
ABSTRACT
Transparency and stability to UV light are important and desirable properties for modern tunable optical elements and active soft robots. A library of novel reactive mesogens for liquid crystal polymer networks resilient and transparent to UV light has been synthetised and characterised. Phase behaviours of the reactive mesogens have been determined by polarised optical microscopy and differential scanning calorimetry. Liquid crystal polymer networks based on the combination of these novel reactive mesogens have been evaluated and compared to those based on common commercially available compounds. The results showed a twofold increase in transparency in a broad UV spectral region (200–400 nm) and importantly showed no degradation upon prolonged UV exposure contrary to the networks composed from commercial counterparts.
GRAPHICAL ABSTRACT
1. Introduction
Liquid crystalline materials have recently reached beyond applications in display technology, to extend to smart materials for advanced optics such as tunable filters, retarders, diffraction gratings and sensors [Citation1–5]. The design and synthesis of sophisticated liquid crystal polymer networks (LCPNs) have also opened new perspectives for soft robotics and the development of light-driven soft robots [Citation6,Citation7].
In liquid crystal systems, light responsiveness is promoted by the use of photoactive molecules – either by doping native liquid crystals or by coupling them covalently to a polymer network. Light-induced molecular changes such as isomerisation, cyclisation and cycloaddition cause structural changes in these photoactive molecular switches, and these are transmitted to the material, which modifies its properties [Citation8] and allows performing work, as anticipated by de Gennes in 1997 [Citation9]. Photoswitches (azobenzenes, hydrazones, stilbenes, etc.) [Citation10–13] and molecular motors [Citation14,Citation15] are the most widely used active molecules in LCPNs, and they usually absorb strongly in the UV. However, up-to-date LCPNs fabricated from commercially available reactive mesogens partially absorb light in the same spectral range as the photoswitches, thus limiting their operation and narrowing down the range of light-responsive switches that can be embedded due to two main reasons: (i) low transparency of LCPNs in the desired UV spectral range; (ii) low photostability of LCPNs to UV light. Low transparency sufficiently decreases the efficiency of photochemical processes since most of the light is absorbed by the predominant liquid crystal molecules of LCPNs and dissipated as heat. Regarding the issue of stability, LCPNs are mostly degraded or irremediably modified by photo-Fries rearrangement [Citation16], the homolytic photo-cleavage followed by recombination which usually takes place in phenyl esters but that can also occur with various molecular moieties like diphenyl ether, amide, acetanilide, etc. [Citation17].
In this work, we have overcome these two limitations in order to create materials which are transparent and resilient to UV light, including UV-A (315–400 nm) and UV-B (280–315 nm), and therefore are suitable as host media for UV photoactive molecules (switches and molecular motors). We describe the synthesis and characterisation of 12 reactive mesogens (mono- and diacrylates), which can be used as building blocks for LCPNs with enhanced photostability and UV transparency unmatched by commercially available analogues.
2. Experimental
2.1. Materials and instruments
All reactions are carried out under N2. Tetrahydrofuran (THF) was purified and dried over Braun solvent purification system (MB-SPS-800), and other solvents (DMSO, DMF, CH2Cl2, heptane and ethanol) were purchased dry from Sigma-Aldrich and used without further purification. The chemical reagents were purchased from Synthon, abcr and Sigma-Aldrich and used as received. Analytical thin-layer chromatography was carried out on Merck silica gel 60 F254. Products were revealed by ultraviolet light (254 or 366 nm) and stained with dyeing reagents (potassium permanganate aqueous solution). Flash chromatography was performed on Combiflash® Companion or with Merck silica gel 60 (230–400 mesh). 1H and 13C NMR spectra were recorded at ambient temperature on Bruker Ascend™ 400 spectrometers operating at 400 MHz 1H. 13C nucleus was observed with 1H decoupling. Solvent residual signals were used as internal standard. Chemical shifts (δ) and coupling constants (J) are given in ppm and Hz, respectively. The peak patterns are indicated as the following format multiplicity (s: singlet; d: doublet; t: triplet; q: quartet; sept: septuplet; m: multiplet; dd: doublet of doublet; dt: doublet of triplet; dm: doublet of multiplet, etc.). The prefix br. indicates a broadened signal. Mass spectrometry was performed on MSVision spectrometer (micromass LCT). UV-vis spectra were measured on HR2000 + High-Resolution spectrometer (Ocean Optics). The phase behaviour was studied with a polarised optical microscope BX51 (Olympus) equipped with a heating stage (Instec). Differential scanning calorimetry (DSC) study was performed using Netzsch DCS-214 machine with heating/cooling speed 10 K/min.
2.2. Synthesis
General procedure 1 (GP1)
Phenol compound (1 eq.), alkyl halogen compound (1 eq.), K2CO3 (2.8 eq.) and KI (cat.) were dissolved in absolute ethanol (5 mL/1 mmol) and heated to reflux for 48 h. The organic phase was washed with a saturated solution of NaOH in water (5 mL/1 mmol), distilled water (5 mL/1 mmol) and brine (5 mL/1 mmol). The organic phase was dried over MgSO4, and the solvent was evaporated under reduced pressure. The product was purified by flash chromatography on silica with the appropriate eluent.
General procedure 2 (GP2)
The alcohol (1 eq.) and NEt3 (1.5 eq.) were dissolved in THF (5 mL/1 mmol) and stirred at 0°C. The acyl chloride (1.2 eq.) was added slowly while stirring at 0°C. After 1 h, the reaction was allowed to warm up to r.t. and was stirred for an additional 16 h. The resulting suspension was filtrated, and the filtrate was dried under reduced pressure. The residue was dissolved with CH2Cl2 (10 mL/1 mmol) and washed successively with distilled water (2× 10 mL/1 mmol) and brine (10 mL/1 mmol). The organic phase was dried over MgSO4, and the solvent was evaporated under reduced pressure. The product was purified by flash chromatography on silica with the appropriate eluent.
General procedure 3 (GP3)
The alkyl halogen (1 eq.), potassium acrylate (2 eq. × number of halogen function), KI (0.4 eq. × number of halogen function) and 4-methoxyphenol (2 crystals) were dissolved in DMSO (5 mL/1 mmol) and stirred at 52°C for 72 h. The reaction was cooled down to r.t., and the product was precipitated with distilled water. The precipitate was filtrated and washed with distilled water. The solid was dissolved in CH2Cl2 (10 mL/1 mmol) and washed with distilled water (2× 10 mL/1 mmol) and brine (10 mL/1 mmol). The organic phase was dried over MgSO4, and the solvent was evaporated under reduced pressure. The product was purified by flash chromatography on silica with the appropriate eluent.
6-(4ʹ-((1s,4ʹr)-4-Propylcyclohexyl)phenoxy)hexan-1-ol (1).
1 was synthesised according to GP1. Trans-4-(4-propylcyclohexyl)phenol (5 g, 23 mmol) and 6-bromohexanol were used, respectively, as phenol and alkyl halogen compound. Flash chromatography with CH2Cl2/MeOH (99:1) as eluent yields 4.1 g of pure compound.
Yield = 56%. Rf (CH2Cl2/EtOAc, 8:2) = 0.44.
1H NMR (CDCl3, 400 MHz) δ = 0.86 (t, 3H, J = 7.25 Hz, -CH3), 1.00 (2H, m, -CH2-CH3), 1.15–1.49 (11H, m, -CH-CH2-, -CH2-CH2-CH-, -CH2-CH2-CH2-), 1.56–1.83 (8H, m, -CH-CH2-CH2-CH-), 2.36 (1H, tt, J = 12.20, 3.23 Hz, Ar-CH), 3.60 (2H, t, J = 6.57 Hz, CH2-OH), 3.89 (2H, t, J = 6.48 Hz, Ar-O-CH2), 6.77 (2H, d, J = 8.55 Hz, Ar-H), 7.07 ppm (2H, d, J = 8.57 Hz, Ar-H).
6-(4ʹ-((1s,4ʹr)-4-Pentylcyclohexyl)phenoxy)hexan-1-ol (2).
2 was synthesised according to GP1. Trans-4-(4-pentylcyclohexyl)phenol (5 g, 20 mmol) and 6-bromohexanol were used, respectively, as phenol and alkyl halogen compound. Flash chromatography with CH2Cl2/MeOH (99:1) as eluent yields 5 g of pure compound.
Yield = 71%. Rf (CH2Cl2/EtOAc, 8:2) = 0.44.
1H NMR (CDCl3, 400 MHz) δ = 0.89 (t, 3H, J = 6.98 Hz, -CH3), 1.03 (2H, m, -CH2-CH3), 1.18–1.48 (15H, m, -CH-CH2-, -CH2-CH2-CH-, -CH2-CH2-CH2-), 1.56–1.89 (8H, m, -CH-CH2-CH2-CH-), 2.40 (1H, tt, J = 12.18, 3.21 Hz, Ar-CH), 3.66 (2H, t, J = 6.55 Hz, CH2-OH), 3.93 (2H, t, J = 6.48 Hz, Ar-O-CH2), 6.82 (2H, d, J = 8.63 Hz, Ar-H), 7.11 ppm (2H, d, J = 8.61 Hz, Ar-H).
6-(4ʹ-((1s,4ʹr)-4-Heptylcyclohexyl)phenoxy)hexan-1-ol (3).
3 was synthesised according to GP1. Trans-4-(4-heptylcyclohexyl)phenol (2.8 g, 10 mmol, 1 eq.) and 6-bromohexanol were used, respectively, as phenol and alkyl halogen compound. Flash chromatography with CH2Cl2/MeOH (99:1) as eluent yields 2.5 g of pure compound.
Yield = 67%. Rf (CH2Cl2/EtOAc, 8:2) = 0.44.
1H NMR (CDCl3, 400 MHz) δ = 0.92 (t, 3H, J = 6.59 Hz, -CH3), 1.04 (2H, m, -CH2-CH3), 1.20–1.53 (19H, m, -CH-CH2-, -CH2-CH2-CH-, -CH2-CH2-CH2-), 1.61–1.91 (8H, m, -CH-CH2-CH2-CH-), 2.43 (1H, m, Ar-CH), 3.65 (2H, t, J = 6.57 Hz, CH2-OH), 3.95 (2H, t, J = 6.46 Hz, Ar-O-CH2), 6.84 (2H, d, J = 8.19 Hz, Ar-H), 7.13 ppm (2H, d, J = 8.20 Hz, Ar-H).
(1r,4ʹr)-4ʹ-(4-(Cyclohexyloxy)phenyl)cyclohexan-1-ol (4).
4 was synthesised according to GP1. Trans-4-(4-cyclohexyloxy)phenol (2 g, 10 mmol, 1 eq.) and bromohexane were used, respectively, as phenol and alkyl halogen compound. 2.7 g of the pure compound was obtained without further purification.
Yield = 97.5%. Rf (CH2Cl2/EtOAc, 8:2) = 0.40.
1H NMR (CDCl3, 400 MHz) δ = 0.90 (t, 3H, J = 6.57 Hz, -CH3), 1.32–1.52 (8 H, m, -CH2-CH2-CH2-, -CH2-CH2-CH3), 1.57–2.10 (8 H, m, -CH-CH2-CH2-CH-OH), 2.44 (1 H, m, Ar-CH), 3.67 (1 H, m, CH-OH), 3.92 (2 H, t, J = 6.58 Hz, Ar-O-CH2), 6.82 (2 H, d, J = 8.11 Hz, Ar-H), 7.10 ppm (2 H, d, J = 8.16 Hz, Ar-H).
(1r,4ʹr)-4-(4ʹ-((6-Bromohexyl)oxy)phenyl)cyclohexan-1-ol (5).
5 was synthesised according to GP1. Trans-4-(4-cyclohexyloxy)phenol (2.2 g, 11 mmol, 1 eq.) and 1,6-dibromohexane (5 mL, 33 mmol, 3 eq) were used, respectively, as phenol and alkyl halogen compound. Instead of column chromatography, 200 mL of heptane was added to the oily residue to yield 2 g of the pure compound after filtration.
Yield = 51%. Rf (CH2Cl2/EtOAc, 8:2) = 0.42.
1 H NMR (CDCl3, 400 MHz) δ = 1.38–1.55 (8H, m, -CH2-CH2-CH2-), 1.78–2.10 (8H, m, -CH-CH2-CH2-CH-), 2.44 (1H, m, Ar-CH), 3.42 (2H, t, J = 6.81 Hz, CH2-Br), 3.67 (1H, m, CH-OH), 3.93 (2H, t, J = 6.34 Hz, Ar-O-CH2), 6.82 (2H, d, J = 8.69 Hz, Ar-H), 7.10 ppm (2H, d, J = 8.67 Hz, Ar-H).
1-((6-Bromohexyl)oxy)-4-((1r,4ʹr)-4ʹ-(6-bromohexyloxy)cyclohexyl)benzene (6).
Trans-4-(4-cyclohexyloxy)phenol (0.7 g, 3.6 mmol, 1 eq.), 1,6-dibromohexane (11 mL, 73 mmol, 20 eq.) and NaH (0.26 g, 11 mmol, 3 eq.) were dissolved in 20 mL of THF and heated to reflux for 72 h. The reaction was quenched with a 15 mL solution of NH4Cl (sat) in water and extracted with Et2O (2× 50 mL). The organic phase was washed with 2× 50 mL of brine, the organic phase was dried over MgSO4 and the solvent was evaporated under reduced pressure. The crude material was purified by flash chromatography on silica with hexane/EtOAc (9:1) as eluent yielding 0.9 g of the pure compound.
Yield = 48%. Rf (CH2Cl2) = 0.47.
1H NMR (DMSO, 400 MHz) δ = 1.26–1.53 (16H, m, -CH2-CH2-CH2-), 1.77–2.07 (8H, m, -CH-CH2-CH2-CH-), 2.39 (1H, m, Ar-CH), 3.22 (1H, m, -O-CH) 3.41 (2H, t, J = 6.41 Hz, CH2-Br), 3.90 (2H, t, J = 6.43 Hz, Ar-O-CH2), 6.81 (2H, d, J = 8.70 Hz, Ar-H), 7.10 ppm (2H, d, J = 8.69 Hz, Ar-H).
4-(4ʹ-Hydroxycyclohexyl)benzonitrile (7).
4-(4ʹ-Oxocyclohexyl)benzonitrile (1 g, 5 mmol, 1 eq.) and NaBH4 (0.17 g, 4.5 mmol, 0.9 eq.) were dissolved in 50 mL of dry MeOH at 0°C. The mixture was allowed to warm up to r.t. and was stirred for 90 min. After this time, the reaction was quenched with 20 mL of distilled water, the methanol was removed under vacuum and 50 mL of CH2Cl2 was added. The organic phase was successively washed with 50 mL of NH4Cl (sat) solution in water and 2× 50 mL of brine. The organic phase was dried over MgSO4, and the solvent was evaporated under reduced pressure to give 0.9 g of the compound without further purification.
Yield = 89%. Rf (CH2Cl2/EtOAc, 8:2) = 0.3.
1H NMR (CDCl3, 400 MHz) δ = 1.51–2.17 (8H, m, -CH-CH2-CH2-CH-), 2.59 (1H, m, Ar-CH), 3.72 (1H, m, CH-OH), 7.31 (2H, d, J = 7.95 Hz, Ar-H), 7.61 ppm (2H, d, J = 7.87 Hz, Ar-H).
(1r,4ʹr)-4-(4ʹ-(Hexyloxy)phenyl)cyclohexyl 6-bromohexanoate (8).
8 was synthesised according to GP2. 4 (2.6 g) and 6-bromohexanoyl chloride were used, respectively, as alcohol and acyl chloride compound. Flash chromatography with hexane/CH2Cl2 (1:1) as eluent yields 2.17 g of the pure compound.
Yield = 51%. Rf (CH2Cl2) = 0.56.
1H NMR (CDCl3, 400 MHz) δ = 0.91 (t, 3H, J = 6.78 Hz, -CH3), 1.21–1.70 (14H, m, -CH2-CH2-CH2-, -CH2-CH2-CH3), 1.77–2.12 (8H, m, -CH-CH2-CH2-CH-), 2.35 (2H, t, J = 7.41 Hz, CH2-COO-), 2.49 (1H, m, Ar-CH), 3.45 (2H, t, J = 6.77 Hz, CH2-Br), 3.95 (2H, t, J = 6.59 Hz, Ar-O-CH2), 4.81 (1H, m, CH-O-), 6.86 (2H, d, J = 8.62 Hz, Ar-H), 7.13 ppm (2H, d, J = 8.61 Hz, Ar-H).
4-(4ʹ-Cyanophenyl)cyclohexyl 6-bromohexanoate (9).
9 was synthesised according to GP2. 7 (0.9 g) and 6-bromohexanoyl chloride were used, respectively, as alcohol and acyl chloride compound. Flash chromatography with EtOAc/CH2Cl2 (2:8) as eluent yields 1.2 g of the pure compound.
Yield = 70%. Rf (CH2Cl2/EtOAc, 9:1) = 0.2.
1H NMR (CDCl3, 400 MHz) δ = 1.27–1.65 (6H, m, -CH2-CH2-CH2-), 1.69–2.14 (8H, m, -CH-CH2-CH2-CH-), 2.33 (2H, t, J = 7.40 Hz, CH2-COO-), 2.59 (1H, m, Ar-CH), 3.42 (2H, t, J = 6.72 Hz, CH2-Br), 4.79 (1H, m, CH-O-), 7.30 (2H, d, J = 8.32 Hz, Ar-H), 7.59 ppm (2H, d, J = 8.32 Hz, Ar-H).
(1r,4ʹr)-4-(4ʹ-((6-Bromohexyl)oxy)phenyl)cyclohexyl 6-bromohexanoate (10).
10 was synthesised according to GP2. 5 (1.3 g) and 6-bromohexanoyl chloride were used, respectively, as alcohol and acyl chloride compound. Flash chromatography with EtOAc/CH2Cl2 (2:8) as eluent yields 0.85 g of the pure compound.
Yield = 45%. Rf (CH2Cl2) = 0.56.
1H NMR (CDCl3, 400 MHz) δ = 1.45–1.77 (14H, m, -CH2-CH2-CH2-), 1.80–2.10 (8H, m, -CH-CH2-CH2-CH-), 2.33 (2H, t, J = 7.42 Hz, CH2-COO-), 2.46 (1H, m, Ar-CH), 3.42 (4H, t, J = 6.77 Hz, CH2-Br), 3.93 (2H, t, J = 6.37 Hz, Ar-O-CH2), 4.78 (1H, m, CH-O-), 6.82 (2H, d, J = 8.62 Hz, Ar-H), 7.10 ppm (2H, d, J = 8.58 Hz, Ar-H).
6-(4-((1r,4ʹr)-4ʹ-Hydroxycyclohexyl)phenoxy)hexyl acrylate (11).
11 was synthesised according to GP3. 5 (1 g) was used as alkyl halogen compound. Flash chromatography with EtOAc/CH2Cl2 (2:8) as eluent yields 1 g of the pure compound.
Yield = 51%. Rf (CH2Cl2/EtOAc, 9:1) = 0.32.
1H NMR (CDCl3, 400 MHz) δ = 1.38–1.56 (8H, m, -CH2-CH2-CH2-), 1.70–1.79 (4H, m, -CH-CH2-CH2-CH-), 1.82–2.10 (4H, m, -CH-CH2-CH2-CH-), 2.44 (1H, m, Ar-CH), 3.67 (1H, m, CH-OH), 3.93 (2H, t, J = 6.41 Hz, Ar-O-CH2), 4.16 (2H, t, J = 6.66 Hz, CH2-OOC), 5.81 (1H, dd, J = 10.42, 1.51 Hz, Acr-H), 6.12 (1H, dd, J = 17.31, 10.41 Hz, Acr-H), 6.40 (1 H, dd, J = 17.32, 1.46 Hz, Acr-H), 6.82 (2 H, d, J = 8.62 Hz, Ar-H), 7.10 ppm (2H, d, J = 8.64 Hz, Ar-H). 13C NMR (CDCl3, 100 MHz) δ = 25.89 (CH2), 25.92 (CH2), 28.70 (CH2), 29.35 (CH2), 32.81 (CH2), 36.12 (CH2), 42.66 (CH2), 64.68 (C-O), 67.87 (C-O), 70.81 (C-OH), 114.45 (C=C), 127.70 (C=C), 128.72 (C=C), 130.65 (C=C), 138.72 (C=C), 157.48 (C=C), 166.47 ppm (C=O). MS(ESI) (m/z): calcd for C21H30NaO4+ (M+Na+): 369.2036, found: 369.2020. See Figure S1.
6-(4-((1s,4ʹr)-4ʹ-Propylcyclohexyl)phenoxy)hexyl acrylate (12).
12 was synthesised according to GP2. 1 (3.8 g) and acryloyl chloride were used, respectively, as alcohol and acyl chloride compound. Flash chromatography with heptane/EtOAc (9:1) as eluent yields 4 g of the pure compound.
Yield = 89%. Rf (CH2Cl2) = 0.51.
1H NMR (CDCl3, 400 MHz) δ = 0.82 (t, 3H, J = 7.26 Hz, -CH3), 1.05–1.50 (13H, m, -CH-CH2-, -CH2-CH2-CH-, -CH2-CH2-CH2-), 1.56–1.88 (8 H, m, -CH-CH2-CH2-CH-), 2.41 (1H, m, Ar-CH), 3.93 (2H, t, J = 6.42 Hz, Ar-O-CH2), 4.17 (2H, t, J = 6.67 Hz, CH2-OOC), 5.81 (1H, dd, J = 10.41, 1.53 Hz, Acr-H), 6.12 (1H, dd, J = 10.41, 17.32 Hz, Acr-H), 6.40 (1H, dd, J = 17.33, 1.5 Hz, Acr-H), 6.81 (2H, d, J = 8.63 Hz, Ar-H), 7.11 ppm (2H, d, J = 8.63 Hz, Ar-H). 13 C NMR (CDCl3, 100 MHz) δ = 14.56 (CH3), 20.18 (CH2), 25.90 (CH2), 25.94 (CH2), 28.71 (CH2), 29.37 (CH2), 33.77 (CH2), 34.72 (CH2), 37.17 (CH2), 39.88 (CH2), 43.87 (CH2), 64.69 (C-O), 67.84 (C-O), 114.36 (C=C), 127.72 (C=C), 128.73 (C=C), 130.62 (C=C), 140.14 (C=C), 157.27 (C=C), 166.44 ppm (C=O). MS(ESI) (m/z): calcd for C24H36NaO3+ (M+Na+): 395.2557, found: 395.2551. See Figure S2.
6-(4-((1s,4ʹr)-4ʹ-Pentylcyclohexyl)phenoxy)hexyl acrylate (13).
13 was synthesised according to GP2. 2 (2.8 g) and acryloyl chloride were used, respectively, as alcohol and acyl chloride compound. Flash chromatography with heptane/EtOAc (9:1) as eluent yields 2.8 g of the pure compound.
Yield = 87%. Rf (CH2Cl2) = 0.51.
1H NMR (CDCl3, 400 MHz) δ = 0.87 (t, 3 H, J = 6.94 Hz, -CH3), 1.04 (2 H, m, -CH2-CH3), 1.18–1.38 (11 H, m, -CH-CH2-, -CH2-CH2-CH-, -CH2-CH2-CH2-), 1.40–1.57 (4 H, m, -CH-CH2-, -CH2-CH2-CH-, -CH2-CH2-CH2-), 1.65–1.92 (8 H, m, -CH-CH2-CH2-CH-), 2.40 (1 H, m, Ar-CH), 3.91 (2 H, t, J = 6.40 Hz, Ar-O-CH2), 4.16 (2 H, t, J = 6.67 Hz, CH2-OOC), 5.81 (1 H, dd, J = 10.35, 1.39 Hz, Acr-H), 6.12 (1 H, dd, J = 17.35, 10.46 Hz, Acr-H), 6.40 (1 H, dd, J = 17.30, 1.54 Hz, Acr-H), 6.81 (2 H, d, J = 8.55 Hz, Ar-H), 7.10 ppm (2 H, d, J = 8.61 Hz, Ar-H). 13 C NMR (CDCl3, 100 MHz) δ = 14.27 (CH3), 22.86 (CH2), 25.91 (CH2), 25.94 (CH2), 26.81 (CH2), 28.71 (CH2), 29.38 (CH2), 32.36 (CH2), 33.81 (CH2), 34.73 (CH2), 37.46 (CH2), 37.54 (CH2), 43.88 (CH2), 64.69 (C-O), 67.85 (C-O), 114.36 (C=C), 127.73 (C=C), 128.73 (C=C), 130.63 (C=C), 140.15 (C=C), 157.27 (C=C), 166.46 ppm (C = O). MS(ESI) (m/z): calcd for C26H40NaO3+ (M+Na+): 423.2870, found: 423.2868. See Figure S3.
6-(4-((1 s,4ʹr)-4ʹ-Heptylcyclohexyl)phenoxy)hexyl acrylate (14).
14 was synthesised according to GP2. 3 (2.5 g) and acryloyl chloride were used, respectively, as alcohol and acyl chloride compound. Flash chromatography with heptane/EtOAc (9:1) as eluent yields 2.6 g of the pure compound.
Yield = 91%. Rf (CH2Cl2) = 0.51.
1 H NMR (CDCl3, 400 MHz) δ = 0.90 (t, 3 H, J = 6.75 Hz, -CH3), 1.04–1.52 (21 H, m, -CH-CH2-, -CH2-CH2-CH-, -CH2-CH2-CH2-), 1.62–1.96 (8 H, m, -CH-CH2-CH2-CH-), 2.41 (1 H, m, Ar-CH), 3.94 (2 H, t, J = 6.40 Hz, Ar-O-CH2), 4.17 (2 H, t, J = 6.67 Hz, CH2-OOC), 5.82 (1 H, dd, J = 10.35, 1.39 Hz, Acr-H), 6.12 (1 H, dd, J = 17.32, 10.42 Hz, Acr-H), 6.40 (1 H, dd, J = 17.33, 1.52 Hz, Acr-H), 6.82 (2 H, d, J = 8.65 Hz, Ar-H), 7.11 ppm (2 H, d, J = 8.59 Hz, Ar-H). 13 C NMR (CDCl3, 100 MHz) δ = 14.28 (CH3), 22.85 (CH2), 25.90 (CH2), 25.94 (CH2), 27.16 (CH2), 28.71 (CH2), 29.38 (CH2), 29.54 (CH2), 30.12 (CH2), 32.07 (CH2), 33.81 (CH2), 34.73 (CH2), 37.47 (CH2), 37.60 (CH2), 43.88 (CH2), 64.69 (C-O), 67.85 (C-O), 114.36 (C=C), 127.72 (C=C), 128.73 (C=C), 130.62 (C=C), 140.15 (C=C), 157.27 (C=C), 166.45 ppm (C = O). MS(ESI) (m/z): calcd for C28 H44NaO3+ (M+ Na+): 451.3183, found: 451.3183. See Figure S4.
(1 r,4ʹr)-4-(4ʹ-(Hexyloxy)phenyl)cyclohexyl 6-(acryloyloxy)hexanoate (15).
15 was synthesised according to GP3. 8 (1 g) was used as alkyl halogen compound. Flash chromatography with EtOAc/CH2Cl2 (2:8) as eluent yields 0.47 g of the pure compound.
Yield = 48%. Rf (CH2Cl2) = 0.29.
1 H NMR (CDCl3, 400 MHz) δ = 0.90 (t, 3 H, J = 6.92 Hz, -CH3), 1.27–1.64 (14 H, m, -CH-CH2-, -CH2-CH2-CH-, -CH2-CH2-CH2-), 1.91–2.08 (8 H, m, -CH-CH2-CH2-CH-), 2.31 (2 H, t, J = 7.42 Hz, CH2-COO), 2.45 (1 H, m, Ar-CH), 3.92 (2 H, t, J = 6.58 Hz, Ar-O-CH2), 4.16 (2 H, t, J = 6.61 Hz, CH2-O), 5.82 (1 H, dd, J = 10.42, 1.51 Hz, Acr-H), 6.12 (1 H, dd, J = 17.34, 10.43 Hz, Acr-H), 6.41 (1 H, dd, J = 17.32, 1.56 Hz, Acr-H), 6.83 (2 H, d, J = 8.64 Hz, Ar-H), 7.10 ppm (2 H, d, J = 8.66 Hz, Ar-H). 13 C NMR (CDCl3, 100 MHz) δ = 14.28 (CH3), 22.85 (CH2), 25.90 (CH2), 25.94 (CH2), 27.16 (CH2), 28.71 (CH2), 29.38 (CH2), 29.54 (CH2), 30.12 (CH2), 32.07 (CH2), 33.81 (CH2), 34.73 (CH2), 37.47 (CH2), 37.60 (CH2), 43.88 (CH2), 64.69 (C-O), 67.85 (C-O), 114.36 (C=C), 127.72 (C=C), 128.73 (C=C), 130.62 C=C), 140.15 (C=C), 157.27 (C=C), 166.45 ppm (C = O). MS(ESI) (m/z): calcd for C27H40NaO5+ (M+ Na+): 467.2768, found: 467.2775. See Figure S5.
4-(4ʹ-Cyanophenyl)cyclohexyl 6-(acryloyloxy)hexanoate (16).
16 was synthesised according to GP3. 9 (1.2 g) was used as alkyl halogen compound. Flash chromatography with EtOAc/CH2Cl2 (2:8) as eluent to give 1 g of the pure desired compound.
Yield = 85%. Rf (CH2Cl2) = 0.29.
1 H NMR (CDCl3, 400 MHz) δ = 1.38–1.58 (6H, m, -CH2-CH2-CH2-), 1.62–1.44 (4 H, m, -CH-CH2-CH2-CH-), 1.90–2.13 (4 H, m, -CH-CH2-CH2-CH-), 2.32 (2 H, t, J = 7.44 Hz, CH2-COO-), 2.58 (1 H, m, Ar-CH), 4.16 (2 H, t, J = 7.44 Hz, CH2-O), 4.78 (1 H, m, CH-O-), 5.82 (1 H, dd, J = 10.42, 1.53 Hz, Acr-H), 6.11 (1 H, dd, J = 17.33, 10.42 Hz, Acr-H), 7.28 (2 H, d, J = 8.26 Hz, Ar-H), 7.58 ppm (2 H, d, J = 8.22 Hz, Ar-H). 13 C NMR (CDCl3, 100 MHz) δ = 24.80 (CH2), 25.63 (CH2), 28.46 (CH2), 31.90 (CH2), 31.94 (CH2), 34.61 (CH2), 43.61 (CH2), 64.48 (C-O), 72.41 (C-COO), 110.23 (C=C), 119.12 (CN), 127.77 (C=C), 128.67 (C=C), 130.71 (C=C), 132.46 (C=C), 151.75 (C=C), 166.40 (C = O), 173.16 ppm (C = O). MS(ESI) (m/z): calcd for C22 H27NNaO4+ (M+ Na+): 392.1832, found: 392.1829. See Figure S6.
(1 r,4ʹr)-4-(4ʹ-((6-(Acryloyloxy)hexyl)oxy)phenyl)cyclohexyl hexanoate (17).
17 was synthesised according to GP2. 11 (0.1 g) and hexanoyl chloride were used, respectively, as alcohol and acyl chloride compound. Flash chromatography with heptane/EtOAc (9:1) as eluent yields 0.11 g of the pure compound.
Yield = 86%. Rf (CH2Cl2) = 0.20.
1 H NMR (CDCl3, 400 MHz) δ = 0.90 (3 H, t, J = 6.88 Hz, CH3), 1.26–1.56 (14 H, m, -CH2-CH2-CH2-, -CH2-CH2-CH3), 1.56–1.83 (4 H, m, -CH-CH2-CH2-CH-), 1.91–2.09 (4 H, m, -CH-CH2-CH2-CH-), 2.28 (2 H, t, J = 7.54 Hz, CH2-COO), 2.47 (1 H, m, Ar-CH), 3.94 (2 H, t, J = 6.39 Hz, Ar-O-CH2), 4.16 (2 H, t, J = 6.66 Hz, CH2-OOC), 4.78 (1 H, m, CH-OOC), 5.81 (1 H, dd, J = 10.33, 1.42 Hz, Acr-H), 6.12 (1 H, dd, J = 17.33, 10.41 Hz, Acr-H), 6.40 (1 H, dd, J = 17.32, 1.49 Hz, Acr-H), 6.82 (2 H, d, J = 8.66 Hz, Ar-H), 7.10 ppm (2 H, d, J = 8.66 Hz, Ar-H). 13 C NMR (CDCl3, 100 MHz) δ = 14.08 (CH3), 22.47 (CH2), 24.92 (CH2), 25.90 (CH2), 25.93 (CH2), 28.71 (CH2), 29.35 (CH2), 31.44 (CH2), 32.23 (CH2), 32.59 (CH2), 34.85 (CH2), 42.59 (CH2), 64.68 (C-O), 67.88 (C-O), 72.82 (C-COO), 114.49 (C=C), 127.69 (C=C), 128.73 (C=C), 130.64 (C=C), 138.43 (C=C), 157.54 (C=C), 166.45 (C = O), 173.61 ppm (C = O). MS(ESI) (m/z): calcd for C27H40NaO5+ (M+ Na+): 467.2768, found: 467.2750. See Figure S7.
(1 r,4 r)-4-(4-(Hexyloxy)phenyl)cyclohexyl acrylate (18).
18 was synthesised according to GP2. 3 (0.05 g) and acryloyl chloride were used, respectively, as alcohol and acyl chloride compound. Flash chromatography with heptane/EtOAc (9:1) as eluent to give 50 mg of the pure compound.
Yield = 84%. Rf (CH2Cl2) = 0.40.
1H NMR (CDCl3, 400 MHz) δ = 0.90 (3 H, t, J = 7.03 Hz, -CH3), 1.20–1.54 (8 H, m, -CH2-CH2-CH2-, CH2-CH3), 1.57–1.84 (4 H, m, -CH-CH2-CH2-CH-), 1.87–2.21 (4 H, m, -CH-CH2-CH2-CH-), 2.47 (1 H, m, Ar-CH), 3.93 (2 H, t, J = 6.56 Hz, Ar-O-CH2), 4.86 (1 H, m, CH-OOC), 5.82 (1 H, dd, J = 10.41, 1.58 Hz, Acr-H), 6.12 (2 H, dd, J = 17.29, 10.38 Hz, Acr-H), 6.41 (2 H, dd, J = 17.32, 1.51 Hz, Acr-H), 6.83 (2 H, d, J = 8.67 Hz, Ar-H), 7.11 ppm (2H, d, J = 8.63 Hz, Ar-H). 13 C NMR (CDCl3, 100 MHz) δ = 14.19 (CH3), 22.77 (CH2), 25.90 (CH2), 29.46 (CH2), 31.75 (CH2), 32.20 (CH2), 32.85 (CH2), 42.59 (CH2), 68.15 (C-O), 73.32 (C-O), 114.53 (C=C), 127.68 (C=C), 129.18 (C=C), 130.46 (C=C), 138.29 (C=C), 157.67 (C=C), 165.95 ppm (C = O). MS(ESI) (m/z): calcd for C21H30NaO3+ (M+ Na+): 353.2087, found: 353.2110. See Figure S8.
(1 r,4ʹr)-4-(4ʹ-((6-(Acryloyloxy)hexyl)oxy)phenyl)cyclohexyl 6-(acryloyloxy)hexanoate (19).
19 was synthesised according to GP3. 10 (0.8 g) was used as alkyl halogen compound. Flash chromatography with CH2Cl2/MeOH (99:1) as eluent yields 315 mg of the pure compound.
Yield = 41%. Rf (CH2Cl2) = 0.11.
1H NMR (CDCl3, 400 MHz) δ = 1.40–1.69 (14 H, m, -CH2-CH2-CH2-), 1.69–2.14 (8 H, m, -CH-CH2-CH2-CH-), 2.32 (2H, t, J = 7. 24 Hz, CH2-COO), 2.46 (1H, m, Ar-CH), 3.92 (2H, t, J = 6.44 Hz, Ar-O-CH2), 4.16 (4H, t, J = 6.63 Hz, CH2-OOC), 4.78 (1H, m, CH-OOC), 5.81 (2H, d, J = 10.42 Hz, Acr-H), 6.11 (2H, dd, J = 17.33, 10.43 Hz, Acr-H), 6.40 (2H, dd, J = 17.25 Hz, Acr-H), 6.82 (2H, d, J = 8.61 Hz, Ar-H), 7.10 ppm (2H, d, J = 8.63 Hz, Ar-H). 13C NMR (CDCl3, 100 MHz) δ = 24.84 (CH2), 25.64 (CH2), 25.89 (CH2), 25.92 (CH2), 28.47 (CH2), 28.71 (CH2), 29.35 (CH2), 32.22 (CH2), 32.57 (CH2), 34.67 (CH2), 42.57 (CH2), 64.51 (C-O), 64.68 (C-O), 67.88 (C-O), 72.98 (C-O), 114.49 (C=C), 127.68 (C=C), 128.68 (C=C), 128.72 (C=C), 130.65 (C=C), 130.69 (C=C), 138.38 (C=C), 157.55 (C=C), 166.41 (C = O), 166.46 (C = O), 173.22 ppm (C = O). MS(ESI) (m/z): calcd for C30H42NaO7+ (M+ Na+): 537.2823, found: 537.2805. See Figure S9.
6-(4-((1 r,4ʹr)-4ʹ-((6-(acryloyloxy)hexyl)oxy)cyclohexyl)phenoxy)hexyl acrylate (20).
20 was synthesised according to GP3. 6 (0.2 g) was used as alkyl halogen compound. Flash chromatography with heptane/EtOAc (9:1) as eluent yields 157 mg of the pure compound.
Yield = 80%. Rf (CH2Cl2) = 0.20.
1H NMR (CDCl3, 400 MHz) δ = 1.33–1.68 (16H, m, -CH2-CH2-CH2-), 1.69–2.20 (8H, m, -CH-CH2-CH2-CH-), 2.45 (1H, m, Ar-CH), 3.25 (1H, m, CH-O), 3.48 (2H, t, J = 6.63 Hz,), 3.93 (2H, t, J = 6.41 Hz, Ar-O-CH2), 4.16 (4H, m, CH2-OOC), 5.82 (2H, d, J = 10.39 Hz, Acr-H), 6.11 (2H, dd, J = 17.34, 10.42 Hz, Acr-H), 6.40 (2H, d, J = 17.34 Hz, Acr-H), 6.81 (2H, d, J = 8.66 Hz, Ar-H), 7.10 ppm (2H, d, J = 8.66 Hz, Ar-H). 13 C NMR (CDCl3, 100 MHz) δ = 25.90 (CH2), 25.94 (CH2), 25.97 (CH2), 26.08 (CH2), 28.72 (CH2), 28.75 (CH2), 29.37 (CH2), 30.25 (CH2), 32.89 (CH2), 32.94 (CH2), 43.01 (CH2), 64.69 (C-O), 64.75 (C-O), 67.88 (C-O), 68.15 (C-O), 114.43 (C=C), 127.69 (C=C), 128.73 (C=C), 128.77 (C=C), 130.60 (C=C), 130.65 (C=C), 138.97 (C=C), 157.45 (C = O), 166.47 ppm (C = O). MS(ESI) (m/z): calcd for C30H44NaO6+ (M+ Na+): 523.3030, found: 523.3023. See Figure S10.
(1 r,4ʹr)-4-(4ʹ-((6-(Acryloyloxy)hexyl)oxy)phenyl)cyclohexyl 4-((6-(acryloyloxy)hexyl)oxy)benzoate (21).
11 (80 mg, 0.23 mmol, 1 eq.), N,N′-dicyclohexylcarbodiimide (143 mg, 0.7 mmol, 3 eq.), DMAP (3 mg, 0.023 mmol, 0.1 eq) and 4-((6-(acryloyloxy)hexyl)oxy)benzoic acid (67 mg, 0.23 mmol, 1 eq) were dissolved in 5 mL of dry CH2Cl2 and stirred 24 h at 30°C. 20 mL of CH2Cl2 were added to the solution before washing with 2× 20 mL of distilled water and 20 mL of brine. The organic phase was dried with MgSO4, and the solvent was evaporated under reduced pressure. The crude material was then purified by flash chromatography on silica with heptane/EtOAc (8:2) as eluent to yield 60 mg of the pure compound.
Yield = 42%. Rf (CH2Cl2) = 0.13.
1H NMR (CDCl3, 400 MHz) δ = 1.42–1.71 (16H, m, -CH2-CH2-CH2-), 1.72–1.89 (4H, m, -CH-CH2-CH2-CH-), 1.89–2.27 (4H, m, -CH-CH2-CH2-CH-), 2.52 (1H, m, Ar-CH), 3.94 (2H, t, J = 6.40 Hz, Ar-O-CH2), 4.01 (2H, t, J = 6.42 Hz, CH2-O), 4.18 (4H, m, CH2-O), 4.99 (1H, m, COO-CH), 5.81 (2H, d, J = 10.42 Hz, Acr-H), 6.12 (2H, dd, J = 17.31, 10.41 Hz, Acr-H), 6.40 (2H, d, J = 17.31 Hz, Acr-H), 6.84 (2H, d, J = 8.67 Hz, Ar-H), 6.90 (2H, d, J = 8.92 Hz, Ar-H), 7.13 (2H, d, J = 8.43 Hz, Ar-H), 7.99 ppm (2H, d, J = 8.86 Hz, Ar-H). 13 C NMR (CDCl3, 100 MHz) δ = 25.85 (CH2), 25.87 (CH2), 25.91 (CH2), 25.94 (CH2), 28.72 (CH2), 28.69 (CH2), 28.72 (CH2), 29.15 (CH2), 29.36 (CH2), 32.36 (CH2), 32.64 (CH2), 42.66 (CH2), 64.62 (C-O), 64.69 (C-O), 67.89 (C-O), 68.10 (C-O), 73.34 (C-O), 114.10 (C=C), 114.51 (C=C), 123.21 (C=C), 127.72 (C=C), 128.70 (C=C), 128.73 (C=C), 130.65 (C=C), 130.70 (C=C), 131.68 (C=C), 138.47 (C=C), 157.56 (C=C), 162.88 (C = O), 166.05 (C = O),166.46 ppm (C = O). MS(ESI) (m/z): calcd for C37H48NaO8+ (M+ Na+): 643.3241, found: 643.3243. See Figure S11.
(1 r,4ʹr)-4-(4ʹ-((6-(Acryloyloxy)hexyl)oxy)phenyl)cyclohexyl 4-((6-(acryloyloxy)hexyl)oxy)-2-methylbenzoate (22).
4-((6-(acryloyloxy)hexyl)oxy)-2-methylbenzoic acid (221 mg, 0.72 mmol, 1 eq.), triethylamine (0.3 mL, 2.2 mmol, 3 eq.) and 2,4,6-trichlorobenzoyl chloride (0.14 mL, 0.87 mmol, 1.2 eq.) were dissolved in 3 mL of dry THF and stirred 2 h at r.t. The reaction mixture was filtrated under inert atmosphere. 11 (0.3 g, 0.87 mmol, 1.2 eq.) and DMAP (0.133 g, 1.1 mmol, 1.5 eq.) were dissolved in 3 mL of dry THF and added to the first mixture. The reaction was stirred for 16 h at r.t. and filtrated before being condensed under reduced pressure. The residue was dissolved in 100 mL of CH2Cl2 and washed with 2× 100 mL of distilled water and 100 mL of brine. The organic phase was then dried with MgSO4, and the solvent was evaporated under reduced pressure. The crude material was then purified by flash chromatography on silica with CH2Cl2/EtOAc (9:1) as eluent to yield 220 mg of the pure desired compound.
Yield = 48%. Rf (CH2Cl2) = 0.13.
1H NMR (CDCl3, 400 MHz) δ = 1.38–1.75 (16H, m, -CH2-CH2-CH2-), 1.75–1.87 (4H, m, -CH-CH2-CH2-CH-), 1.94–2.27 (4H, m, -CH-CH2-CH2-CH-), 2.53 (1H, m, Ar-CH), 2.60 (3H, s, CH3) 3.94 (2H, t, J = 6.38 Hz, Ar-O-CH2), 3.99 (2H, t, J = 6.41 Hz, CH2-O), 4.18 (4H, m, CH2-O), 4.97 (1H, m, COO-CH), 5.82 (2H, d, J = 10.41 Hz, Acr-H), 6.12 (2H, dd, J = 17.34, 10.42 Hz, Acr-H), 6.40 (2H, d, J = 17.31 Hz, Acr-H), 6.73 (2H, m, Ar-H), 6.83 (2H, d, J = 8.68 Hz, Ar-H), 7.13 (2H, d, J = 8.43 Hz, Ar-H), 7.92 ppm (H, m, Ar-H). 13 C NMR (CDCl3, 100 MHz) δ = 22.53 (CH2), 25.84 (CH2), 25.86 (CH2), 25.90 (CH2), 25.93 (CH2), 28.69 (CH2), 28.71 (CH2), 29.17 (CH2), 29.35 (CH2), 32.42 (CH2), 32.66 (CH2), 42.66 (CH2), 64.62 (C-O), 64.68 (C-O), 67.88 (C-O), 67.90 (C-O), 73.15 (C-O), 111.45 (C=C), 114.50 (C=C), 117.53 (C=C), 122.40 (C=C), 127.71 (C=C), 128.70 (C=C), 128.73 (C=C), 130.64 (C=C), 130.68 (C=C), 133.00 (C=C), 138.47 (C=C), 142.92 (C=C), 157.55 (C=C), 161.81 (C = O), 166.45 (C = O), 166.89 ppm (C = O). MS(ESI) (m/z): calcd for C38H50NaO8+ (M+ Na+): 657.3398, found: 657.3409. See Figure S12.
6-(4-((1 r,4 r)-4-(Acryloyloxy)cyclohexyl)phenoxy)hexyl acrylate (23). (GP2)
23 was synthesised according to GP2. 11 (100 mg) and acryloyl chloride were used, respectively, as alcohol and acyl chloride compound. Flash chromatography with heptane/EtOAc (9:1) as eluent yields 100 mg of the pure desired compound.
Yield = 89%. Rf (CH2Cl2) = 0.24.
1H NMR (CDCl3, 400 MHz) δ = 1.39–1.62 (8H, m, -CH2-CH2-CH2-), 1.66–1.84 (4H, m, -CH-CH2-CH2-CH-), 1.88–2.18 (4H, m, -CH-CH2-CH2-CH-), 2.49 (1H, m, Ar-CH), 3.93 (2H, t, J = 6.42 Hz, Ar-O-CH2), 4.16 (2H, t, J = 6.67 Hz, CH2-OOC), 4.85 (1H, m, CH-OOC), 5.81 (2H, dd, J = 10.42, 1.5 Hz, Acr-H), 6.12 (2H, dd, J = 17.31, 10.40 Hz, Acr-H), 6.40 (2H, dm, J = 17.32 Hz, Acr-H), 6.82 (2H, d, J = 8.70 Hz, Ar-H), 7.11 ppm (2H, d, J = 8.65 Hz, Ar-H). 13 C NMR (CDCl3, 100 MHz) δ = 25.89 (CH2), 25.92 (CH2), 28.70 (CH2), 29.34 (CH2), 32.18 (CH2), 32.56 (CH2), 42.57 (CH2), 64.67 (C-O), 67.88 (C-O), 73.28 (C-O), 114.50 (C=C), 127.68 (C=C), 128.72 (C=C), 129.16 (C=C), 130.45 (C=C), 130.63 (C=C), 138.37 (C=C), 157.56 (C=C), 165.91 (C = O), 166.44 ppm (C = O). MS(ESI) (m/z): calcd. for C24H32NaO5+ (M+ Na+): 423.2142, found: 423.2111. See Figure S13.
Preparation of LCPNs
Three photopolymerisable LC mixtures (TN, RN1 and RN2) were prepared by mixing monomers (see ) with 0.5 wt% of the photo-initiator (Irgaqure 651) in dichloromethane. Dichloromethane was slowly evaporated, and mixtures were dried at 80°C overnight. The mixtures TN, RN1 and RN2 form nematic phase with clearing temperature 31°C, 62°C and 57°C, respectively.
Table 1. Summary of phase behaviour, enthalpy of LC phase transition and maximum absorption wavelength (λmax) of the synthesised reactive mesogens. Monotropic LC phases are indicated in the brackets
Table 2. Chemical compositions (in wt.%) of the test network (TN) and reference networks (RN1 and RN2) and their total transparencies in different UV spectral regions
The mixture was introduced into a 2-µm-thick quartz cell by capillary forces in the isotropic state and cooled down to polymerisation temperature which was 25°C for TN and 50°C for RN1 and RN2. Unidirectional LC alignment has been achieved by poly(vinyl alcohol) (~31 kDa, from Sigma-Aldrich) layers coated on quartz substrates and rubbed unidirectionally with a velvet cloth. Once filled, the cells were cross-polymerised by exposing to UV light (LED 365 nm, intensity ~100 mW/cm2) for 5 min. The cells were finally post-cured at 60°C overnight.
3. Results and discussion
3.1. Design of the reactive mesogens
We have designed and synthesised a number of LC monomers having minimal aromatic conjugation in order to shift the absorption band hypsochromically. Moreover, we have excluded from the design any moieties that could lead to photo-Fries rearrangement. We have used cyclohexylbenzene as rigid mesomorphic core since its absorbance is sufficiently blue-shifted, and the aromatic moiety improves the compatibility with various organic compounds. Bicyclohexyl-based LC core has not been used in the general design because of poor compatibility with aromatic compounds despite its largely blue-shifted absorbance band. We have also designed a monomer with a nitrile moiety 16 which has a higher dipole moment and therefore is more sensitive to external electric fields. It is also worth noting that during the synthesis of targeted molecules we have synthetised nine new intermediates which can be used as starting materials to build up libraries of new reactive mesogens. Overall, we have designed and synthetised eight new monomers in gram scale and five new cross-linkers suitable for LCPN synthesis which are transparent and resilient to UV light.
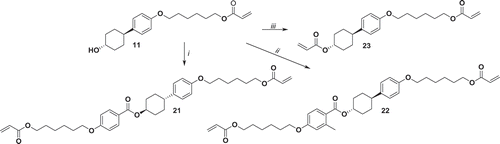
3.2. Synthesis of the reactive mesogens
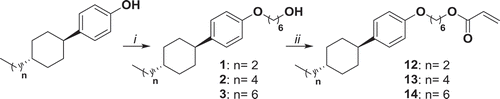
Cyclohexylbenzene-based LC monomers 12, 13 and 14 have been synthetised starting, respectively, from 4-(4-propylcyclohexyl)phenol, 4-(4-pentylcyclohexyl)phenol and 4-(4-heptylcyclohexyl)phenol. These compounds have been synthesised by Williamson reaction with 6-bromohexanol to yield 1, 2 and 3 which have been transformed into polymerisable acrylates by the reaction with acryloyl chloride to yield 12, 13 and 14 in gram scale ().
Figure 1. Synthesis of compounds 1, 2, 3, 12, 13 and 14. i: K2CO3, KI, 6-bromohexanol, EtOH, reflux 48 h; ii: NEt3, acryloyl chloride, THF, 0°C 1 h, r.t. 16 h
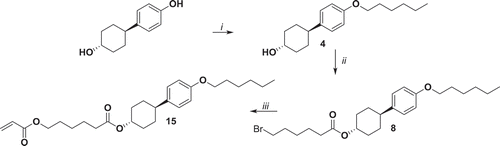
Compound 15 has been synthesised starting from 4-(4ʹ-hydroxycyclohexyl)phenol which gives 4 in quantitative yield after the Williamson reaction with bromohexane. Here, we took advantage of the reduced reactivity of the secondary alcohol compared to phenol in order to asymmetrically functionalise 4-(4ʹ-hydroxycyclohexyl)phenol with an alkyl tail. Compound 4 has been transferred to acrylic monomer consequently by the reaction with bromohexanoyl chloride yielding 8 and then with potassium acrylate yielding monomer 15 in gram scale ().
Figure 2. Synthesis of compound 15. i: K2CO3, KI, bromohexane, EtOH, reflux 48 h; ii: 6-bromohexanoyl chloride, NEt3, THF, 0°C 1 h, r.t. 16 h; iii: potassium acrylate, KI, DMSO, 52°C 72 h
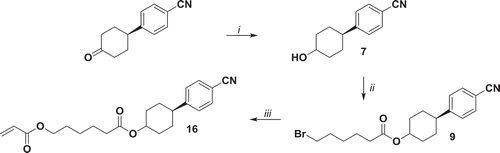
To avoid the reduction of the nitrile group, compound 7 has been synthesised by reduction of 4-(4ʹ-oxocyclohexyl)benzonitrile in mild conditions. The resulting alcohol has been converted into monoacrylate 16 in gram scale following the procedure for compound 15 ().
Figure 3. Synthesis of compound 16. i: NaBH4, MeOH, r.t. 90 min. ii: 6-bromohexanoyl chloride, NEt3, THF, 0°C 1 h, r.t. 16 h; iii: potassium acrylate, KI, DMSO, 52°C 72 h
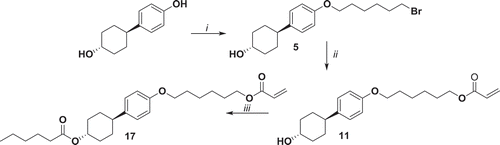
Compound 17 has been synthetised in the same way as compound 15. 4-(4ʹ-Hydroxycyclohexyl)phenol reacts with 1,6-dibromohexane to give 5 which further reacts with potassium acrylate leading to 11. Compound 11 was then acylated with hexanoyl chloride, resulting in 17 with a 86% yield (). Notably, this approach allows for gram-scale production of the intermediate 11 which is a versatile functional precursor for the synthesis of mono- and diacrylates.
Figure 4. Synthesis of compound 17. i: K2CO3, KI, 1,6-dibromohexane, EtOH, reflux 48 h; ii: potassium acrylate, KI, DMSO, 52°C 72 h; iii: hexanoyl chloride, NEt3, THF, 0°C 1 h, r.t. 16 h
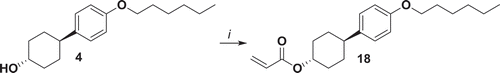
Compound 18 has been synthetised starting from 4 which has been acylated with acryloyl chloride to yield the desired compound with 84% yield ().
Figure 5. Synthesis of 18. i: NEt3, acryloyl chloride, THF, 0°C 1 h, r.t. 16 h
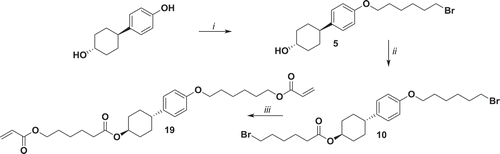
Cross-linker 19 has been synthetised by acylation of compound 5, with bromohexanoyl chloride followed by reaction with potassium acrylate ().
Figure 6. Synthesis of 19. i: K2CO3, KI, 1,6-dibromohexane, EtOH, reflux 48 h; ii: 6-bromohexanoyl chloride, NEt3, THF, 0°C 1 h, r.t. 16 h; iii: potassium acrylate, KI, DMSO, 52°C 72 h
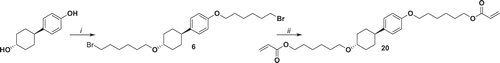
Cross-linker 20 has been synthetised starting with 4-(4ʹ-hydroxycyclohexyl)phenol which has been deprotonated by NaH in order to allow the reaction of both alcohol with 1,6-dibromohexane to give 6. Compound 6 has been reacted with potassium acrylate, resulting in 20 in 80% yield ().
Figure 7. Synthesis of 20. i: NaH, 1,6-dibromohexane, THF reflux 78 h; ii: potassium acrylate, KI, DMSO, 52°C 72 h
Cross-linkers 21, 22 and 23 have been all synthetised starting from compound 11. Compound 21 has been produced with a 42% yield by Steglich esterification starting from 4-((6-(acryloyloxy)hexyl)oxy)benzoic acid (). The same reaction has been carried out under similar conditions with 4-((6-(acryloyloxy)hexyl)oxy)-2-methylbenzoic acid resulting in 22 with only 1–2% yield, likely due to the hindrance of the carboxylic acid. To overcome this limitation, we have used Yamaguchi esterification of 4-((6-(acryloyloxy)hexyl)oxy)-2-methylbenzoic acid with the Yamaguchi reagent. The resulting anhydride has been reacted with 11 in the presence of a stoichiometric amount of DMAP, which is used as an acyl transfer agent, to produce 22 with 48% yield (). The cross-linker 23 has been obtained in 89% yield by acylation of 11 with acryloyl chloride ().
Figure 8. Synthesis of 21, 22 and 23. i: DCC, DMAP, 4-((6-(acryloyloxy)hexyl)oxy)benzoic acid, CH2Cl2, 30°C 24 h. ii: 4-((6-(acryloyloxy)hexyl)oxy)-2-methylbenzoic acid, triethylamine, 2,4,6-trichlorobenzoyl chloride, DMAP THF 18 h r.t.; iii: NEt3, acryloyl chloride, THF, 0°C 1 h, r.t. 16 h
3.3. Characterisation of the reactive mesogens
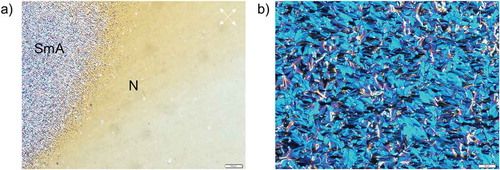
Phase behaviour of the synthesised mono- and diacrylates has been determined by polarised optical microscopy, and enthalpies of phase transitions have been measured by DSC and gathered in . Monoacrylates 12–14 and 18 melt to isotropic liquid; however, nematic monotropic mesophase has been observed upon cooling. Upon cooling, compound 14 forms smectic A phase (). Diacrylates 20–22 form smectic phases, while the others melt to isotropic liquids. Diacrylate 20 forms monotropic smectic C phase upon cooling as revealed by schlieren texture in the cell with homeotropic boundary conditions (Figure S14(a)). Compound 21 forms smectic A phase with characteristic focal conic texture (Figure S14(b)). Diacrylate 22 first melts to disordered smectic X phase (Figure S14(c)) and then to nematic phase existing in quite broad temperature range. It should be noted that despite the fact that some of the compounds are not liquid crystalline, they can be used in mixtures with the other LC monomers or low molar mass liquid crystals to form UV-transparent LCPNs. To demonstrate the improvement of phase behaviour, we designed few monomeric compositions (see Table S1) that are in nematic state at room temperature, e.g. a mixture of monomers 13 and 14 (1:1 by weight) forms nematic phase up to 27.5°C. Moreover, such monomers can be polymerised into side-chain LC polymers where their monomeric phase behaviour will completely altered by the fact that the monomers are now connected in the macromolecule.
Figure 9. Polarised optical microscopy images of LC textures of compound 14 demonstrating the transition from smectic A to nematic (a) upon cooling, and fan-like texture of smectic A obtained at 22°C (b). Planar boundary conditions. Scale bars correspond to 100 and 50 µm, respectively. Polariser and analyser are shown as white arrows
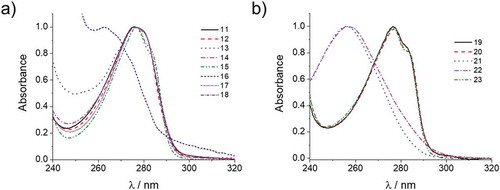
shows the absorbance spectra of all reactive mesogens synthesised in the work. It is clearly seen from the spectra that all compounds are transparent in the UV-A spectral region. Absorbance in UV-B is still present which is mostly associated with the presence of aromatic benzene rings in the structure of reactive mesogens; however, electron-withdrawing substitution like nitril (for compound 16) or carboxylic groups (for compounds 21 and 22) sufficiently shifts the maxima of absorbance bathochromically by 14–19 nm.
Figure 10. Normalised absorbance spectra of mono- (a) and diacrylates (b). Spectra are measured in CH2Cl2 at room temperature
3.4. UV-transparent LCPNs
We have designed a monomeric mixture containing monoacrylates 12–14 and cross-linker 22 to demonstrate the advantages of LCPNs prepared from the new library of reactive mesogens. This monomeric mixture is liquid crystalline at room temperature forming a nematic mesophase with clearing temperature 31°C. Fluidity at r.t. significantly simplifies processing of LC mixtures, and we envision that it will be particularly attractive for microfluidic production of LCPN droplets and shells. The test network (TN) with unidirectional planar alignment (Figure S15) has been produced by photopolymerisation at room temperature. Reference networks have been prepared from widely used commercially available reactive mesogens ()). Reference network 1 (RN1) consists of cyanobiphenyl-based monoacrylate and cross-linker RM-257. Reference network 2 (RN2) is composed of alkyloxyphenyl benzoate-based monoacrylates and RM-257.
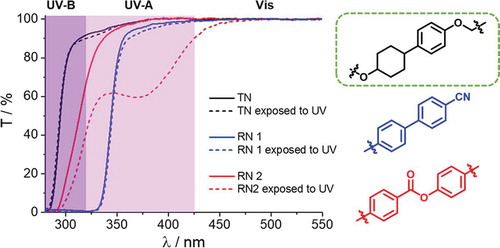
Figure 11. (a) Chemical structures of commercially available reactive mesogens used for the production of reference networks RN1 and RN2. (b) Transmittance spectra of the studied LCPNs before and after exposure to UV light (312 nm, 5.8 mWcm−2) for 1 h
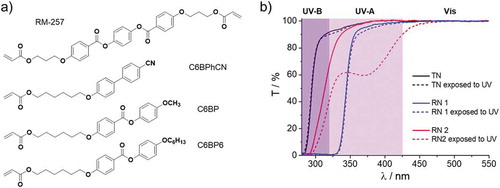
Spectroscopic characterisation of RN1 shows that the network acts as a cut-off filter below 340 nm, due to the absorption of the cyanobiphenyl fragments of the network leading to only 62.8% of transmittance in UV-A region and complete blocking of UV-B light (transmittance here is calculated as the area under the spectral curve in the given spectral range) (), ). Photostability test performed at 312 nm demonstrates the resilience of RN1 to UV light likely due to its high absorption. Network RN2 is characterised by overall higher transparency, 92.6% and 18.6% in UV-A and UV-B, respectively. Nevertheless, RN2 degrades upon UV exposure (312 nm) by photo-Fries rearrangement () dashed line). The TN network, consisting of the newly synthesised reactive mesogens presented in this work, displays the best optical performances associated with 96.7% and 48.9% transparency in UV-A and UV-B, respectively, and high stability to UV light ()). We believe that the optical window of networks composed of the monomers described here will enable effective use of broad range of photoactive dopants (cinnamates, stilbenes, overcrowded alkenes, etc.), and generally, it provides a new toolbox for UV-stable and UV-transparent optical materials.
4. Conclusion
A new library of reactive mesogens (mono- and diacrylates) as building blocks for liquid crystal polymers and networks has been synthesised and characterised. The library has been used for the design and fabrication of UV-transparent LCPNs with remarkable stability to UV light. The use of these novel reactive mesogens sufficiently increases the transparency of the LCPNs in a broad UV spectral region (280–400 nm) in comparison with LCPNs based on commercially available compounds and results in materials resistant to prolonged UV exposure. Overall, the LC materials we designed are an attractive media for the integration of photoactive compounds (photoswitches and molecular motors) while preserving their effective photochemical and photophysical performance. Moreover, LCPNs based on these reactive mesogens can also find applications for a wide range of optical elements and devices (e.g. retarders, filters, coatings, etc.) where UV stability and transparency are critical.
Supp_info_Reactive_mesogens_for_UV_transparent_liquid_crystal_polymer_networks.docx
Download MS Word (3.7 MB)Acknowledgements
The authors thank Professor Nathalie Katsonis for useful discussions. The authors acknowledge the ERC (Consolidator Grant, Morpheus, 772564) and the Volkswagen Foundation (Integration of Molecular Components in Functional Macroscopic Systems, 93424) for funding.
Disclosure statement
No potential conflict of interest has been reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Li Q. Liquid crystals beyond displays: chemistry, physics, and applications. Kent (OH): John Wiley & Sons; 2012.
- Meier G, Sackmann E, Grabmaier JG. Applications of liquid crystals. New York (NY): Springer Science & Business Media; 2012.
- Ryabchun A, Bobrovsky A. Cholesteric liquid crystal materials for tunable diffractive optics. Adv Opt Mat. 2018;6(15):1800335.
- Schenning A, Crawford GP, Broer DJ. Liquid crystal sensors. Boca Raton (FL): CRC Press; 2017.
- Mulder DJ, Schenning APHJ, Bastiaansen CWM. Chiral-nematic liquid crystals as one dimensional photonic materials in optical sensors. J Mater Chem C. 2014;2(33):6695–6705.
- White TJ, Broer DJ. Programmable and adaptive mechanics with liquid crystal polymer networks and elastomers. Nat Mater. 2015;14(11):1087–1098.
- Hao Z, Wasylczyk P, Wiersma DS, et al. Light robots: bridging the gap between microrobotics and photomechanics in soft materials. Adv Mater. 2018;30(24):1703554.
- Lancia F, Ryabchun A, Katsonis N. Life-like motion driven by artificial molecular machines. Nat Rev Chem. 2019;3(9):536–551.
- de Gennes P. Un muscle artificiel [An artificial muscle]. Comptes Rendus Acad Sci. 1997;324(5):343–348. French.
- Ryabchun A, Li Q, Lancia F, et al. Shape-persistent actuators from hydrazone photoswitches. J Am Chem Soc. 2019;141(3):1196–1200.
- Ryabchun A, Bobrovsky A, Shibaev V, et al. Fluorescent and photooptical properties of H-bonded LC composites based on stilbazole derivative. J Photochem Photobiol A. 2011;221(1):22–29.
- Ryabchun A, Raguzin I, Stumpe J, et al. Cholesteric polymer scaffolds filled with azobenzene-containing nematic mixture with phototunable optical properties. ACS Appl Mater Interfaces. 2016;8(40):27227–27235.
- Iamsaard S, Anger E, Aßhoff SJ, et al. Fluorinated azobenzenes for shape-persistent liquid crystal polymer networks. Angew Chem Int Ed. 2016;55(34):9908–9912.
- Feringa BL, Browne WR. Molecular switches. Weinheim (DE): Wiley-vch; 2001.
- Eelkema R, Pollard MM, Katsonis N, et al. Rotational reorganization of doped cholesteric liquid crystalline films. J Am Chem Soc. 2006;128(44):14397–14407.
- Anderson JC, Reese CB. Photo-induced fries rearrangements. Proc Chem Soc. 1960;VIII:217.
- Zerong W. Comprehensive organic name reactions and reagents. Hoboken (NJ): John Wiley & Sons; 2010.