apoptosis downstream from stress stimuli [Citation15]. MLK3 induces not only activation of JNK but negatively regulates activation of ERK [Citation16]. TAK1 was originally found to function in transforming growth factor-β (TGF-β)-mediated MAPK activation [Citation17]. However, the mechanism of how JNK is activated by heat is largely unknown.
We previously reported that hyperthermia decreased the levels of MAP3Ks such as TAK1, RAF1, and MEKK2, without reduction of the downstream components in the ERK cascades [Citation7]. In the present study, we evaluated the effect of hyperthermia on the expression of MAPK family members consisting of JNK signaling cascade, including ASK1, MLK3, SEK1, and JNK. Based on our findings, we further investigated the molecular mechanisms underlying the hyperthermia-induced JNK phosphorylation and their impact on cell death.
Materials and methods
Cell culture and stimulation
Human cervix epithelial carcinoma cell line HeLa was purchased from the Japanese Collection of Research Bioresources Cell Bank (Ibaraki, Osaka). These cells were cultured in Dulbecco’s modified Eagle’s medium/F-12 (1:1) (Sigma, St. Louis, MO) supplemented with 10% fetal bovine serum (Hyclone, South Logan) and 1% penicillin/streptomycin. Human lung giant cell carcinoma cell line LU99 cells was cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Hyperthermic treatment was carried out at 41–43 °C for 60 min by inserting a catheter equipped with an 8-MHz capacitive heating device (Thermotron RF-8; Yamamoto Vinita Co., Ltd., Osaka) into the culture flask with an average power of 4.3–5.1 W. The indicated temperature was maintained with a precision of ± 0.1 °C in the flask using a micro thermo probe (IT-18; Physitemp, Clifton, NJ). HeLa cells were pretreated with a potent proteasome inhibitor MG132 (Sigma) or a potent calpain inhibitor ALLN (Sigma) at 5–10 µM for 60 min and then subjected to hyperthermia for the indicated durations. To assay cell death or apoptosis, the cells were left untreated or heated at 43 °C for 60 min, and assayed 24 h later by staining with either trypan blue or using a Mitochondrial Transmembrane Potential Apoptosis Detection Kit (Abcam, Cambridge), respectively. Mitochondrial transmembrane potential was determined by counting mitochondrial capture reagent (red) positive cells.
Western blot analysis
Equivalent amounts of total cell lysates were separated by SDS-PAGE and then transferred onto Immobilon PVDF membranes (Millipore, Bedford, MA). The membranes were blocked in Tris-buffered saline containing 0.05% Tween 20 (TBS-T) and 2% nonfat dry milk or Blocking One-P (Nacalai Tesque, Kyoto) for phosphoprotein detection. The blots were then incubated at 1:1000 dilution overnight at 4 °C with one of the following antibodies: anti-ASK1 (Santa Cruz Biotechnology, Santa Cruz, CA), anti-phospho-ASK1(Gene Tex, Irvine, CA), anti-MLK3 (Abcam, Cambridge), anti-phospho-MLK3 (Gene Tex), anti-SEK1/MKK4 (Cell Signaling Technology, Beverly, MA), anti-phospho-SEK1 (Cell Signaling Technology), anti-JNK (Cell Signaling Technology), anti-phospho-JNK (Cell Signaling Technology), anti-DUSP10 (Santa Cruz Biotechnology), anti-DUSP16 (Proteintech, Rosemont, IL), anti-CDK2 (Santa Cruz Biotechnology), anti-GAPDH (MBL), anti-cleaved Caspase-3 (Proteintech), anti-HSP90 (Cell Signaling Technology), or anti-β-actin (Sigma). The blots were washed three times with TBS-T and incubated with secondary peroxidase-conjugated antibodies (Dako, Glostrup). The resulting signals were detected on X-ray films (GE Healthcare, Buckinghamshire) using an enhanced chemiluminescence detection system (GE Healthcare). The captured images were analyzed using ImageJ (NIH, Bethesda) and quantified by measuring the density of each protein band.
In vitro kinase assays
For in vitro kinase assays, GST-recombinant SEK1 (r-SEK1) or r-JNK was incubated in the kinase buffer (20 mM Tris-HCl [pH 7.5], 10 mM MgCl2, 1 mM DTT) containing 0.37 MBq ml−1 [γ-32P] ATP for 30 min at 42-43 °C. The kinase reaction products were subjected to SDS-PAGE and analyzed with a phosphoimaging device (BAS-2000; GE Healthcare, Buckinghamshire).
shRNAs and transfection
pRP-Puro-U6 plasmid was used for expression of DUSP16 shRNA (Vector Builder, Yokohama). HeLa cells were transfected with the control or DUSP16-specific shRNA expression vector using FuGENE HD (Roche, Indianapolis, IN). Twenty-four hours after transfection, the medium was replaced with fresh medium containing 2 μg/ml puromycin (Invivogen, San Diego, CA) for selection of the transformants. Following an additional incubation for 72 h, the cells were treated with heat, and harvested for Western blotting analysis.
Semi-quantitative RT-PCR analysis
Total RNA was isolated from cells treated with hyperthermia or those that were untreated using the ISOGEN II RNA Isolation System (Nippongene, Tokyo). RNA was converted to cDNA using SuperScript III reverse transcriptase (Invitrogen) and oligo(dT)12-18 (GE Healthcare), and the cDNA was amplified by polymerase chain reaction (PCR) using PrimeSTAR DNA Polymerase (Takara Bio) and specific primers (Santa Cruz Biotechnology).
Results
Heat treatment stimulates the phosphorylation of SEK1 and JNK with a reduction in MAP3K protein levels
Our previous report demonstrated that hyperthermia results in a decrease in the expression levels of MAP3K members (i.e., TAK1, RAF1, and MEKK2) but not the downstream members MAP2K and MAPK in the ERK signaling cascade in the HeLa and LU99 cells [Citation7]. In this report, we further examined the effects of heat on the expression of other MAP3Ks, especially those belonging to the JNK signaling pathway. Hyperthermia, induced using a Thermotron RF-8, caused a rapid decrease in the expression of a JNK kinase kinase, namely ASK1 or MLK3, among MAP3Ks, with increases in the temperature (41–43 °C) in HeLa cells (). The heat-induced reduction in the levels of MAP3Ks was also observed in LU99 cells (Supplementary Figure S1(A)). Thermotherapy with radio frequency was more efficient than water bath heating in terms of the effect on MAP3K (Supplementary Figure S1(B)). Heat treatment slightly increased the protein level of a JNK kinase, specifically SEK1, among MAP2Ks but did not decrease the expression of JNK. The level of phosphorylated MLK3 decreased in a temperature-dependent manner (). The phosphorylation of ASK1 showed a transient several-fold increase at 41 °C but decreased at and above 42 °C. SEK1 phosphorylation increased up to 42 °C but decreased at 43 °C. Unlike the phosphorylation status change in the MAP3Ks and MAP2K, phosphorylation of the downstream JNK increased in a temperature-dependent manner. These results suggest that the phosphorylation status of JNK after hyperthermia does not necessarily correspond to that of the upstream MAP3Ks or MAP2Ks.
Figure 1. Hyperthermia increases SEK1 and JNK phosphorylation despite the reduction and inactivation of upstream MAP3Ks. (A) HeLa cells were heated at 41–43 °C for 1 h using a Thermotoron-RF-8 and harvested. Cell lysates were prepared and subjected to western blot analysis with antibodies against the indicated proteins. (B) The activity of ASK1, MLK3, SEK1, or JNK was analyzed via western blotting with respective phospho-specific antibodies. (C) HeLa cells were pretreated with DMSO, 10 μM MG132, or ALLN for 1 h and then heated to 43 °C for 1 h. Cell lysates were prepared and subjected to western blot analysis with antibodies against the indicated proteins. For MLK3, cell extracts were centrifuged, and the presence of the soluble or insoluble fraction was determined. CDK2 was used as a loading control. Relative levels of MAPK-related proteins were determined from the western blots using Image J. Data are presented as means ± standard deviations of three independent experiments. Statistical significance was determined using the Student’s t-test (*P < 0.05), versus the hyperthermia (HT) only group.
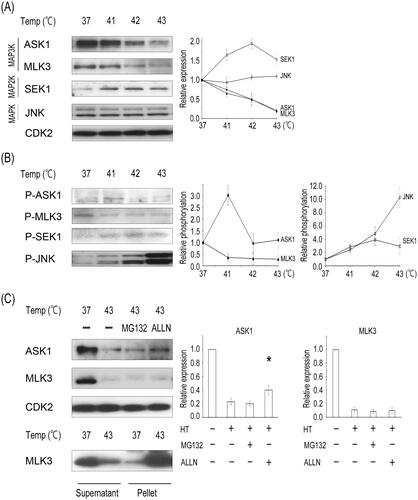
We next hypothesized that the hyperthermia-induced decreases in MAP3K expression might be due to protein instability or the downregulation of gene expression. Thermal stress also causes protein unfolding [Citation3]. These unfolded proteins are either refolded via molecular chaperones or are broken down if the protein structure cannot be restored [Citation1]. Accordingly, we examined the two potential pathways through which heat-induced degradation occurs, namely the ubiquitin-proteasome pathway and calpain degradation. The addition of a calpain inhibitor, ALLN, partially suppressed the heat-induced degradation of ASK1; however, the addition of a 26S proteasome inhibitor, MG132, did not (). Moreover, hyperthermia resulted in the prominent downregulation of ASK1 mRNA expression at 43 °C (Supplementary Figure S1(C)), suggesting that the reduction in ASK1 levels could be attributed to transcriptional downregulation and protein lability. However, hyperthermia did not affect MLK3 mRNA levels. Neither MG132 nor ALLN rescued the degradation of MKL3 after heat treatment. After heat treatment at 43 °C, MLK3 disappeared from the soluble fraction and appeared in a pellet, indicating that the reduction in MLK3 levels can be attributed to protein insolubilization. Although ASK1 and MLK3 have a serine-threonine kinase domain, they each belong to a different MAP3K cluster (MEKK or MLK) based on kinase domain homology. These differences among MAP3Ks might result in differences in the protein structure and responses to heat.
Heat treatment reduces DUSP16 protein levels
JNK was activated by hyperthermia despite a decrease in or the inactivation of upstream MAP3Ks. There are several possibilities for this; one is increased autophosphorylation associated with increased amounts of SEK1 and JNK. We hypothesized that the hyperthermia-induced increase in SEK1 or JNK phosphorylation might be due to the upregulation of SEK1 gene expression. We, therefore, assessed the effect of heat on the gene expression levels of SEK1 or JNK via semi-quantitative RT-PCR. As shown in , the mRNA expression levels of SEK1 slightly decreased in response to heat treatment, with no effect on JNK1 mRNA levels. Therefore, the increase in the total protein content of SEK1 does not appear to be due to an increase in SEK1 gene expression but rather an increase in the phosphorylated SEK1 protein. The second possibility is enhanced SEK1 and JNK autophosphorylation activity induced by hyperthermia. JNK2 is known to undergo trans-autophosphorylation through dimer formation and subsequent autoactivation [Citation18]. However, the in vitro kinase assay demonstrated that heat did not stimulate the autophosphorylation of either recombinant SEK1 (r-SEK1) or r-JNK1/2 (), suggesting that the increased phosphorylation of SEK1 by hyperthermia is caused by post-translational modifications by other factors. The third possibility is the involvement of their phosphatases. Kinases function as phosphorylating enzymes, and phosphatases dephosphorylate their target substrates. The proper balance between activation and inactivation of the JNK pathway needs to be tightly regulated for the maintenance of cellular homeostasis. Dual specificity phosphatases (DUSPs) dephosphorylate both threonine/serine and tyrosine residues in their substrates. Members of a subfamily of DUSPs contain the MAP kinase-binding motif or the kinase-interacting motif, which is involved in regulating the magnitude and duration of signal transduction via the MAPK/JNK pathway through the dephosphorylation of their substrates [Citation19,Citation20]. Especially, DUSP8, DUSP10, and DUSP16 preferentially inactivate JNK [Citation20]. Another previous report demonstrated that the JNK phosphatase M3/6, namely DUSP8, is inactivated by heat shock at 45 °C [Citation21]. Thus, we assessed the effect of heat on the expression levels of several JNK phosphatases. The expression of DUSP8 was not detected via western blotting analysis using several specific antibodies in our system. As shown in , the expression of DUSP16 was decreased upon thermal treatment above 42 °C; however, DUSP10 expression was not decreased from 41 °C to 43 °C. The heat-induced reduction in DUSP16 was rescued by the addition of either MG132 or ALLN (). Our finding is consistent with a report indicating that DUSP16 protein levels are rapidly decreased through ubiquitination and subsequent proteasomal degradation in quiescent cells [Citation22,Citation23]. Thus, our results suggest that DUSP16 is degraded by proteases, such as the proteasome or calpain, during hyperthermia.
Figure 2. Heat treatment decreases the expression of DUSP16, a JNK phosphatase. (A) HeLa cells were heated at 41–42 °C for 1 h, as described in . Total RNA was prepared from treated and untreated cells, and levels of MAPK-related mRNAs were estimated using semi-quantitative RT-PCR. (B) An in vitro kinase reaction was performed by incubating GST-recombinant SEK1 (r-SEK1), GST-JNK1β2, or JNK2β2 (r-JNK1/2) for 30 min at 42–43 °C in the presence of [γ-32P] ATP. The kinase reaction products were subjected to SDS-PAGE and then visualized via autoradiography (32P). (C) HeLa cells were heated at 41–43 °C for 1 h and harvested. Cell lysates were prepared and subjected to western blot analysis with antibodies against the indicated proteins. (D) HeLa cells were pretreated with DMSO, 10 μM MG132, or ALLN for 1 h and then heated to 43 °C for 1 h. Cell lysates were prepared and subjected to western blot analysis with antibodies against the indicated proteins. Relative levels of DUSP-related proteins were determined from the western blot using ImageJ. Data are presented as means ± standard deviations of three independent experiments. Statistical significance was determined using the Student’s t-test (*p < 0.05), versus the HT only group.
![Figure 2. Heat treatment decreases the expression of DUSP16, a JNK phosphatase. (A) HeLa cells were heated at 41–42 °C for 1 h, as described in Figure 1(A). Total RNA was prepared from treated and untreated cells, and levels of MAPK-related mRNAs were estimated using semi-quantitative RT-PCR. (B) An in vitro kinase reaction was performed by incubating GST-recombinant SEK1 (r-SEK1), GST-JNK1β2, or JNK2β2 (r-JNK1/2) for 30 min at 42–43 °C in the presence of [γ-32P] ATP. The kinase reaction products were subjected to SDS-PAGE and then visualized via autoradiography (32P). (C) HeLa cells were heated at 41–43 °C for 1 h and harvested. Cell lysates were prepared and subjected to western blot analysis with antibodies against the indicated proteins. (D) HeLa cells were pretreated with DMSO, 10 μM MG132, or ALLN for 1 h and then heated to 43 °C for 1 h. Cell lysates were prepared and subjected to western blot analysis with antibodies against the indicated proteins. Relative levels of DUSP-related proteins were determined from the western blot using ImageJ. Data are presented as means ± standard deviations of three independent experiments. Statistical significance was determined using the Student’s t-test (*p < 0.05), versus the HT only group.](/cms/asset/877225de-d722-4b74-bbd7-7f4276af5f34/ihyt_a_2335199_f0002_b.jpg)
DUSP16 depletion enhances heat-induced ASK1–SEK1–JNK signaling and apoptosis
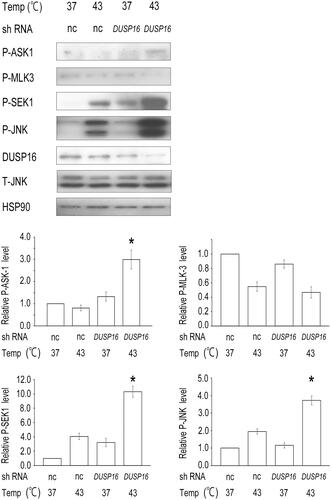
DUSP16 preferentially inactivates JNK [Citation20,Citation24], and a recent report demonstrated that DUSP16 inhibits the activation of JNK, thereby reducing apoptosis and promoting cancer chemoresistance [Citation25]. To clarify the biological significance of DUSP16 degradation after hyperthermia, we examined the effects of DUSP16 knockdown on JNK signaling and cell death. Transient transfection with shRNA targeting DUSP16 significantly enhanced the heat-induced phosphorylation of ASK1, SEK1, and JNK (), indicating that DUSP16 regulates heat-induced JNK signaling. However, the phosphorylation of MLK3 was not stimulated by DUSP16 knockdown or the combination with heat.
Figure 3. DUSP16 depletion enhances the heat-induced activation of JNK signaling. HeLa cells were transfected with the negative control (nc) or DUSP16-specific shRNA expression vector. After selection with puromycin for 96 h, the cells were treated with hyperthermia at 43 °C for 1 h or left untreated as controls and harvested. The cell lysates were subjected to western blotting with the indicated antibodies. HSP90 was used as a loading control. Relative levels of P-ASK1, P-MLK3, P-SEK1, or P-JNK were determined from the western blot results using ImageJ. The data are representative of three independent experiments. Data are presented as means ± standard deviations of three independent experiments. Statistical significance was determined using the Student’s t-test (*p < 0.05), versus the negative control shRNA + HT group. T-JNK, total JNK.
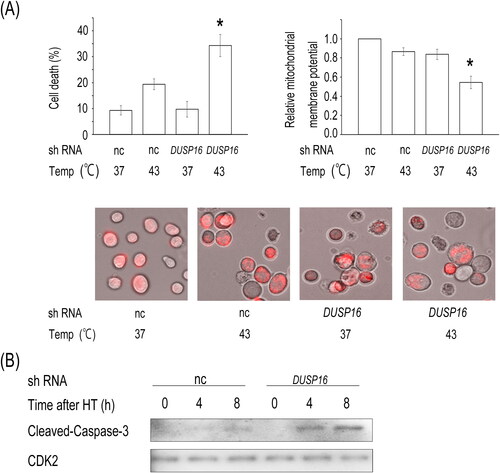
The JNK signaling pathway has a critical role in stress-induced apoptosis [Citation26]. Thus, we determined whether the knockdown of DUSP16 enhances heat-induced cell death. When exposed to heat, cell death increased markedly in DUSP16 shRNA-expressing HeLa cells (34.3%) compared to that in cells expressing control shRNA (19.3%) (, upper left panel). Importantly, the combination of DUSP16 shRNA and heat significantly decreased the mitochondrial membrane potential and enhanced the cleavage of Caspase-3, which are considered initial and irreversible steps in apoptosis, compared to that observed in the heat-treated HeLa cells expressing control shRNA ( upper right, lower panels, and (B)). The enhanced phosphorylation of JNK mediated by DUSP16 knockdown correlates with increased cell death during hyperthermia. Together with these results, DUSP16 knockdown sensitizes cells to hyperthermia by enhancing ASK1–SEK1–JNK signaling and subsequently promoting apoptosis.
Figure 4. DUSP16 depletion enhances heat-induced apoptosis. (A) HeLa cells were transfected with the negative control (nc) or DUSP16-specific shRNA expression vector and selected as described previously herein. The shRNA-transfected cells were heated at 43 °C for 1 h, and then, cell death was assessed 24 h later (upper left panel). Mitochondrial transmembrane potential was determined using the Mitochondrial Capture reagent, which results in a decrease in red fluorescence upon the loss of mitochondrial potential (upper right panel). Representative images of the Mitochondrial Capture reagent (red) in heat-treated HeLa cells (lower panels). (B) Caspase-3 cleavage. HeLa cells were transfected with the nc or DUSP16-specific shRNA expression vector and selected as described. The shRNA-transfected cells were heated at 43 °C for 1 h and then harvested at the indicated times. The cell lysates were subjected to western blotting with the indicated antibodies. CDK2 was used as a loading control. The data are representative of three independent experiments. Data are presented as means ± standard deviations of three independent experiments. Statistical significance was determined using the Student’s t-test (*p < 0.05), versus negative control shRNA + HT group.
Discussion
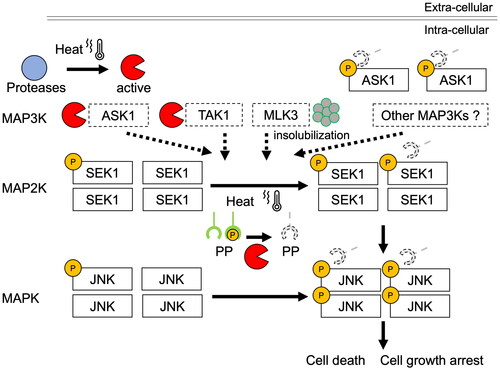
In this study, we found that hyperthermia activates JNK in a temperature-dependent manner, with the degradation of some MAPK3s belonging to the JNK signaling pathway (). The degradation of MAP3K, induced by radiofrequency hyperthermia was more effective than that with water bath heating (Supplementary Figure S1(B)), which is consistent with reports on the effects of radiofrequencies on cells [Citation27,Citation28]. Other report demonstrated that exposure of cells to heat shock strongly reduced the rate of JNK dephosphorylation without stimulation by upstream kinases [Citation29]. After the degradation of MAP3Ks in the JNK cascade, they might be incapable of transducing apoptotic stimuli into activation signals and/or activating each downstream target through phosphorylation, leading to the inhibition of JNK signaling and apoptosis induction. However, we demonstrated that DUSP16 is degraded in response to hyperthermia and that the knockdown of DUSP16 enhances JNK phosphorylation ( and ). Although DUSP16 can bind all three MAPKs, it preferentially inactivates JNK1/2, followed by p38α/β MAPK (JNK1/2≫p38 MAPK > ERK) [Citation24], suggesting that hyperthermia might prevent DUSP16 from dephosphorylating JNK. Moreover, the degree of JNK phosphorylation induced by the combination of hyperthermia and DUSP16 knockdown is greater than the additive effects of each treatment alone. Some members of the DUSP family also regulate signaling upstream of JNK. DUSP14 directly dephosphorylates TAB1, leading to inhibition of the TAB1–TAK1 complex and sequential inactivation of TAK1 and downstream JNK activity [Citation30]. DUSP16 knockdown also enhanced the heat-induced phosphorylation of ASK1, among MAP3Ks, and SEK1, among MAP2Ks. Our results also suggest that DUSP16 is involved in multiple dephosphorylation events in the JNK signaling cascade. The enhanced phosphorylation of SEK1 by the depletion of DUSP16 might be triggered via a reduction in its ability to directly dephosphorylate ASK1 and/or SEK1. Regarding MLK3, no effect of DUSP16 knockdown was observed, suggesting its low capacity to interact with DUSP16 and poor homology as a substrate.
Further, there are some other possibilities regarding heat-induced JNK activation. One possibility is the involvement of MAP3Ks other than those kinases. Since there are 21 characterized MAP3Ks that activate known MAP2Ks [Citation31], other heat-resistant MAP3Ks might stimulate MAP2Ks and MAPKs. SEK1 phosphatases could also be linked to JNK activation. In addition, protein phosphatases 2A and 2C dephosphorylate SEK1 [Citation32,Citation33]. Therefore, if hyperthermia results in inactivation of the SEK1 phosphatases, it could lead to JNK activation. Together, hyperthermia could cause dysregulation of the proper balance between the activation and inactivation of JNK signaling through a reduction in JNK phosphatase functions, resulting in the amplification of JNK signaling and enhanced apoptosis ().
Figure 5. Schematic showing the effects of hyperthermia on JNK signaling pathway. Major JNK signaling pathways activated by the MAP3K members ASK1, MLK3, and TAK1 are shown. The MAP3K cascades are involved in the regulation of cell growth and apoptosis. Hyperthermia decreases the expression levels of MAP3Ks but increases the phosphorylation of ASK1, the downstream JNK kinase SEK1, and JNK through the suppression of JNK phosphatase functions. This deregulated JNK activation could be one of the mechanisms underlying the heat-induced death of cancer cells. P: phosphorylation; PP: protein phosphatase.
In conclusion, our study demonstrates that degradation of DUSP16 enhanced the heat-induced activation of JNK signaling. However, the detailed mechanism requires more extensive studies.
Inhibition of DUSP16 could enhance stress-induced apoptosis. Thus, targeting DUSP16 may provide a more effective approach for cancer therapy.
Supplemental Material
Download Zip (1.3 MB)Acknowledgements
We greatly appreciate Tetsuya Sekiguchi for support with the use of the RF-8 heating device.
Disclosure statement
TF and AY belong to the Social Cooperation Program, Department of Clinical Cannabinoid Research, supported by the Japan Cosmetic Association and Japan Federation of Medium &Small Enterprise Organizations. No potential conflict of interest is reported by the other authors.
Data availability statement
The datasets produced and/or analyzed during this study are available from the corresponding author on reasonable request.
Additional information
Funding
References
- Roti Roti JL. Cellular responses to hyperthermia (40–46 °C): cell killing and molecular events. Int J Hyperthermia. 2008;24(1):1–10. doi:10.1080/02656730701769841.
- Hou CH, Lin FL, Hou SM, et al. Hyperthermia induces apoptosis through endoplasmic reticulum and reactive oxygen species in human osteosarcoma cells. Int J Mol Sci. 2014;15(10):17380–17395. doi:10.3390/ijms151017380.
- Ahmed K, Zaidi SF, Mati-Ur-Rehman MR et al. Hyperthermia and protein homeostasis: cytoprotection and cell death. J Therm Biol 2020;91:102615. doi:10.1016/j.jtherbio.2020.102615.
- Luo GJ, Sun X, Hasselgren PO. Hyperthermia stimulates energy-proteasome-dependent protein degradation in cultured myotubes. Am J Physiol Regul Integr Comp Physiol. 2000;278(3):R749–R756. doi:10.1152/ajpregu.2000.278.3.R749.
- Ciechanover A. Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol. 2005;6(1):79–87. doi:10.1038/nrm1552.
- Enomoto A, Fukasawa T. The role of calcium-calpain pathway in hyperthermia. Front. Mol. Med. 2022;2:1005258. doi:10.3389/fmmed.2022.1005258.
- Enomoto A, Fukasawa T, Terunuma H, et al. Decrease in MAP3Ks expression enhances the cell death caused by hyperthermia. Int J Hyperthermia. 2022;39(1):200–208. doi:10.1080/02656736.2021.2024281.
- van den Tempel N, Odijk H, van Holthe N, et al. Heat-induced BRCA2 degradation in human tumours provides rationale for hyperthermia-PARP-inhibitor combination therapies. Int J Hyperthermia. 2018;34(4):407–414. doi:10.1080/02656736.2017.1355487.
- Chang L, Karin M. Mammalian MAP kinase signalling Cascades. Nature. 2001;410(6824):37–40. doi:10.1038/35065000.
- Cobb MH, Goldsmith EJ. How MAP kinases are regulated. J Biol Chem. 1995;270(25):14843–14846. doi:10.1074/jbc.270.25.14843.
- Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80(2):179–185. doi:10.1016/0092-8674(95)90401-8.
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103(2):239–252. doi:10.1016/s0092-8674(00)00116-1.
- Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81(2):807–869. doi:10.1152/physrev.2001.81.2.807.
- Zeke A, Misheva M, Reményi A, et al. JNK signaling: regulation and functions based on complex protein-protein partnerships. Microbiol Mol Biol Rev. 2016;80(3):793–835. doi:10.1128/MMBR.00043-14.
- Saitoh M, Nishitoh H, Fujii M, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17(9):2596–2606. doi:10.1093/emboj/17.9.2596.
- Teramoto H, Coso OA, Miyata H, et al. Signaling from the small GTP-binding proteins Rac1 and Cdc42 to the c-Jun N-terminal kinase/stress-activated protein kinase pathway. A role for mixed lineage kinase 3/protein-tyrosine kinase 1, a novel member of the mixed lineage kinase family. J Biol Chem. 1996;271(44):27225–27228. doi:10.1074/jbc.271.44.27225.
- Yamaguchi K, Shirakabe K, Shibuya H, et al. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science. 1995;270(5244):2008–2011. doi:10.1126/science.270.5244.2008.
- Nitta RT, Chu AH, Wong AJ. Constitutive activity of JNK2 alpha2 is dependent on a unique mechanism of MAPK activation. J Biol Chem. 2008;283(50):34935–34945. doi:10.1074/jbc.M804970200.
- Ha J, Kang E, Seo J, et al. Phosphorylation dynamics of JNK signaling: effects of Dual-Specificity Phosphatases (DUSPs) on the JNK pathway. Int J Mol Sci. 2019;20(24):6157. doi:10.3390/ijms20246157.
- Kondoh K, Nishida E. Regulation of MAP kinases by MAP kinase phosphatases. Biochim Biophys Acta. 2007;1773(8):1227–1237. doi:10.1016/j.bbamcr.2006.12.002.
- Palacios C, Collins MK, Perkins GR. The JNK phosphatase M3/6 is inhibited by protein-damaging stress. Curr Biol. 2001;11(18):1439–1443. doi:10.1016/s0960-9822(01)00426-2.
- Katagiri C, Masuda K, Urano T, et al. Phosphorylation of Ser-446 determines stability of MKP-7. J Biol Chem. 2005;280(15):14716–14722. doi:10.1074/jbc.M500200200.
- Chen HF, Chuang HC, Tan TH. Regulation of Dual-Specificity Phosphatase (DUSP) ubiquitination and protein stability. Int J Mol Sci. 2019;20(11):2668. doi:10.3390/ijms20112668.
- Masuda K, Shima H, Watanabe M, et al. MKP-7, a novel mitogen-activated protein kinase phosphatase, functions as a shuttle protein. J Biol Chem. 2001;276(42):39002–39011. doi:10.1074/jbc.M104600200.
- Low HB, Wong ZL, Wu B, et al. DUSP16 promotes cancer chemoresistance through regulation of mitochondria-mediated cell death. Nat Commun. 2021;12(1):2284. doi:10.1038/s41467-021-22638-7.
- Dhanasekaran DN, Reddy EP. JNK signaling in apoptosis. Oncogene. 2008;27(48):6245–6251. doi:10.1038/onc.2008.301.
- Sackenheim MM. Radio frequency ablation: the key to cancer treatment. J Diagn Med Sonogr. 2003;19(2):88–92. doi:10.1177/8756479303251097.
- Wust P, Kortüm B, Strauss U, et al. Non-thermal effects of radiofrequency electromagnetic fields. Sci Rep. 2020;10(1):13488. doi:10.1038/s41598-020-69561-3.
- Meriin AB, Yaglom JA, Gabai VL, et al. Protein-damaging stresses activate c-Jun N-terminal kinase via inhibition of its dephosphorylation: a novel pathway controlled by HSP72. Mol Cell Biol. 1999;19(4):2547–2555. doi:10.1128/MCB.19.4.2547.
- Yang CY, Li JP, Chiu LL, et al. Dual-specificity phosphatase 14 (DUSP14/MKP6) negatively regulates TCR signaling by inhibiting TAB1 activation. J Immunol. 2014;192(4):1547–1557. doi:10.4049/jimmunol.1300989.
- Craig EA, Stevens MV, Vaillancourt RR, et al. MAP3Ks as Central regulators of cell fate during development. Dev Dyn. 2008;237(11):3102–3114. doi:10.1002/dvdy.21750.
- Avdi NJ, Malcolm KC, Nick JA, et al. A role for protein phosphatase-2A in p38 mitogen-activated protein kinase-mediated regulation of the c-Jun NH2-terminal kinase pathway in human neutrophils. J Biol Chem. 2002;277(43):40687–40696. doi:10.1074/jbc.M204455200.
- Takekawa M, Maeda T, Saito H. Protein phosphatase 2Cα inhibits the human stress-responsive p38 and JNK MAPK pathways. EMBO J. 1998;17(16):4744–4752. doi:10.1093/emboj/17.16.4744.