
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Statistical learning of the structures of cellular networks, such as protein signaling pathways, is a topical research field in computational systems biology. To get the most information out of experimental data, it is often required to develop a tailored statistical approach rather than applying one of the off-the-shelf network reconstruction methods. The focus of this paper is on learning the structure of the mTOR protein signaling pathway from immunoblotting protein phosphorylation data. Under two experimental conditions eleven phosphorylation sites of eight key proteins of the mTOR pathway were measured at ten non-equidistant time points. For the statistical analysis we propose a new advanced hierarchically coupled non-homogeneous dynamic Bayesian network (NH-DBN) model, and we consider various data imputation methods for dealing with non-equidistant temporal observations. Because of the absence of a true gold standard network, we propose to use predictive probabilities in combination with a leave-one-out cross validation strategy to objectively cross-compare the accuracies of different NH-DBN models and data imputation methods. Finally, we employ the best combination of model and data imputation method for predicting the structure of the mTOR protein signaling pathway.
1. Introduction
Learning the structures of cellular networks is a topical research field in computational systems biology. Gene regulatory networks, protein signaling cascades and metabolic pathways play important roles in the organization of life, since the interplay of the molecular interactions within those networks govern the functions of living cells.
Over the years many network reconstruction methods have been proposed in the Statistics and Machine Learning literature. For an overview of different model classes, see, e.g. the individual chapters in the recently published textbook on gene regulatory networks [Citation11]. Dynamic Bayesian networks (DBNs) and non-homogeneous DBNs (NH-DBNs) are among the most widely applied network reconstruction models. For an introduction to the class of NH-DBNs we refer to [Citation8].
The scope of this paper is on predicting the structure of the mammalian target of rapamycin (short: mTOR) signaling pathway based on protein phosphorylation data. Two important features of the available data set are (i) that measurements were taken under two different experimental conditions, and (ii) that measurements were taken at non-equidistant time points. Because of these two data features, the assumptions of the well-established off-the-shelf network reconstruction methods are not met, so that there is need for designing a tailor-made statistical approach. As the experimental condition can influence the protein-protein interaction strengths (= the network parameters), we decide for a hierarchical modeling approach, and we propose to take into account that the experimental condition can affect the interactions to different extents (e.g. some interactions might be stable across conditions while others might undergo substantial changes). Therefore we design a new tailor-made hierarchical Bayesian model for the data analysis. Moreover, we propose to apply established data imputation methods to deal with non-equidistant network measurements. As conventional dynamic DBNs and NH-DBNs are constructed such that they cannot deal with non-equidistant observations, we use the data imputation methods to predict the unobserved (missing) equidistant observations.
To derive the new NH-DBN model, we borrow modeling ideas from the recent work by Shafiee Kamalabad and Grzegorczyk [Citation13], in which a sequentially coupled NH-DBN with segment-specific coupling parameters was proposed. We here build on the conceptual framework of [Citation13] to define a complementary model, namely a globally coupled NH-DBN with covariate-specific coupling parameters. The resulting new NH-DBN model extends and improves over two earlier proposed models [Citation12,Citation14]. Unlike the earlier models, our new model does not only distinguish between two groups of interactions, namely between ‘constant and coupled’ [Citation14] or between ‘uncoupled and coupled’ interactions [Citation12], respectively. Our new model offers more flexibility, since each network interaction comes with its own individual coupling parameter, so that the coupling strengths can be fine-tuned individually on continuous scales.
The remainder of this paper is organized as follows: The focus of Section 2 is on the biological background and the available mTOR protein phosphorylation data. All mathematical details have been delegated to Section 3. Most importantly, we propose a new hierarchical Bayesian model along with a Reversible Jump Markov Chain Monte Carlo (RJMCMC) inference algorithm in Sections 3.1–3.5. For objectively cross-comparing different network reconstruction approaches we propose in Section 3.6 to apply a leave-one-out-cross validation (LOOCV) strategy and to approximate the predictive probabilities (PPs) of the left-out data points. In Section 3.7, we briefly review the data imputation methods that we used for predicting the missing equidistant observations. Section 4 is on the simulation details and on three competing NH-DBN models. The empirical results of our study are reported in Section 5. In Section 5.1, we report about the outcome of a comparative evaluation study, which we performed to identify the best combination of network model and data imputation method. Subsequently, in Section 5.2 we use the best combination to predict the structure of the unknown mTOR protein signaling pathway. In Section 6, we summarize our findings with a brief discussion.
2. Protein phosphorylation data from the mTOR signaling pathway
The ‘mammalian Target Of Rapamycin’ (mTOR) pathway is an important cellular protein signaling cascade that regulates the cell cycle in mammalian cells. Because of its direct effect on the cell cycle, the mTOR pathway plays an important role in fundamental cellular processes, such as quiescence, proliferation, cancer, and longevity. Recent studies have shown that the mTOR pathway is also associated with arthritis, insulin resistance and osteoporosis. So called ‘mTOR inhibitors’ are used for targeted therapy for tumors, organ transplantation, rheumatoid arthritis, etc.. For a recent literature review on the mTOR pathway we refer to the work by Zou et al. [Citation16].
Like all protein signaling pathways, the mTOR pathway consists of a set of proteins that act on one another. Proteins are the functional units of the cells, because activated proteins can catalyze biochemical reactions. The biochemical processes that temporarily activate and deactivate proteins are called ‘phosphorylation’ and ‘dephosphorylation’. In particular, an activated (‘phosphorylated’) protein can initiate or contribute to the activation (‘phosphorylation’) of specific other proteins. Any incoming external signal can thus initiate a cascade of protein activations, altogether leading to the required physiological response of the cell. As proteins can have multiple ‘phosphorylation sites’, they can get activated in different ways. The activity of a protein is determined by the phosphorylation states at its sites. This way, proteins can have different functions within the signaling pathway. The immunoblotting protein phosphorylation data from Pezze et al. [Citation2], which we will analyze in our study, contain time course data of eleven phosphorylation sites of eight proteins of the mTOR pathway. Table lists the eight involved proteins along with their measured phosphorylation sites. In total, the states of N = 11 phosphorylation sites (see right column of Table ) were measured in H = 2 experiments that were subject to different experimental treatments, namely:
stimulation of the mTOR pathway with amino acids only (h = 1)
stimulation of the mTOR pathway with amino acids and insulin (h = 2)
Table 1. Proteins and their phosphorylation sites.
In both experiments (h = 1, 2), after the experimental treatment, measurements were taken after minutes, yielding two relatively short time series, each featuring
non-equidistant observations.Footnote1
3. Statistical methods
The standard assumption in DBNs is that all node interactions are subject to a lag of one time unit. Because of this time lag, there is no need for an acyclicity constraint and the task of learning a dynamic network among N nodes is typically separated into N independent regression tasks. In Section 3.1, we propose a new hierarchical Bayesian regression model, and we show how to generate posterior samples using RJMCMC simulations; cf. Sections 3.2–3.4. In Section 3.5, we describe how the new hierarchical regression model from Section 3.1 can be extended to an NH-DBN model. To assess the model performance we follow a leave-one-out cross validation approach and we compute predictive probabilities of the left-out data points; cf. Section 3.6. In Section 3.7, we briefly review widely applied data imputation methods, which we use to predict unobserved equidistant observations.
3.1. Non-homogeneous Bayesian linear regression
Consider H linear regression models indexed by that share the same target variable Y and the same k covariates
. With regard to our application to mTOR protein phosphorylation data (cf. Section 2), each h index might refer to a specific experimental condition, under which data were measured. For each
we have a regression data set with
observations, namely an
-dimensional response vector
and a design matrix
. When the models feature intercepts, the design matrices have a first column of 1's and each model has k + 1 regression coefficients. The design matrices
are then
-by-
dimensional. Let
denote the regression coefficient vector for experimental condition h. We assume for the likelihood:
(1)
(1)
where
is the identity matrix, and
is a shared noise variance parameter, for which we assume as prior an inverse Gamma distribution,
For the intercept and for each covariate,
, we have H condition-specific regression coefficients
and our assumption is that each regression coefficient i has a different sensitivity to the experimental conditions.
Among the most popular priors for regression coefficients are Gaussian and Laplace priors. The conceptual advantage of non-conjugate Laplace priors is that they tend to infer sparser regression models by shrinking the regression coefficients more efficiently towards zero. However, we prefer working with Gaussian priors, since they are the conjugate priors. In particular, in Bayesian models with hierarchical prior distributions the employment of conjugate prior distributions allows the RJMCMC based model inference to be done by much more efficient Gibbs sampling steps. For the proposed hierarchical Gaussian prior (see Equation (Equation2(2)
(2) ) below) the full conditional distributions and the marginal likelihood can be computed analytically; cf. Section 3.2 for more details.
To tune the degree of information exchange (similarity) across the H conditions, we propose the following new hierarchical prior for the regression coefficient vectors:
(2)
(2)
where
is the global mean vector across conditions. We assume that the covariance matrix in Equation (Equation2
(2)
(2) ) is of the form
.Footnote2 And for
we use a diagonal matrix with k + 1 positive diagonal elements
, which can be interpreted as ‘variance multipliers’. Symbolically, we have:
For the individual regression coefficients this implies the univariate Gaussian priors
(3)
(3)
where
is the ith diagonal element of
. Equation (Equation3
(3)
(3) ) reveals that the parameter
regulates the similarity of the ith regression coefficients
(
. Large values
allow the ith regression coefficients to deviate from the global mean
, while low values
enforce the ith regression coefficients to stay similar to
; i.e. to stay similar across conditions. With regard to our application to mTOR data in Section 5, large values
indicate protein-protein interactions that strongly depend on conditions, while low values
indicate protein-protein interactions that are rather constant across conditions. Because of this interpretation, we here refer to the ‘variance multipliers’ as ‘(inverse) coupling (strength) parameters’. The lower the ‘variance multiplier’, the stronger the coupling (similarity) across conditions.
On the coupling parameters we impose another hierarchical prior. For each we use an inverse Gamma distribution with hyperparameters
and
, where
is fixed and
has a Gamma hyperprior:
(4)
(4)
where
and
are fixed hyperhyperparameters.
For the density of the posterior distribution we then have:
(5)
(5)
where the symbols
and
denote the sets of the H regression coefficient vectors and the H response vectors, respectively, and
is the diagonal matrix of the variance multipliers. For the likelihood we have the factorization:
(6)
(6)
and the distribution of
was specified in Equation (Equation1
(1)
(1) ). Moreover we have:
and the distributions of
and
were specified in Equations (Equation2
(2)
(2) ) and (Equation4
(4)
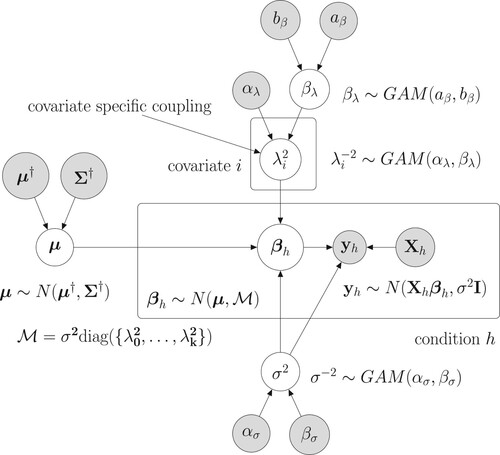
(4) ). A graphical model representation of the coupled Bayesian regression model is provided in Figure .
Figure 1. Graphical model representation of the new hierarchically coupled Bayesian regression model. The mathematical details can be found in Section 3.1. Free (hyper-)parameters are in white circles. The data and fix hyperparameters are in gray circles. The two boxes indicate the experimental conditions and the covariate-specific coupling parameters
.
3.2. Gibbs sampling for given covariates
If the relevant covariates are known (say: like in Section 3.1), they do not have to be inferred from the data. A Gibbs sampling scheme can then be used to generate a posterior sample of the model parameters
. In the supplementary material we derive the required full conditional distributions, from which the model parameters can be successively re-sampled. One Gibbs sampling iteration consists of the following five consecutive sampling steps:
| (G.1) | Sample:
| ||||
| (G.2) | For | ||||
| (G.3) | For | ||||
| (G.4) | Sample:
| ||||
| (G.5) | Sample:
| ||||
We also show in the supplementary material that (
) and
can be integrated out from the likelihood,
, yielding the marginal likelihood:
(7)
(7)
where
and
is the total number of observations. In terms of the marginal likelihood, we have for the posterior density:
(8)
(8)
where
is the diagonal matrix of the coupling parameters.
We note that the RJMCMC moves between covariate sets, which we describe in Section 3.3, make use of the marginal likelihood from Equation (Equation7(7)
(7) ) and the relationship given in Equation (Equation8
(8)
(8) ).
3.3. Metropolis-Hastings sampling of the covariates
In many applications the true covariates are not known and have to be inferred from the data. We assume that there are n potential covariates , and we let
denote a subset of covariates with prior probability
. Moreover, we write
to denote the marginal likelihood implied by the covariate set
, where we do not make explicit that
and
depend on
. For generating a posterior sample of covariate sets we employ the marginal likelihood from Equation (Equation7
(7)
(7) ) and we implement three RJMCMC moves:
| (D) | In the deletion move we randomly choose a covariate | ||||
| (A) | In the addition move we randomly select one covariate | ||||
| (E) | In the exchange move we randomly select one covariate | ||||
Each move yields a new covariate set , and we propose to replace the current
by
. Since the global mean vector
and the diagonal matrix of variance multipliers
refer to the original covariate set
, we have to adjust them for
. Therefore, along with the new covariate set
we also have to propose a new vector
and a new matrix
.
The addition and the exchange move both add a new covariate
to π. For this new covariate we sample the variance multiplier
Given
When randomly selecting the move type, the acceptance probability for the move from to
is:
with the Hastings inverse proposal ratio (HR)
where n is the number of potential covariates, and
denotes the cardinality.Footnote4
3.4. Model inference
For model inference we combine the Gibbs sampling moves from Section 3.2 with the RJMCMC moves from Section 3.3, so as to generate a posterior sample:
where we do not make explicit that each
,
and
depend on the covariate set
. From the RJMCMC sample we can estimate for each of the n covariates a marginal inclusion probability. For example, for covariate
we get:
(9)
(9)
where
if
, and
otherwise. That is, the estimator is the relative frequency of sampled covariate sets that obtained covariate
.
Moreover, we can estimate the predictive probability of a new target vector taken under condition h. Let
denote the corresponding design matrix built from the covariates of the set
. The estimated predictive probability is:
(10)
(10)
where
is the density of the
distribution.
3.5. From Bayesian regression to dynamic Bayesian networks
Consider variables
that were observed under conditions
. Let
be the observation of
under condition h at time point
, where
. The goal is to infer the interactions among the variables, where all interactions are subject to a lag of one time point. That is, each interaction
indicates that
has an effect on
(
and
). Because of the time lag, the network model can be separated into N independent regression models, whose effective sample sizes reduce from
to
.
In the wth regression model takes the role of the target variable, Y, and the remaining
variables
(
) are the potential covariates
. Because of the data structure, the hierarchically coupled Bayesian regression model from Section 3.1 can be employed and covariate sets
for
can be MCMC sampled, as described in Section 3.4. The condition-specific response vectors are given by
while the corresponding design matrices
contain the values of the covariates from
. When
, design matrix
contains the column
. For each variable
a regression model posterior sample can be generated:
The variable-specific covariate sets that were sampled in iteration r, symbolically
, can then be merged to a network
. The rth network
features the regulatory interaction
if
. Henceforth, the marginal covariate inclusion probabilities from Equation (Equation9
(9)
(9) ) can then be interpreted as marginal network interaction probabilities.
Given additional new data points that were measured under condition h and that yield the N variable-specific response vectors the predictive probability from Equation (Equation10
(10)
(10) ) extends to
(11)
(11)
where
is the density of the
distribution.
3.6. Leave-one-out-cross-validation
For objectively comparing the performances of different methods, we propose to apply a leave-one-out-cross-validation (LOOCV) approach and to Monte Carlo approximate the predictive probabilities (PPS) for the left out data points using Equation (Equation11(11)
(11) ).
To this end, we loop through the conditions , and within each condition h we loop through the time points
. For each
we then remove for each variable
(
) the value
from response vector
and run a regression MCMC simulation using the reduced response vector
. Performing a regression MCMC simulation on each reduced response vector
(
) yields N MCMC outputs, which we can use to estimate the predictive probability for the new (left-out) observation
using Equation (Equation11
(11)
(11) ). In total, we get the predictive probabilities for
network data points (cf. Section 3.5), by generating posterior samples for
hierarchical Bayesian regression models (cf. Sections 3.1–3.4).
3.7. Data imputation methods to predict equidistant covariate values
The immunoblotting mTOR protein data have been measured under two conditions ( at
non-equidistant time points,
, cf. Section 2. That is, for each phosphorylation site
(
) we only have the observations
(h = 1, 2 and
) rather than equidistant observations. The experimental design has been in mismatch to one of the key assumptions of dynamic Bayesian networks, namely the assumption that the made observations are equidistant. This mismatch between data and model is typically ignored and the naive simple shift (SH) approach is to treat the data as if they were equidistant. With regard to our mTOR application, the SH approach implies combining measurements that refer to protein phosphorylation processes that took place in substantially different time intervals, ranging from 1 to 60 min.
In this paper we show that the naive SH approach yields inaccurate results, and we propose an alternative approach to deal with non-equidistant network measurements. We consider the observed non-equidistant time series as incomplete, and we propose to make use of established data imputation techniques to predict the required unobserved equidistant preceding covariate values. For example, the last nine non-equidistant observations of site under condition h become the elements of the response vectors:
When
(
) is a covariate, we do not follow the naive SH approach and fill the corresponding column of the design matrix
with the nine observations at the non-equidistant preceding points
but we fill the design matrix
with nine predicted equidistant observations
so that each (predicted) covariate value
is exactly one minute before the corresponding (observed) response value
. We note that the initial covariate values at time point t = 0 have actually been observed and do not need to be estimated by the data imputation methods. That is, instead of estimating
for
, we can use the actually observed covariate value
. This only applies to
, since the first two measurements are subject to a time lag of exactly one minute.
In our study, we compare the naive simple shift approach (SH) with various data imputation techniques, such as a simple linear interpolation (LI), three moving average approaches (SM, LW, EW), smoothing splines (SP), Gaussian processes (GP), Kalman smoothing (KS), and local polynomial regression fitting (LO). For an overview of the 8 data imputation methods we refer to Table . For all imputation methods we used the default settings of the employed R software packages without trying to optimize their performances. For example, we only applied Gaussian processes with the squared exponential kernel. Moreover, we note that we initialized the Kalman smoothing method with the truly observed covariate values at time point .
Table 2. Overview of the 8 data imputation methods that we considered in our study.
4. Competing methods and implementation details
4.1. Competing network models
We compare the performance of the proposed new NH-DBN model from Section 3 with three competing NH-DBN models. The competing models are conceptually similar but base on alternative regression approaches (cf. Section 3.1).
A conventional DBN merges the data from all conditions
An uncoupled non-homogeneous DBN (‘UC’) assumes the regression coefficient vectors to be condition-specific but it does not encourage the vectors to stay similar across conditions. The prior from Equation (Equation2
The globally coupled non-homogeneous DBN (‘GLOBAL’) is similar to the proposed model but less flexible, as it uses the same coupling parameter
4.2. Hyperparameter settings and simulation details
The (hyper-)parameter setting for which we obtained the results reported in Section 5 can be found in Table . Since our new model is an improved version of the globally coupled NH-DBN model from [Citation7,Citation8], which we referred to as ‘GLOBAL’ in Section 4.1, we decided to stay close to the earlier studies and to re-employ the same prior hyperparameters that were already used in the earlier studies; cf. the hyperparameter setting listed in Table .
Table 3. List of the (hyper-)prior distributions along with the selected fix hyperparameters.
For the mTOR data we also performed additional RJMCMC simulations with different (hyper-)parameter settings to test for model robustness. In consistency with the findings of the sensitivity study reported in [Citation7], the alternative hyperparameter settings led to comparable results in terms of the marginal network interaction probabilities. Table 3 of the supplementary material lists the alternative hyperparameter settings and Figure 2 of the supplementary material shows scatter plots of the resulting marginal network interaction probabilities. From the individual scatter plots it can be seen that the hyperparameter settings yield very similar marginal network interaction probabilities, indicating that the newly proposed model is robust with respect to the (hyper-)parameters of the prior distributions.
We run all RJMCMC simulations for 100,000 iterations and we withdraw the samples generated during the first iterations to take a burn-in phase into account. To check for RJMCMC convergence we ran various independent RJMCMC simulations with different random seeds on the mTOR data set. Scatter plots of the marginal network interaction probabilities suggest sufficient convergence after 100,000 RJMCMC iterations. A few example scatter plots are shown in Figure 1 of the supplementary material. From the scatter plots it can be seen that independent and differently seeded RJMCMC simulations lead to (approximately) the same marginal network interaction probabilities. Since we obtain a concrete predicted network by imposing a threshold on those interaction probabilities and showing only those interactions whose probabilities exceed the threshold, we conclude that independent RJMCMC simulations yield exactly the same predicted mTOR pathway; see Figure .
5. Results
To identify the best combination of data imputation method and NH-DBN model, we perform a systematic comparative evaluation study; cf. Section 5.1. We then employ the best combination to statistically learn the structure of the mTOR protein signaling pathway; cf. Section 5.2.
5.1. Comparative evaluation study
In our comparative evaluation study, we follow a LOOCV approach and we compute predictive probabilities (PPs) for the left-out data points. For the mathematical details we refer to Section 3.6. The goal of our study is to cross-compare the performances of the four NH-DBN models (cf. Figure and the three competitors from Section 4.1) and the data imputation methods (cf. Table ). For every combination of NH-DBN model and data imputation method we can compute 18 predictive probabilities (PPs), and we refer to them as the LOOCV-PPs. For each of model-method combination Figure shows a boxplot of the LOOCV-PP value distributions. The black reference lines indicate the median of the combination newly proposed NH-DBN (NEW) with smoothing splines based data imputation (SP). The first obvious finding is that the globally coupled model (GLOBAL) and the newly proposed model (NEW) perform clearly better than the homogeneous DBN and the uncoupled NH-DBN (UC). Furthermore, it can be easily seen from Figure that the data imputation methods also have clear effects on the LOOCV-PP distributions. In Sections 5.1.1 and 5.1.2 we therefore have a closer look at the 9 data imputation methods and at the 4 NH-DBN models, respectively.
Figure 2. Raw data: Boxplots of the logarithmic predictive probabilities (PPs), obtained by leave-one-out-cross validation (LOOCV-PPs). For each of the four NH-DBN models (DBN, UC, GLOBAL and NEW) there is a separate panel. And within each panel there are 9 boxplots that refer to the 9 data interpolation methods for missing data (see Table ). Each of the 36 boxplots displays the distribution of 18 predictive probabilities (one for each left-out data point). The black horizontal lines mark the largest boxplot median, which was achieved by the new NH-DBN model (NEW) in combination with smoothing splines (SP). Relevant pairwise comparisons of the log LOOCV-PPs can be found in Figures and .
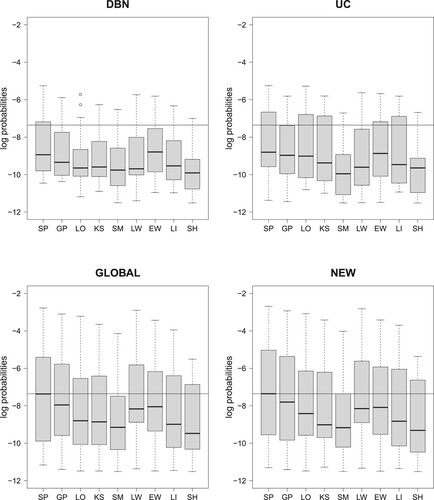
Figure 3. Comparing the data imputation methods; cf. Table . Boxplots of the absolute LOOCV-PP values have been provided in Figure . The 32 boxplots of this figure refer to the relative LOOCV-PP differences in favor of the smoothing splines interpolation method (SP). For each of the four NH-DBN models (DBN, UC, GLOBAL and NEW) there is a separate panel. Within each panel there are 8 boxplots showing the pairwise differences in the logarithmic predictive probabilities in favor of the smoothing splines (SP). The black horizontal lines mark the zero line. For the data imputation methods behind the abbreviations we refer to Table . It can be seen that all differences are consistently in favor of the smoothing splines (SP).
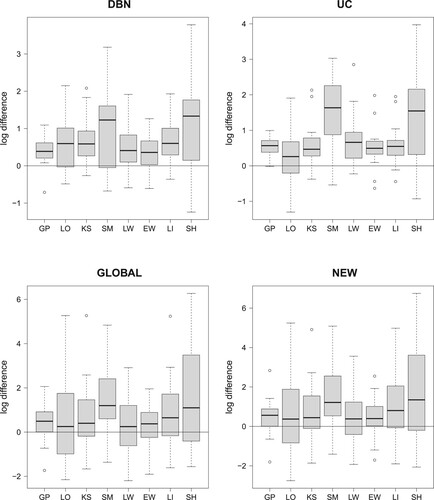
Figure 4. Comparing the four NH-DBN models. Boxplots of the absolute LOOCV-PP values were provided in Figure . The 27 boxplots of this figure refer to the pairwise relative LOOCV-PP differences in favor of the newly proposed NH-DBN (NEW). There are 9 panels, one for each data interpolation method. Within each panel there are three boxplots showing the relative differences in the logarithmic predictive probabilities in favor of the newly proposed NH-DBN model (NEW). Left boxplot: NEW vs. DBN, center boxplot: NEW vs. UC and right boxplot: NEW vs. GLOBAL. In each panel a black horizontal line marks the zero line. It can be seen that all differences are consistently in favor of the newly proposed NH-DBN (NEW).
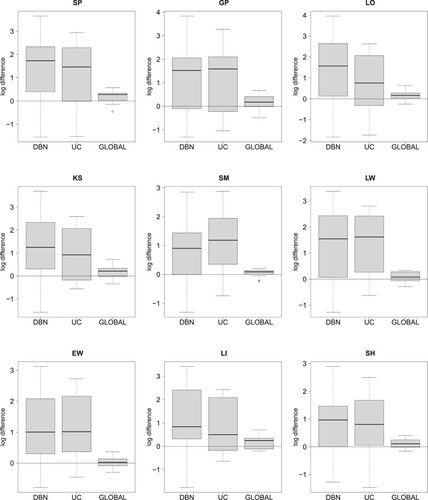
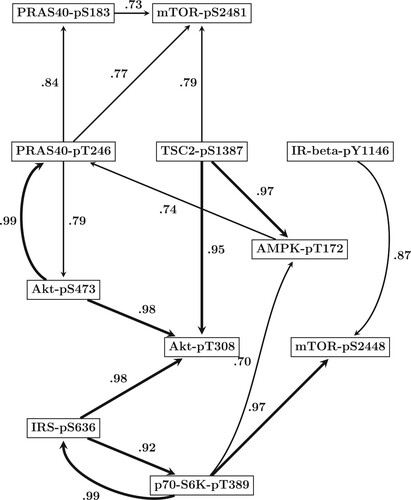
Figure 5. Predicted mTOR protein signaling pathway. Based on the results of our comparative evaluation study in Section 5.1, we employed the newly proposed hierarchical Bayesian model from Section 3.1 (NEW) in combination with smoothing splines (SP) to infer the structure of the mTOR pathway from all data points. By running a RJMCMC for each site we posterior sampled the site-specific regulator sets (subsets of the n = 10 other sites). The marginal network interaction probability for the interaction
refers to the proportion of posterior sampled covariate sets of site
that contained site
as covariate. Shown are the 16 regulatory interactions whose probabilities exceeded the threshold of 0.7. Interactions whose probabilities exceeded the more conservative threshold of 0.9 are displayed by bold edges.
5.1.1. Comparing the data imputation methods
In each of the four panels of Figure the smoothing splines approaches (SP) led to the largest medians. To inspect whether the differences are statistically significant, Figure shows boxplots of the pairwise differences of the log LOOCV-PP values in favor of the SP method. There are 8 pairwise comparisons per panel (per NH-DBN model); i.e. 32 pairwise comparisons in total. The p-values of two-sided Wilcoxon signed-rank tests are provided in Table of the supplementary material. Although the differences are consistently in favor of the smoothing splines (SP), some of the differences are not statistically significant. The main results can be summarized as follows: Most importantly, the smoothing splines method clearly (and significantly) outperforms the widely applied naive simple shift method (SI) as well as the linear interpolation method (LI), which produces jagged (non-smooth) interpolation curves. Most competitive to the smoothing splines (SP) seem to be the LOESS (LO) method (three p-values greater ) and the linear-weighted moving average (LW) method (two p-values greater than
).
5.1.2. Comparing the NH-DBN models
Figure shows boxplots of the pairwise differences of the log LOOCV-PPs in favor of the newly proposed hierarchically coupled NH-DBN (NEW) from Section 3.1. For all 9 data imputation methods the homogeneous DBN and the uncoupled model (UC) perform much worse than the newly proposed model. Also the globally coupled model (GLOBAL) is outperformed, but here the relative differences are smaller. The p-values of 27 two-sided Wilcoxon signed-rank tests are provided in Table of the supplementary material. The p-values for the comparisons with the models DBN and UC are all smaller than , but only for 5 out of 9 interpolation methods the newly proposed model performs significantly better than the globally coupled model. For Kalman Smoothing (KS), for exponential- and linear-weighted moving averages (EW and LW), as well as for Gaussian processes (GPs) the differences are not significant, but yet in favor of the newly proposed model (NEW).
5.2. Learning the structure of the mTOR signaling pathway
The results of our comparative evaluation study from Section 5.1 suggest that the smoothing splines data interpolation method along with the newly proposed hierarchically coupled model from Section 3.1 yields the best network learning performance.
We therefore employ this combination for predicting the structure of the mTOR protein signaling pathway from all data points. To this end, we run RJMCMC simulations for each of the N = 11 phosphorylation sites listed in Table , so as to infer the site-specific regulators. In the wth RJMCMC simulation phosphorylation site takes the role of the response and the remaining n = 10 sites become the potential covariates. Via RJMCMC simulation we then sample the covariate subsets (= regulator sets) of site
from the posterior distribution. The marginal covariate inclusion probabilities are the relative frequencies with which the posterior sampled covariate sets for
contained the corresponding covariates; see Equation (Equation9
(9)
(9) ). By merging all possible inclusion probabilities, we get a probability for each potential network interaction. The marginal network interaction probability for the interaction
is the relative frequency of posterior sampled covariate sets of site
that contained site
as covariate. For a more thorough mathematical description we refer to Section 3.5.
The predicted mTOR pathway is shown in Figure . To avoid a very densely connected network, the figure shows only those 16 protein interactions whose marginal probabilities exceeded the threshold of 0.7.
6. Discussion and conclusions
The focus of this paper has been on predicting the structure of the mTOR protein signaling pathway from protein phosphorylation data. We have proposed a hierarchical Bayesian regression model and we used it as building block for a new NH-DBN model. Since data were measured at non-equidistant time points, we have considered established data imputation methods to predict unobserved equidistant data points. Since most of the interactions of the true mTOR network are still unknown, it was not possible to objectively compare the network reconstruction accuracies of different approaches. Therefore, we have proposed a LOOCV strategy to cross-compare the performances in terms of predictive probabilities (PPs). The empirical results of our comparative evaluation study have shown the following trends. First, the smoothing splines method (SP) performed best in predicting the missing data points, while the widely applied and rather naive simple shift approach (SH), which treats non-equidistant data as if they were equidistant, showed consistently the worst performance. Second, we found that the new NH-DBN model performed better than three earlier proposed models. Since the newly proposed NH-DBN model can be seen as a consensus model between the uncoupled and the globally coupled NH-DBN, we are not surprised that it performed better than the less flexible models. We conclude that the new model seems to be capable of inferring the best trade-off between the earlier models directly from the data. Because of the results of our comparison of the data imputation methods, we strongly advice not to ignore it when observations are non-equidistant. We found that each data interpolation method led to a better performance than the simple shift method, and we would recommend smoothing splines (SP) for future studies. Finally we used the best combination, namely the new NH-DBN model along with smoothing splines to predict the mTOR pathway. A proper biological interpretation of the predicted network is beyond the scope of this paper, but a first literature review revealed that some of the learned protein interactions are consistent with the biological literature.
One advantage of hierarchical Bayesian regression models for non-homogeneous dynamic network learning is the high model flexibility. For the mTOR data from Section 2 we could easily design a tailored regression framework that took the structure of the mTOR data into account (cf. Section 3.1). Another advantage is the scalability. These regression-based models scale very well to larger applications, since the task of learning a dynamic network among N nodes can be decomposed into N independent regression tasks. Although we did not require it for the analysis of the mTOR data, we note that the RJMCMC simulations for the individual regression tasks could be run in parallel (e.g. on separate nodes of a computer cluster) and later be merged into a network sample, as explained in more detail in Section 3.5. But this independence of the individual regression tasks also reveals the potential limitation of the whole framework. There is no possibility for information exchange among the regression models. For example, in addition to the dynamic interactions there might be contemporaneous interactions among the network nodes; i.e. additional regulatory interactions without any time lag. An interesting Bayesian model for simultaneously learning both: dynamic as well contemporaneous edges among the nodes of a network has recently been proposed in [Citation10]. An interesting idea for future research might be to incorporate the new hierarchical prior, proposed here, into the more general modeling framework from [Citation10], so as to combine the strengths of both approaches.
Supplemental Material
Download PDF (464.1 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Notes
1 Note that dynamic Bayesian networks (DBNs) are subject to a time lag of one time point. Henceforth, we will later lose one data point, reducing to the effective sample size
; cf. Section 3.5 for details.
2 Note that re-employing the variance parameter in Equation (Equation2
(2)
(2) ), yields a fully-conjugate prior in both groups of parameters
(
) and
(see, e.g. Sections 3.3 and 3.4 in [Citation5]), so that the marginal likelihood (with both parameter groups integrated out) has a closed form solution.
3 Depending on the move type , we have
.
4 In the acceptance probability, the prior ratio cancels with the HR factor
.
References
- M. Aslam, M. Azam, and C.-H. Jun, A new lot inspection procedure based on exponentially weighted moving average, Int. J. Syst. Sci. 46 (2015), pp. 1392–1400.
- P. Dalle Pezze, S. Ruf, A.G. Sonntag, M. Langelaar-Makkinje, P. Hall, A.M. Heberle, P. Razquin Navas, K. van Eunen, R.C. Tölle, J.J. Schwarz, H. Wiese, B. Warscheid, J. Deitersen, B. Stork, E. Fäßler, S. Schäuble, U. Hahn, P. Horvatovich, D.P. Shanley, and K. Thedieck, A systems study reveals concurrent activation of AMPK and mTOR by amino acids, Nat. Commun. 7 (2016), Article ID 13254.
- C. Erickson, GauPro: Gaussian Process Fitting, 2021. R Package Version 0.2.4.
- N. Gaaloul and N. Daouas, An extended approach of a Kalman smoothing technique applied to a transient nonlinear two-dimensional inverse heat conduction problem, Int. J. Therm. Sci. 134 (2018), pp. 224–241.
- A. Gelman, J. Carlin, H. Stern, and D. Rubin, Bayesian Data Analysis, 2nd ed., Chapman and Hall/CRC, London, 2004.
- M.A. Graham, A. Mukherjee, and S. Chakraborti, Distribution-free exponentially weighted moving average control charts for monitoring unknown location, Comput. Stat. Data Anal. 56 (2012), pp. 2539–2561.
- M. Grzegorczyk and D. Husmeier, Regularization of non-homogeneous dynamic Bayesian networks with global information-coupling based on hierarchical Bayesian models, Mach. Learn. 91 (2013), pp. 105–154.
- M. Grzegorczyk and D. Husmeier, Modelling non-homogeneous dynamic Bayesian networks with piecewise linear regression models, in Handbook of Statistical Genetics, 4th ed., D. Balding, I. Moltke, and J. Marioni, eds., Wiley, New Jersey, 2019, chapter 32, pp. 899–931.
- S. Moritz and T. Bartz-Beielstein, Imputets: Time series missing value imputation in R, R J. 9 (2017), p. 207.
- A. Salam and M. Grzegorczyk, Model averaging for sparse seemingly unrelated regression using Bayesian networks among the errors, Comput. Stat. (2022). Available at https://link.springer.com/article/10.1007/s00180-022-01258-9.
- G. Sanguinetti and V. Huynh-Thu, Gene Regulatory Networks, Methods in Molecular Biology: Vol. 1883, Humana Press, New York, 2019.
- M. Shafiee Kamalabad and M. Grzegorczyk, Non-homogeneous dynamic Bayesian networks with edge-wise sequentially coupled parameters, Bioinform. 36 (2020), pp. 1198–1207.
- M. Shafiee Kamalabad and M. Grzegorczyk, A new Bayesian piecewise linear regression model for dynamic network reconstruction, BMC Bioinform. 22 (2021), pp. 1–24.
- M. Shafiee Kamalabad, A.M. Heberle, K. Thedieck, and M. Grzegorczyk, Partially non-homogeneous dynamic Bayesian networks based on Bayesian regression models with partitioned design matrices, Bioinform. 35 (2019), pp. 2108–2117.
- O.M. Staal, S. Sælid, A. Fougner, and Ø. Stavdahl, Kalman smoothing for objective and automatic preprocessing of glucose data, IEEE J. Biomed. Health Inform. 23 (2018), pp. 218–226.
- Z. Zou, T. Tao, H. Li, and X. Zhu, mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges, Cell. Biosci. 10 (2020), Article ID 31.