
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
We report insights into the behaviour of achiral bent-core liquid crystals using (a) atomistic models of the PnOPIMB series of mesogens and (b) simple coarse-grained models that demonstrate the spontaneous transfer of chirality between molecules by preferential selection of chiral conformations. The models explain the unusual phenomena that chiral interactions can lead to an increase in chirality (decrease in pitch) of a bulk cholesteric liquid crystal when doped with an achiral material. We demonstrate also that chiral interactions are sufficiently strong to induce nanophase separation of an achiral mesogen within a liquid crystal phase, into nanodomains of opposite handedness by preferential selection of conformations that are instantaneously chiral (i.e. nanophase separation of and
conformations of a molecule that is strictly achiral). We suggest that this unusual behaviour contributes to the templating of chiral structures in bulk phases and, hence, to many of the extraordinary properties of bent-core mesogens that include the formations of helical nanofilaments and the chiral dark conglomerate phase.
Graphical Abstract
1. Prologue: a Festschrift for Claudio Zannoni
I was woken at 5.30am by gentle birdsong and the sun streaming through the wooden shutters of my small monastic cell. I awoke with the idea for this paper clearly in my mind: An idea born out of the conversations of the night before over Marsala and wonderful Italian hospitality.
Table 1. Optimised Ryckaert-Bellemans coefficients,
, obtained from fitting to quantum chemical data.
Table 2. Force constants and equilibrium bond lengths and angles for the coarse-grained models used.
I was in the little Italian town of Erice, perched high above the natural harbour of Trapani, on the westernmost part of Sicily, invited by Professor Claudio Zannoni to attend the International School of Liquid Crystals at the world famous Ettore Majorana centre, set in four restored monasteries. I realised that week that Claudio loved Erice (hence, the yearly International School set in such a beautiful place); but, he loved liquid crystals even more. Particularly, he loved the insights into liquid crystalline structure and dynamics provided by molecular simulation models. Over the years, the modelling work (and experiments too) from the Zannoni group has provided extraordinary insights into how liquid crystal molecules pack and arrange themselves in so many subtle ways. Consequently, I am honoured to be asked to write an article for this Festschrift and maybe it is fitting that the idea for this paper arose from discussions in Erice.
Of course, ideas born over Marsala may gloss over some of the following finer details:
How can we produce a coarse-grained mesogenic model that is super quick, quick enough and sensitive enough to be able to provide insights into the effects we are interested in?
How can we do sensible molecular dynamics (MD) by linking together anisotropic units without the integration of the equations of motion becoming intractable?
How can we introduce structural chirality into the model in a way that mimics what might occur in bent-core mesogens?
How can we measure chirality in a convenient way within a simulation model?
Many years later, all these problems are now soluble; and we are at last able to write the paper! Here, (like so many others in the field) I am again indebted to Claudio because the insights that Claudio has provided in numerous papers, over many years, have provided the knowledge and the inspiration to tackle these (and many other) problems. Grazie mille Claudio.
Mark Wilson, Durham, May 2018.
2. Introduction
The importance of chiral interactions in molecular systems has a long history dating back to the pioneering work of Pasteur. Back in 1848, Pasteur was able to separate, by hand, left-handed and right-handed crystals of tartaric acid salts to provide resolution of two enantiomeric forms [Citation1]. Here, chiral interactions at the surface of growing crystals are sufficiently strong to cause spontaneous enantiomeric resolution and the formation of enantiopure compounds.
In liquid crystal systems, chirality is ubiquitous: from chiral mesophases to the humble twisted nematic display. Back in 1888, Friedrich Reinitzer discovered the first liquid crystal from a derivative of cholesterol [Citation2,Citation3]. The cholesteryl benzoate molecules that Reinitzer worked with were chiral; but, it turned out that this ‘molecular chirality’ was transmitted to the whole phase. Hence, the first liquid crystal phase discovered was itself chiral: the cholesteric (chiral nematic) phase.
Intriguingly, liquid crystals can show chirality without the molecules themselves being chiral [Citation4]. Under the right conditions, achiral molecules can give rise to chiral structures [Citation5–Citation8]. This emergent chirality is a fascinating phenomenon and manifests itself in many different ways. For example, doping a cholesteric liquid crystal with an achiral bent-core molecule can lead to an increase in the helical twist (decrease in pitch) of the cholesteric helix [Citation9,Citation10]. Contrary to expectations, the achiral molecule, in this case, behaves like a chiral dopant with a stronger helical twisting power (HTP) than the cholesteric host. In fact, for bent-core dopant molecules, subtle changes, such as the lengthening of an achiral chain, can lead to increases in the twist of the chiral host [Citation11,Citation12]. We know also that achiral bent-core mesogens can form the twist-bend nematic phase [Citation13], where molecules form a short pitch local heliconical organisation, similar to that seen in the
phase of achiral liquid crystal dimers [Citation14–Citation18] and the related
phase [Citation19]. Moreover, for achiral bent-core molecules, the presence of spontaneous saddle splay deformation is sufficient to prevent long-range orientational or translational order, leading to the formation of structures such as the dark conglomerate phase, which exhibits large homochiral domains [Citation20–Citation22]. Whilst spontaneous racemisation of a crystal growing from a solution of two distinct enantiomeric forms seems a beautiful natural manifestation of chiral interactions, chirality arising from strictly achiral molecules in a fluid, in which molecules are able to sample a wide range of different conformations, seems truly remarkable!
This paper seeks to provide insight into such systems from a molecular viewpoint via the use of molecular simulation models. We show that achiral bent-core molecules can possess chiral conformations that have exceptionally high HTPs. We show that the bend angle in the centre of a bent-core mesogen can influence the values of HTP and that achiral chains couple to an instantaneous chirality arising from twist within the molecular core. We demonstrate chirality transfer between molecules, leading to the preferential selection of chiral conformers of one handedness over their mirror images. We also show that nanophase segregation can occur into chiral nanodomains, mediated by preferential chiral interactions.
3. Computational
3.1. Atomistic modelling
Atomistic MD simulations were carried out using the GROMACS 4.5.5 package [Citation23]. The calculations use our own modified version of the GAFF force field (GAFF-LCFF), which has been developed in two recent publications [Citation24,Citation25], including for bent-core mesogens [Citation25].
The energy function employed in the MD simulations is given by
where
are, respectively, natural bond lengths and angles,
and
are, respectively, bond, angle and torsional force constants,
and
are the usual Lennard-Jones parameters and
are partial electronic charges. Changes in
arising from deviations in improper dihedral angles,
, are represented by cosine functions using the force constants,
, the harmonic coefficients,
, and the phase angles
. The standard Lorentz-Berthelot mixing rules,
and
, have been applied throughout this work.
The Antechamber software from AmberTools 1.4 was used to generate GAFF topologies, with the point charges derived from the AM1-BCC method. GAFF-LCFF force field parameters from [Citation24,Citation25] were added in this stage. The GAFF topologies and coordinate files were converted into the GROMACS format using the ACPYPE script [Citation26]. Atomistic simulations were carried out for one molecule in the gas phase using a stochastic dynamics (SD) methodology, a 1 fs time-step and a cutoff of 1.2 nm. Typical run lengths were in the region of 500 ns ( steps). Further, SD calculations employing restraints on different parts of the molecule were carried out also, as described in Section 4.1.
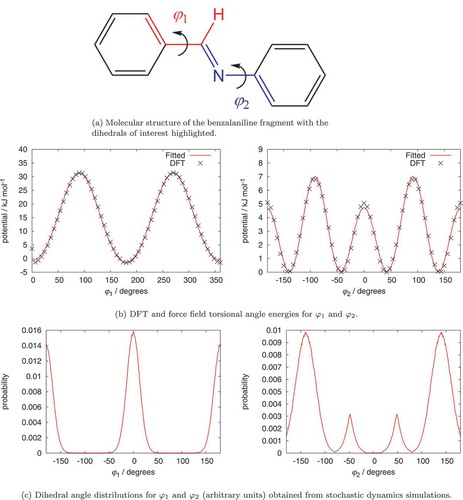
The original GAFF force field parameters do not accurately represent the two dihedral angles and
within the benzalaniline (N-benzylideneaniline) fragment, as shown in . Moreover, GAFF-LCFF [Citation24,Citation25] has not yet been extended to include these. To rectify this, we calculated torsional parameters from electronic structure theory calculations and fitted these to atomistic potentials. The quantum chemical potentials were calculated using the Gaussian 09 package [Citation27]. The B3LYP hybrid functional was used to calculate the energies in combination with a 6–311+G* basis set. The resulting potential (after subtraction of contributions from the rest of the force field) was fitted to the Ryckaert-Bellemans function in Eq. (2) by minimisation of the squared differences between quantum chemical and force field energies. The fitted parameters are given in table 1. As shown in ,
has minima at
and
resulting in a preferred planar arrangement of the connecting group with the phenyl ring. Whilst
prefers angles of
and
corresponding to a nonplanar arrangement. This nonplanar preference coupled with a preferred nonplanar arrangement of linking ester groups [Citation25,Citation28] gives rise to twisted conformations for many bent-core molecules, which, following Kawauchi et al. [Citation28], we suggest is important in contributing to the nature of the neighbour-neighbour interactions exhibited by these molecules.
Figure 1. (Colour online) Fitted dihedrals for a benzalaniline (N-benzylideneaniline) molecular fragment.
3.2. Coarse-grained modelling
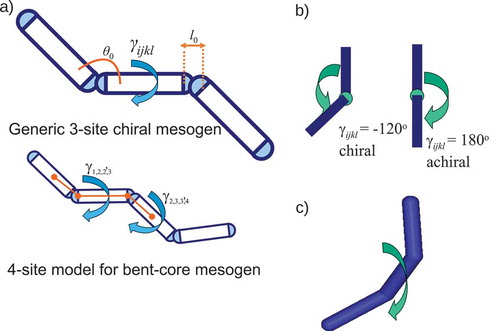
A coarse-grained simulation model was designed in which a mesogen was represented by spherocylinders of width
, joined together to form a continuous ‘bent tube’ with flexible joints, as shown in . We choose two models: one with four spherocylinders arranged to resemble a bent-core liquid crystal molecule, and a generic model composed of three spherocylinders. The latter is chosen as the minimum number of sites needed to generate true structural chirality within a ‘rigid’ bent-core framework (i.e. a bent-molecular core linked to one additional site, which can sample different orientations relative to the bent-core, as shown in .
Figure 2. (Colour online) (a) Schematic representation of the three-spherocylinder and four-spherocylinder coarse-grained models. The diagram for the four-spherocylinder model shows the position of the four points used for the definition of the dihedral angle . (b) A periodic dihedral potential around a spherocylinder is employed to favour desired equilibrium dihedral angles and provide energetic control over conformational chirality. (c) Three-dimensional representation of an instantaneously chiral conformation.
Adjacent spherocylinders were bonded together through harmonic springs, , linking the ends of the spherocylinder line segments. Additional harmonic potentials,
, were used to constrain the angles of terminal spherocylinders with respect to the axis vector of the central spherocylinder for the three-site model and with respect to neighbouring spherocylinders for the four-site model. Equilibrium values for bond lengths and angles are given in . For this phenomenological model, which is akin to a DPD model for a liquid crystal mesogen [Citation29] (but with anisotropic sites), force constants are chosen to be sufficiently large to maintain the molecular geometry for the chosen simulation conditions and the soft-core spherocylinder potential described in the following, whilst sufficiently small to conserve energy for the chosen time step,
. As usual, nonbonded interactions were excluded between directly bonded spherocylinder sites.
To control the conformational chirality of the model molecule, a ‘dihedral’ potential was employed to control rotation around a spherocylinder, as defined by the angle . Two dihedral potentials were considered,
and
Here, the parameter controls the height of the barrier between the consecutive wells and
is used to change the equilibrium dihedral angle (and hence the chirality). The integer
sets the number of wells in Eq. (2) and controls the width of the two wells in Eq. (3). In our calculations, for
we measure the torsional angle between ends of spherocylinder line segments for a central spherocylinder and the centres of mass of the two adjacent spherocylinders (i.e.
would be the single dihedral defined for the three-spherocylinder model) and integrate the forces and torques that arise from the chosen potential (Eqs. (2) or (3)). For the three-site model, there is just one dihedral angle defined,
. However, for the four-site model, there are two dihedral angles:
and
as shown in . Note that, for the three-spherocylinder model, choosing values of
or
results in an achiral (planar) molecule; and, for the four-spherocylinder model, choosing both
and
equal to values of either
or
similarly results in an achiral (planar) molecule.
Nonbonded interactions between spherocylinders were described by a soft-core spherocylinder model [Citation30,Citation31] (see in the following) with a parametrisation,
. This parameterisation is known to lead to nematic stability over a wide density range for single site mesogens [Citation30,Citation31]. We employed the MD methodology of reference [Citation30] using the GBMOL program [Citation32–Citation34]. Simulations were carried out at
in the nematic phase, with typical runs employing 2–4 million MD steps.
The soft-core spherocylinder interaction potential is given by
where is the interaction energy of fully overlapped particles,
is the line segment distance and
is the well depth.
, the magnitude of the attractive component is angle dependent, i.e. a function of the orientation of the vector from a centre of mass,
, and orientations of particles i and j (
and
). This potential can be easily tuned for different liquid crystal behaviours by altering the four parameters:
,
,
and
[Citation30,Citation31] in a similar way to the well-known Gay-Berne potential [Citation35].
For the three-site spherocylinder model, molecules were simulated in a cuboidal box (
and
) with periodic boundaries in
and
directions. A repulsive wall was employed in the
-direction, acting on the
-component of the centre of mass of spherocylinders,
, beyond the top or bottom of the simulation box:
with
and
.
3.3. Importance of choosing soft-core interaction potentials
The key advantage of the coarse-grained DPD-like model used in this work is the speed of equilibration. The model is sufficiently repulsive to promote liquid crystal behaviour through anisotropic excluded volume interactions but sufficiently soft to allow rapid movement through phase space, avoiding the jamming which is sometimes seen with harder particle coarse-grained models. The benefits of this type of model have been demonstrated by Zannoni and co-workers [Citation36,Citation37] and used to look at changes in molecular organisation in the actuation cycle of a liquid crystal elastomer [Citation38]. We also note the similarity of our four-site model to the Monte Carlo model of Peroukidis et al. [Citation39]. In their model, they used spherocylinders of different sizes for the core and ‘tail’ of a bent-core mesogen, allowing the molecule to adopt both chiral and achiral conformations. They observed the formation of clusters exhibiting local smectic order, which were either orthogonal or tilted, together with homochiral domains through a helical arrangement of the tilted smectic clusters.
We note in passing that the use of a soft repulsive wall together with our soft-core spherocylinder model leads to very weak anchoring at the wall surface by design. This means that the wall does not restrict the rotation of molecules in the plane, allowing a twist induced on a nematic director in the
plane to easily overcome any influence that can otherwise be introduced by the presence of harder walls. In addition, the weak anchoring ensures that smectic layers are not induced at the wall surface. An interesting alternative will be the use of soft walls in combination with self-determined boundary conditions, as in the recent work of Kuhnhold and Schilling [Citation40].
3.4. Quantifying molecular twist in the bulk
To quantify the effect of molecular chirality on the twist of the bulk phase, for boxes with walls in the -direction, the simulation box was divided into layers with thickness of 1.7
along the
-axis. A local nematic director was calculated for each layer,
from diagonalisation of the ordering tensor for the central spherocylinders. This allowed the calculation of the twist angle,
, along the
-axis. For linear increases in
along
, the bulk pitch,
, can be obtained by a linear fit,
, as
.
3.5. Calculation of HTPs
Ferrarini et al. [Citation41–Citation44] showed that molecular isosurfaces could be used to evaluate the chirality order parameter, , which is proportional to the HTP of a mesogen in a nematic phase. To calculate
, we use the step-by-step approach described in detail by Earl and Wilson [Citation45]. Specifically, we first calculate a molecular surface from an atomistic molecular conformation (or a coarse-grained representation in the case of the coarse-grained models). We used an isosurface corresponding to the van der Waals surface accessible to a spherical probe with a diameter of 5 Å (in the case of coarse-grained models the probe was placed in contact with the surface of the spherocylinders). Isosurfaces were generated using the simple invariant molecular surface routine of Vorobjev and Hermans [Citation46] with a resolution of 10 dots per Å
. We next calculated a normal vector at each surface point and then calculated the surface tensor
, the helicity tensor
and the ordering matrix
(see Eqs. (3), (4) and (6) in [Citation45]) in order to evaluate
.
values were determined for
conformations for atomistic simulations and for selected snapshots for coarse-grained models.
4. Results and discussion
4.1. Conformational distribution functions
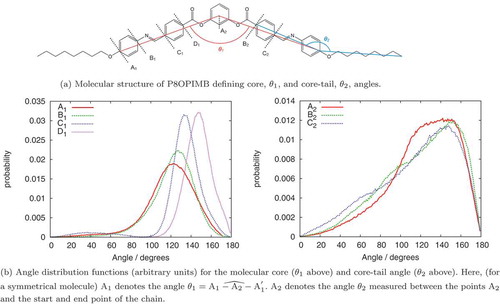
Angular distribution functions, obtained from the SD simulations, are plotted in for the bend angle of the molecular core and the angle between the core and chain. As shown in , the measured distributions are highly dependent on the exact definition of the angle. However, in the most commonly used definition of the ‘molecular core’ ( in ) the angle distribution function is peaked at 120
commensurate with that expected from a bent structure originating from a central phenyl ring. The distribution function for
is, however, quite broad, reflecting the flexibility of the ester and benzalaniline linkages in the core. Consequently, the idealised concept of a ‘rigid bent-core’ is certainly not true. The core-chain angle has a particularly wide distribution function, which is peaked between
but which extends to quite low angles arising from a long-flexible chain, which is able to bend back on itself. Within a nematic phase, we would expect these distribution functions to be somewhat less broad, with the angle distributions being coupled to the field of surrounding molecules. The phenomenon is known well for chain conformations in
-alkylcyanobiphenyls, where preferential selection occurs for conformations that enhance alignment with the nematic mean field [Citation47,Citation48].
Figure 3. (Colour online) Angle distributions for P8OPIMB from single-molecule stochastic dynamics calculations at 300 K using the GAFF-LCFF force field.
4.2 χ values for PnOPIMB
Recently, Kim et al. [Citation12] have calculated values for various members of the PnOPIMB series. Here (), we repeat that work for
using the new GAFF-LCFF force field, and extend the calculation to the core only
homologue. In addition, we carry out SD calculations with MD restraints applied to the chain (giving an all-trans chain), to the ester group (giving a planar arrangement for the three central rings) and to the whole core (giving a planar arrangement for the whole core). Several features are apparent from :
Figure 4. (Colour online) Top: distribution functions (arbitrary units) for a PnOPIMB molecule from stochastic dynamics calculations at 300 K. Middle:
distribution functions for P12OPIMB employing chain, ester and core restraints. Bottom: An instantaneous twisted conformation for P8OPIMB with
showing the molecular surface used to calculate
.
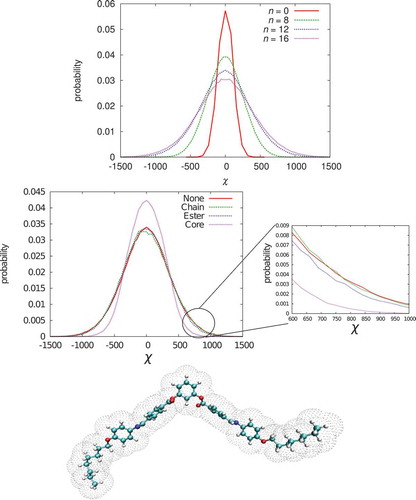
Excellent conformational sampling within the SD calculations leads to distribution functions that are symmetrical and centred on zero; demonstrating, as expected, no net chirality.
The wings of the distribution function extend to exceptionally high values of
, around 50
The effect of the chain length is significant, with increased chain lengths leading to a widening of the distribution function. In contrast, the calculations with no chain give a sharply peaked distribution. (We note that with GAFF-LCFF, which has an improved description of the ester and benzalaniline linkages, and the alkoxy chain, that a widening of the
For P12OPIMB, applying restraints to the whole core (giving an approximately planar core), leads to a big reduction in the higher values of
We suggest that the huge instantaneous values of seen in the wings of the distribution are responsible for the increase in HTP when a chiral nematic is doped with PnOPIMB. Highly twisting conformations will be preferentially selected in a chiral molecular field. Moreover, it appears that the main origin of the high HTP values arises from twisted (chiral) conformations of the core and if these are eliminated, by using a planar core, the
distribution is sharpened up considerably. We note, however, that even in this case, chiral conformations of the chains can occur and their HTP is amplified by the presence of the bent-core shape. Constraining the chains to an all-trans conformation does not reduce HTP significantly for P12OPIMB. This is because the twist of the core still leads to an overall twisted shape for the molecule, with the two chains pointing in different directions, and hence a large HTP is obtained. Interestingly, if the chains are extended to
, applying chain restraints leads to a further widening of the distribution function. This suggests that, in this case, it is not the addition of further ‘instantaneously chiral’ chain conformations that is most important but rather the length of the chain that contributes to an overall enhanced chiral shape for the molecule as a whole. We explore these ideas further with the use of idealised coarse-grained models (see the following).
4.3 χ values for coarse-grained models
We carried out single-molecule dynamics calculations for the three-site and four-site models, described in Section 3.2, using an equilibrium bond angle of and an equilibrium dihedral of
but with a very low force constant (
) to allow easy sampling of twisted conformations arising from other values of this dihedral. In addition, we removed the two terminal spherocylinders from the four-site model, leaving only a ‘bent-core’ (
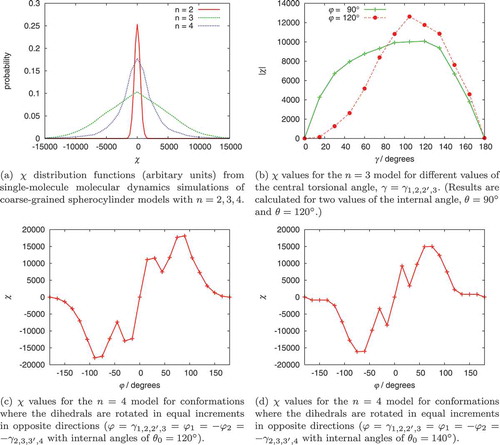
model). The results are given in (a).
Intriguingly, the system appears to be the model that exhibits chiral conformations with the largest HTP. However, the four-site model captures the same shaped
distribution that we saw from atomistic modelling. Not surprisingly, removing the two terminal spherocylinders to leave a
system leads to a distribution strongly peaked about
with only configurations where the spherocylinders fluctuate out of plane contributing significantly to
. For the
and
models, the
values in the wings of the distribution are an order of magnitude larger than those obtained with the highest HTP conformations seen in the atomistic simulations. This provides hope that, for these models, one might be able to see induced helical twists on the scale of molecular simulations (see Section 4.4).
Further insights are provided by examining how varies with molecular conformation. For the three-site model ((b)) the value of
peaks for a dihedral of
and varies smoothly as a function of this dihedral. The values of
are also shown to depend on the internal angle
. For the four-site model, with internal angles of 120
, the highest HTP occurs at
, and there is an unexpected dip in HTP for
((c)). Moreover, changing the internal angle to
((d)), as would occur in an oxadiazole-based bent-core mesogen [Citation49], subtly changes the form of the curve, with the maxima shifting to
and the local minima shifting to
. We note that, for the four-site model, conformations where
(which, for an achiral molecule, always have identical torsional energies to conformations with
) have superimposable mirror images and hence
.
Figure 5. (Colour online) values for single molecule coarse-grained models.
4.4 Bulk simulations of the three-site coarse-grained model
The effect of molecular-level chirality on a bulk phase was initially determined via six simulations using the dihedral potential of Eq. (2); using parameters and
,
,
,
,
,
. These correspond to equilibrium dihedrals
,
,
,
,
,
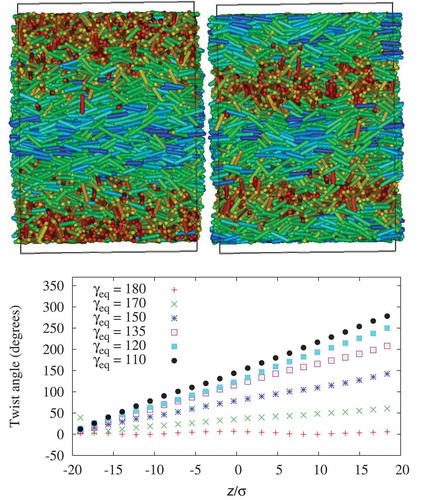
. The simulations used the soft repulsive wall boundary conditions described in Section 3.2, leading to macroscopic twists across the simulation box, corresponding to a chiral nematic phase with a small pitch length. We quantify the twist by calculating the order parameter,
, and the director,
, along the
-axis of the simulation box (as described in Section 3.4), and by plotting the change in angle of the director with changing
, as shown in . The overall twist,
, is seen to increase from,
, for the planar (achiral) molecules
to
for the highest chiral strength
. The mean nematic order parameter remains well inside the nematic region, ranging from
for the achiral system to
for the system with the highest chirality. A small local increase in
near the wall was observed (maximum of 17%) which drops to the bulk value over the width of one histogram bin
.
Figure 6. (Colour online) Top: Snapshots for for the three-site spherocylinder model. Snapshots are viewed along the
-axis of the simulation box for two chiral nematic systems, with
(top left) and
(top right). Bottom: The effect of molecular chirality on the twist angle between a reference nematic director,
, at the base of the simulation box and the local nematic director,
, along the
-axis of the simulation box.
To model the effects of adding achiral dopants (with the possibility of chiral conformations) to a system, achiral molecules were designed with two symmetrical potential wells at , separated by a barrier of height
(corresponding to
and
in Equation (3)). Two host systems with 10% doping were considered: a chiral host phase with
and, as a control (to check statistics) a second host achiral host phase
was also employed. Both cases used a maximum barrier height of
.
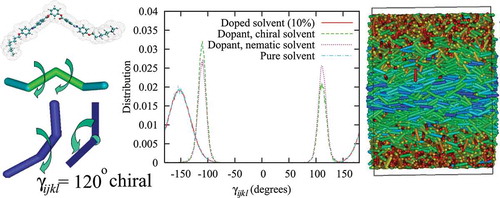
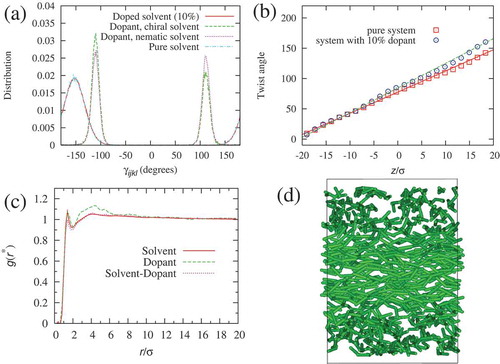
shows the dihedral angle distributions, , obtained for the solvent and dopant molecules. The preferred handedness of the dopant molecules can be obtained from the relative populations of right- and left-handed molecules,
and
, provided by numerical integration of
. For the test case, i.e. dopant molecules in the achiral nematic phase (dotted line in (a)),
within a statistical error of 1%. This level of sampling is only possible because of the speed of the coarse-grained model we employ, which means that it is able to fully equilibrate the system and sample the conformations to a high degree of statistical accuracy.
When the same dopants are added to the chiral host phase (dot-dashed line in (a)) preferential selection of conformations occurs for the conformer with the same handedness as the host phase. Here, relative populations of 0.5% and
were observed for a left-handed host solvent. This same observation is found for a series of values of different
for the dopant system. In the case of
, the dopants have conformations with higher twisting power than the host phase. A subsequent plot of bulk twist angle in (b) shows an increase in the twist of the system with 10% achiral dopants in comparison to the pure chiral solvent. From the linear fits, pitch lengths were estimated as
and
for the doped and pure systems, respectively. This corresponds to a 17% decrease of the pitch length (increase in the twist).
Figure 7. (Colour online) (a) Dihedral angle distributions from MD simulations. Chiral solvent (solid line), dopant in chiral solvent (dashed line), dopant in nematic solvent (dotted line) and solvent without dopant molecules (dot-dashed line). (b) Twist angle for nematic director along the -axis: pure melt (squares) and system with 10% doping of achiral solute. The lines are least-squares fits of the linear function
. (c) Radial distribution functions,
, for solvent molecules (solid line), dopant molecules (dashed line) and solvent-dopant cross distribution (dotted line). (d) Snapshot of the dopant molecules, where solvent molecules are removed for clarity.
Three possible mechanisms can be evoked to explain the increase in the bulk twist of the doped sample.
(1) Dopant molecules could increase the preferred dihedral angles of the solvent molecules, thereby increasing chirality of the solvent molecules.
(2) Dopant molecules are distributed evenly in the system but the preferential selection of high twisting conformations of the same handedness as the solvent leads to an overall increase of the bulk twist.
(3) The dopant molecules could undergo microphase separation to form highly twisting clusters leading to an overall increase in the twist across the system.
The first of these mechanisms can be immediately ruled out. The calculated dihedral angle distributions for the solvent with 10% doping (solid line in ) and the pure solvent (dot-dashed line) are in agreement within the line width. We expect also that this mechanism is not experimentally important, i.e. most chiral hosts are chiral due to the presence of chiral centres or in-built structural chirality, and do not tend to have highly twisting conformations, which would come into play only when preferentially selected through interaction with an achiral dopant. From the results in , the preferential selection of high HTP conformers of the dopant mechanism clearly does occur. However, this might be seen in conjunction with the third mechanism. To test this, we looked at the spatial distribution of dopant molecules by calculating partial radial distribution functions, for all pairs of species ((c)). Each
shows a peak at a separation of
corresponding to nearest neighbour interactions. However, the
for the dopant-dopant interactions shows the growth of a slight shoulder between separations of
and
, indicating a slight preference for the formation of small clusters at this distance. A snapshot picture ((d)) confirms that at the 10% dopant level the dopant molecules are uniformly distributed across the simulation box, apart from some evidence for the formation of very small local clusters. Hence, it appears that preferential interaction between chiral conformations of neighbouring dopants is likely to contribute to an increase in overall HTP.
Further evidence of chiral selectivity can be observed in related simulations. In , we show a snapshot picture taken from a uniform nematic phase for the three-site spherocylinder model. Here, a system of strictly achiral molecules (with a preference for no chirality ensured from ) has been simulated and a colour coding has been applied according to the handedness of the molecules. From this, it can be seen how locally the molecules prefer the same handedness as their neighbours leading to nanophase segregation into small domains with opposite handedness. In our model, within a nematic phase, the chiral domains are quite small as opposed to experimental findings in more complex phases [Citation20,Citation21,Citation50,Citation51] where large homochiral domains are sometimes found, enhanced by strong neighbour packing effects. Nonetheless, the preference for chiral interactions seen in our model is clear.
Figure 8. (Colour online) A snapshots showing chiral nanophase segregation in an achiral nematic phase () where the colour coding has been applied as blue (red) left (right) handed conformations.

5. Conclusions
We have studied chirality arising from achiral bent-core liquid crystal molecules via atomistic and coarse grained-simulation models. We demonstrate huge HTPs for individual conformations of three bent-core molecules, PnOPIMB (), with the HTP values increasing with alkoxy chain length. We show that these conformational effects can be captured using simple three-site and four-site soft spherocylinder models. Further, we show, with a three-site spherocylinder model, that chiral induction can take place within a bulk nematic phase.
For achiral molecules our coarse-grained simulations show a uniform nematic phase; but, when conformational chirality is introduced, a chiral nematic phase is formed with the measured twist angle of the bulk phase increasing as the internal twist angle of individual molecules is increased. Moreover, the simulations demonstrate a decrease in pitch length (increased twist) for a chiral nematic, when the system is doped with an achiral dopant, in agreement with surprising experimental results for bent-core molecules [Citation9,Citation11]. Here, we see the preferential selection of high twisting enantiomeric conformations over their mirror images.
Finally, we note that we see evidence for local chiral segregation into nanodomains of opposite handedness within a bulk nematic phase. It will be interesting to explore such effects further with a view to understanding the formation of helical nanofilaments and larger chiral domains within more exotic liquid crystal mesophases.
Acknowledgments
MRW and MW would like to thank EPSRC for funding under grants EP/J004413/1 and EP/P007864/1. JSL would like to acknowledge funding from an IdEx (Initiative d’Excellence) Bordeaux Junior Chair. We would like to thank Professor Claudio Zannoni for valuable discussions over many years, and for his insights into the simulation of liquid crystal systems. We gratefully acknowledge conversations with Professors Vanakaras and Photinos in Erice and elsewhere, which have provided many insights into the behaviour of bent-core systems.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Gal J. Pasteur and the art of chirality. Nat Chem. 2017;9:604–605.
- Reinitzer F. Beitrge zur Kenntniss des Cholesterins. Monatshefte Für Chemie. 1888;9:421.
- Reinitzer F. Contributions to the knowledge of cholesterol. Liq Cryst. 1989;5(1):7–18.
- Heppke G, Moro D. Chiral Order from Achiral Molecules. Science. 1998;279(5358):1872–1873.
- Horsch MA, Zhang Z, Glotzer SC. Self–assembly of Polymer–tethered Nanorods. Phys Rev Lett. 2005;95(5):056105.
- Yan F, Hixson CA, Earl DJ. Self–assembled Chiral Superstructures Composed of Rigid Achiral Molecules and Molecular Scale Chiral Induction by Dopants. Phys Rev Lett. 2008;101(15):157801.
- Chakrabarti D, Wales DJ. Tilted and Helical Columnar Phases for an Axially Symmetric Discoidal System. Phys Rev Lett. 2008;100(12):127801.
- Walker M, Wilson MR. Formation of complex self–assembled aggregates in non–ionic chromonics: dimer and trimer columns, layer structures and spontaneous chirality. Soft Matter. 2016;12(41):8588–8594.
- Thisayukta J, Niwano H, Takezoe H, et al. Enhancement of Twisting Power in the Chiral Nematic Phase by Introducing Achiral Banana–shaped Molecules. J Am Chem Soc. 2002;124(13):3354–3358.
- Earl D, Osipov M, Takezoe H, et al. Induced and spontaneous deracemization in bent-core liquid crystal phases and in other phases doped with bent-core molecules. Phys Rev E. 2005;71(2):021706.
- Jo SY, Kim BC, Jeon SW, et al. Enhancement of the helical twisting power with increasing the terminal chain length of nonchiral bent-core molecules doped in a chiral nematic liquid crystal. RSC Adv. 2017;7:1932–1935.
- Kim BC, Walker M, Jo SY, et al. Effect of terminal chain length on the helical twisting power in achiral bent-core molecules doped in a cholesteric liquid crystal. RSC Adv. 2018;8:1292–1295.
- Chen D, Nakata M, Shao R, et al. Twist–bend heliconical chiral nematic liquid crystal phase of an achiral rigid bent-core mesogen. Phys Rev E. 2014;89(2):022506.
- Ivšic′ T, Baumeister U, Dokli I, et al. Sensitivity of the NTB phase formation to the molecular structure of imino–linked dimers. Liq Cryst. 2017;44(1):93–105.
- Abberley JP, Jansze SM, Walker R, et al. Structureproperty relationships in twist–bend nematogens: the influence of terminal groups. Liq Cryst. 2017;44(1):6883.
- Archbold CT, Andrews JL, Mandle RJ, et al. Effect of the linking unit on the twist-bend nematic phase in liquid crystal dimers: a comparative study of two homologous series of methylene- and ether-linked dimers .Liquid Crystals. 2017;44(1):84–92.
- Chen D, Porada JH, Hooper JB, et al. Chiral heliconical ground state of nanoscale pitch in a nematic liquid crystal of achiral molecular dimers. Proc Natl Acad Sci. [2013];110(40):15931–15936.
- Borshch V, Kim YK, Xiang J, et al. Nematic twist–bend phase with nanoscale modulation of molecular orientation. Nat Commun. 2013;4:2635.
- Abberley JP, Killah R, Walker R, et al. Heliconical smectic phases formed by achiral molecules. Nat Commun. 2018;9(1):228.
- Hough LE, Jung HT, Krerke D, et al. Helical Nanofilament Phases. Science. 2009;325(5939):456–460.
- Hough LE, Spannuth M, Nakata M, et al. Chiral Isotropic Liquids from Achiral Molecules. Science. 2009;325(5939):452–456.
- Nagaraj M. Dark conglomerate phases of bent-core liquid crystals. Liq Cryst. 2016;43(13–15):2244–2253.
- Pronk S, Páll S, Schulz R, et al. Gromacs 4.5: a high–throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29(7):845–854.
- Boyd NJ, Wilson MR. Optimization of the GAFF force field to describe liquid crystal molecules: the path to a dramatic improvement in transition temperature predictions. Phys Chem Chem Phys. 2015;17(38):24851–24865.
- Boyd NJ, Wilson MR. Validating an optimized GAFF force field for liquid crystals: TNI predictions for bent-core mesogens and the first atomistic predictions of a dark conglomerate phase. Phys Chem Chem Phys. 2018;20(3):1485–1496.
- Aws DS, Vranken WF. ACPYPE–antechamber python parser interface. BMC Res Notes. 2012;5(1):367.
- Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 09. Wallingford CT: Revision A.1. Gaussian, Inc; 2009.
- Kawauchi S, Choi SW, Fukuda K, et al. Why Achiral Rod–like Compound with Ester Group Amplifies Chiral Power in Chiral Mesophase. Chem Lett. 2007;36(6):750–751.
- Walker M, Wilson MR. Simulation insights into the role of antiparallel molecular association in the formation of smectic A phases. Soft Matter. 2016;12(43):8876–8883.
- Lintuvuori JS, Wilson MR. A new anisotropic soft–core model for the simulation of liquid crystal mesophases. J Chem Phys. 2008;128(4):044906.
- Lintuvuori JS, Wilson MR. A coarse–grained simulation study of mesophase formation in a series of rod–coil multiblock copolymers. Phys Chem Chem Phys. 2009;11(12):2116–2125.
- Wilson MR. GBMOL: A replicated data molecular dynamics program to simulate combinations of Gay–berne and Lennard–jones sites. Author: Mark R. Wilson. University of Durham; 1996.
- Wilson MR, Allen MP, Warren MA, et al. Replicated data and domain decomposition molecular dynamics techniques for simulation of anisotropic potentials. J Comput Chem. 1997;18(4):478–488.
- Wilson MR. Molecular dynamics simulations of flexible liquid crystal molecules using a Gay–berne/Lennard–jones model. J Chem Phys. 1997;107(20):8654–8663.
- Berardi R, Emerson APJ, Zannoni C. Monte Carlo investigations of a GayBerne liquid crystal. J Chem Soc, Faraday Trans. 1993;89(22):4069–4078.
- Berardi R, Zannoni C, Lintuvuori JS, et al. A soft–core GayBerne model for the simulation of liquid crystals by Hamiltonian replica exchange. J Chem Phys. 2009;131(17):174107.
- Berardi R, Lintuvuori JS, Wilson MR, et al. Phase diagram of the uniaxial and biaxial softcore GayBerne model. J Chem Phys. 2011;135(13):134119.
- Skaej G, Zannoni C. Molecular simulations elucidate electric field actuation in swollen liquid crystal elastomers. PNAS. 2012;109(26):10193–10198.
- Peroukidis SD, Vanakaras AG, Photinos DJ. Molecular simulation of hierarchical structures in bent-core nematic liquid crystals. Phys Rev E. 2011;84(1):010702.
- Kuhnhold A, Schilling T. Isotropic–nematic transition and cholesteric phases of helical Yukawa rods. J Chem Phys. 2016;145(19):194904.
- Ferrarini A, Moro GJ, Nordio PL. A shape model for the twisting power of chiral solutes in nematics. Liq Crys. 1995;19(3):397–399.
- Ferrarini A, Moro GJ, Nordio PL. Shape model for ordering properties of molecular dopants inducing chiral mesophases. Molec Phys. 1996;87(2):485–499.
- Ferrarini A, Gottarelli G, Nordio PL, et al. Determination of absolute configuration of helicenes and related biaryls from calculation of helical twisting powers by the surface chirality model. J Chem Soc, Perkin Trans 2. 1999;(3):411–418.
- Di Matteo A, Sm T, Gottarelli G, et al. Correlation between Molecular Structure and Helicity of Induced Chiral Nematics in Terms of Short–range and Electrostatic––induction Interactions. The Case of Chiral Biphenyls. J Am Chem Soc. 2001;123(32):7842–7851.
- Earl DJ, Wilson MR. Predictions of molecular chirality and helical twisting powers: a theoretical study. J Chem Phys. 2003;119(19):10280–10288.
- Vorobjev YN, Hermans J. SIMS: computation of a smooth invariant molecular surface. Biophys J. 1997;73(2):722–732.
- Emsley JW, Luckhurst GR, Stockley CP. A theory of orientational ordering in uniaxial liquid crystals composed of molecules with alkyl chains. Proc R Soc Lond A. 1982;381(117):28741.
- Wilson MR. Molecular modelling of liquid crystal systems: an internal coordinate Monte Carlo approach. Liq Cryst. 1996;21(3):437–447.
- Srigengan S, Nagaraj M, Ferrarini A, et al. Anomalously low twist and bend elastic constants in an oxadiazole–based bent-core nematic liquid crystal and its mixtures; contributions of spontaneous chirality and polarity. J Mater Chem C. 2018;6(5):980–988.
- Gortz V, Goodby JW. Enantioselective segregation in achiral nematic liquid crystals. Chem Commun. 2005;26:3262–3264.
- Gortz V, Southern C, Roberts NW, et al. Unusual properties of a bent-core liquid–crystalline fluid. Soft Matter. 2009;5:463–471.