
ABSTRACT
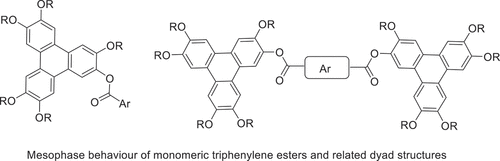
Ester linkages are frequently employed in the design of discotic liquid crystals, and indeed the first examples of discotic liquid crystals reported by Chandrasekhar, Sadashiva and Suresh were hexaesters of benzene. Within the wide range of liquid crystalline triphenylene systems so far reported, the symmetrical hexa(aryl) esters are particularly noteworthy because they form the relatively rare and technologically important discotic nematic mesophase. An alternative strategy for inducing nematogenic behaviour is to build linked and twinned structures, and here we report examples of materials that combine the two design features. Pentahexyloxy triphenylenes bearing a single aryl ester retain the columnar mesophase. Linked dyad structures promote nematic phase formation and stability is influenced by the link type and bonding arrangement within isomeric series (phthalates) and related constructs.
Graphical Abstract
Introduction
The first examples of discotic liquid crystals reported by Chandrasekhar, Sadashiva and Suresh were hexaesters of benzene (1) [Citation1] and ester linkages are common in liquid crystal science. Triphenylene based liquid crystals were reported soon after the initial report [Citation2,Citation3] and the triphenylene nucleus has become the most widely studied and versatile core [Citation4–6] in discotic systems [Citation7]. Within the symmetrical, hexasubstituted series, the heavily studied hexaethers (2) form stable columnar mesophases [Citation4,Citation5], while hexa(aryl) esters (3) form the much rarer discotic nematic phase [Citation8–10]. New synthetic chemistry has given relatively straightforward access to complex, often unsymmetrical derivatives, for both applications and to probe liquid crystal behaviour. Our group has made a number of contributions to this area and we find that breaking the symmetry can have a significant effect on mesophase stability and range. The overall observation, however, is that columnar mesophase formation dominates in monomeric systems [Citation11–15], and in systems linked by flexible spacers of appropriate length [Citation16–18]. More recently we have turned our attention to exploring structural features that support discotic nematic mesophase formation. We and others have found that nematic mesophase formation becomes favourable when triphenylene mesogens are linked via rigid spacers to form dyads (5) (single link point) [Citation19] or rigid twins (e.g. 6, 7) (two link points to give a macrocycle) [Citation20–22]. Selected examples are shown in . Here we report the initial results of combining the two known features for inducing nematic behaviour – triphenylene esters and dyad formation.
Figure 1. Examples of discotic liquid crystals, including dyads and twins.
Discussion
Triphenylene monoesters
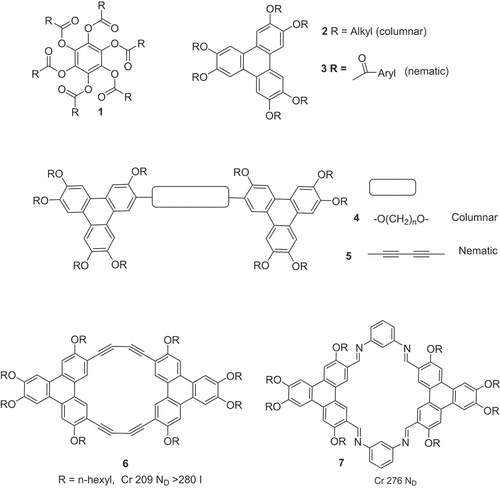
The starting material for all monoester syntheses was 2-hydroxy-3,6,7,10,11-pentakis(hexyloxy) triphenylene (MHT – ‘monohydroxytriphenylene’). It was prepared by two known routes, either directly from dihexyloxybenzene (oxidative trimerisation followed by hydrolysis under the reaction conditions) [Citation23] or by oxidative trimerisation of a mixture of dihexyloxybenzene and hexyloxyphenol [Citation24,Citation25]. MHT was isolated by column chromatography and used immediately in ester synthesis. Ester formation was achieved by direct reaction of MHT with an excess of either the appropriate acid chloride, or by coupling with the partner carboxylic acid using DCC ().
The mesophase behaviour of the triphenylene mono(aryl) esters is also shown in . It is immediately apparent that the structure of the single aryl-ester has only a small effect on the mesophase stability of the materials. In most cases the unsymmetrical materials were isolated pure as preciptates that cleared to isotropic liquids at ~150℃ – significantly higher than the symmetrical hexa(hexyloxy)triphenylene HAT6 (~100℃). Columnar hexagonal mesophases (assigned based on the characteristic textures) are observed on cooling. Introduction of a single ester group therefore does not on its own lead to the onset of nematic behaviour, but it does prevent crystallisation in the examples investigated and the columnar phase persists down to room temperature as a glass.
Scheme 1. Synthesis, structures and mesophase behaviour of penta(hexyloxy)triphenylene monoesters 8 (Hx = n-hexyl).
Triphenylene ester dyads and a triad
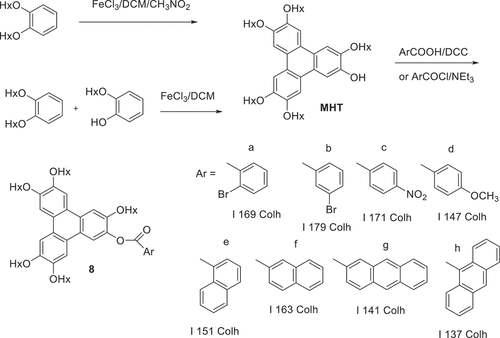
Triphenylene diesters linked by aryl dicarboxylates were selected because the joining unit has rigidity and geometry (conformation) constraints that lie between the fully flexible alkyloxy spacers (columnar) and the linear, rigid di-alkyne bridges (nematic). The isomeric phthalic acid series was selected, with the 1,2-, 1,3- and 1,4- arrangements each providing unique triphenylene-triphenylene preferred orientation. The syntheses of these twinned structures via the DCC coupling route proved more challenging than the corresponding simple monoesters. In the former case, employment of excess carboxylic acid (or acid chloride) allowed efficient conversion and isolation of the ester product. For dyad structures it was not practical to employ a large excess of MHT and reactions were complicated by the formation of side-products containing a single triphenylene and urea fragment originating from DCC (illustrated for one representative synthesis in ). Separation by column chromatography was challenging but pure samples were eventually obtained for the three isomers. Two further examples were initially selected. Thiophene-2,5-dicarboxylic acid was coupled with MHT using DCC. The reaction was again complicated by competitive urea formation, but pure dyad was obtained after chromatography. The diester from naphthalene-2,7-dicarboxylic acid was most conveniently prepared via its acid chloride (prepared by treatment of the diacid with oxalyl chloride) ().
Scheme 2. The synthesis of triphenylene ester dyads and an example of the unwanted side products produced during DCC coupling (Hx = n-hexyl).
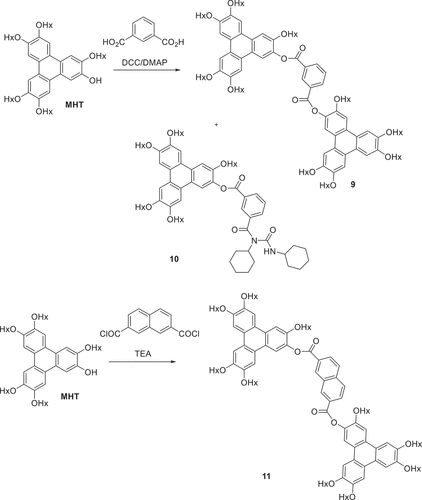
Scheme 3. The synthesis of triphenylene ester triad 16 (Hx = n-hexyl).
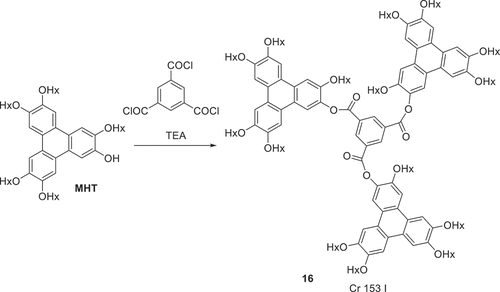
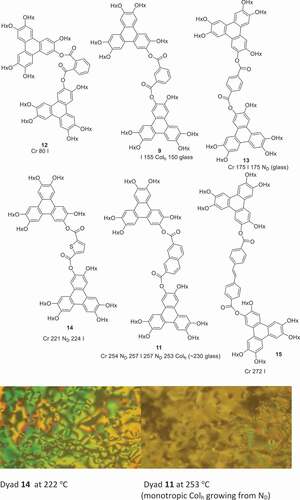
The thermal behaviour of the dyad structures was also investigated by polarising optical microscopy and is summarised in . Unsurprisingly, triphenylene dyad 12 linked by the benzene 1,2-positions did not show any mesophase behaviour. It has a low (80℃) melting point, no doubt reflecting the difficulty such a bent molecular system has in achieving efficient packing that maximises intermolecular interactions, in either a crystal or liquid crystal phase. Isophthalate (1,3-) 9 and terephthalate (1,4-) 13 dyads have similar melting points. Isophthalate dyad 9 is isolated as an amorphous solid that clears fully at 155℃ and reforms a columnar hexagonal phase on cooling (based on its characteristic fan texture) which forms a glass almost immediately (150℃). Linear terephthalate diester 13 melts directly to the isotropic liquid at 175℃. On cooling, regions of nematic and isotropic glass form simultaneously. It is noteworthy that a derivative, differing only by addition of a single vinyl substitutent on the phthalate, has been reported to show stable columnar and nematic phases, perhaps emphasising the importance of symmetry-breaking in the systems to disfavour crystallisation [Citation24]. For one sample the material was subsequently removed from the slide and re-examined by 1 H NMR spectroscopy which showed no evidence of decomposition during the heating cycle. The separation between triphenylene components was increased (in a linear manner) by linking through a stilbene (diester 15). Stilbene ester 15 melts directly to an isotropic liquid at 272℃. Bent dyads linked by 2,5-thiophene 14 and 2,7-naphthalene 11 each displayed narrow range enantiotropic nematic mesophases while the latter also showed monotropic columnar hexagonal phase formation before forming a glass. Finally, triad 16 was synthesised by reaction of MHT with the acid chloride derived from benzene-1,3,5-tricarboxylic acid. The triad shows no mesophase behaviour, and melts directly to isotropic liquid at 153℃.
Figure 2. (Colour online) Structures and mesophase behaviour of triphenylene ester dyads (Hx = n-hexyl).
Experimental
(2-Bromobenzoate) ester 8a
MHT (150 mg, 0.0002 mol), 2-bromo benzoyl chloride (0.052 g, 0.00024 mol, 1.2 eq) and NEt3 (1 ml) were stirred in CH2Cl2 (25 ml) at 0℃ for 2 h. Methanol was added and the crude solid was collected by filtration and reprecipitated from DCM-methanol to give ester 8a as an off-white solid (0.03 g, 16.6%). IR (thin film, cm−1) 3534, 2972, 2855, 1746,1616, 1512,1433, 1392,1258. 1H NMR (400 MHz, Chloroform-d) δ 8.24 (dd, J = 8.0, 2.0 Hz, 1H), 8.21 (s, 1H), 7.91 (s, 1H), 7.88 (s, 1H), 7.84 (s, 1H), 7.83 (s, 1H), 7.81 (s, 1H), 7.78 (dd, J = 8.0, 2.0 Hz, 1H), 7.51–7.41 (m, 2 H), 4.34–4.17 (m, 10 H), 2.02–1.88 (m, 8 H), 1.88–1.79 (m, 2 H), 1.66–1.52 (m, 10 H), 1.50–1.33 (m, 16 H), 1.28 (qt, J = 6.5, 3.4 Hz, 4 H), 1.00–0.89 (m, 12 H), 0.88–0.81 (m, 3 H). 13C NMR (101 MHz, CDCl3) δ 164.09, 149.82, 149.42, 149.28, 148.99, 148.84, 139.71, 134.73, 133.20, 132.35, 131.11, 128.18, 127.23, 124.74, 123.52, 123.20, 123.02, 122.68, 116.82, 108.09, 107.34, 106.98, 106.64, 106.08, 77.34, 77.02, 76.70, 69.95, 69.83, 69.53, 69.28, 68.94, 31.70, 31.65, 31.60, 30.93, 29.45, 29.40, 29.33, 25.87, 25.82, 25.78, 22.67, 22.56, 14.06, 13.98. MS (MALDI): m/z calculated for C55H75BrO7 (M+, 100%): 928.10 found 928.00.
(3-Bromobenzoate) ester 8b
Prepared from MHT and 3-bromobenzoyl chloride following the procedure described for 8a, giving dyad 8b as an off-white solid (0.07 g, 39%). IR (thin film, cm−1) 2956,2920,2855,2364,1736,1620, 1517,1434,1393,1281,1245. 1H NMR (500 MHz, Chloroform-d) δ 8.45 (t, J = 2.0 Hz, 1H), 8.22 (dt, J = 8.0, 2.0 Hz, 2 H), 8.19 (s, 1H), 7.91 (s, 1H), 7.88 (s, 1H), 7.83 (s, 1H), 7.82 (s, 1H), 7.81–7.78 (m, 2 H), 7.43 (t, J = 8.0 Hz, 1H), 4.32–4.16 (m, 10 H), 2.06–1.90 (m, 8 H), 1.86–1.72 (m, 2 H), 1.71–1.50 (m, 10 H), 1.48–1.36 (m, 16 H), 1.28–1.18 (m, 4 H), 1.02–0.88 (m, 12 H), 0.82 (t, J = 7.0 Hz, 3 H). 13C NMR (126 MHz, Chloroform-d) δ 163.82, 149.81, 149.37, 148.97, 148.83, 139.86, 136.45, 133.25, 131.58, 130.16, 128.87, 128.18, 124.71, 123.46, 123.25, 123.20, 122.98, 122.68, 116.70, 107.99, 107.31, 106.93, 106.37, 69.90, 69.83, 69.52, 69.18, 69.03, 31.72, 31.66, 31.51, 29.46, 29.42, 29.34, 29.24, 25.89, 25.87, 25.83, 25.72, 22.69, 22.66, 22.56, 14.08, 14.06, 13.95. MS (MALDI): m/z calculated for C55H75BrO7 (M+, 100%): 928.10 found 928.00.
(4-Nitrobenzoate) ester 8 c
Prepared from MHT and 4-nitrobenzoyl chloride following the procedure described for 8a, giving dyad 8 c as an off-white solid (0.058 g, 32%). IR (thin film, cm−1) 2924,2856,1740, 1614, 1514, 1435, 1387, 1345, 1258. 1H NMR (500 MHz, Chloroform-d) δ 8.48 (d, J = 8.8 Hz, 2 H), 8.40 (d, J = 8.8 Hz, 2 H), 8.22 (s, 1H), 7.93 (s, 1H), 7.88 (s, 1H), 7.84 (s, 1H), 7.83 (s, 1H), 7.79 (s, 1H), 4.29–4.15 (m, 10 H), 2.00–1.88 (m, 8 H), 1.79–1.72 (m, 2 H), 1.63–1.52 (m, 10 H), 1.47–1.34 (m, 16 H), 1.26–1.19 (m, 4 H), 1.08–0.88 (m, 12 H), 0.80 (t, J = 7.0 Hz, 3 H). 13C NMR (126 MHz, Chloroform-d) δ 163.30, 150.88, 149.91, 149.30, 149.09, 148.87, 139.57, 135.05, 131.39, 128.34, 124.77, 123.73, 123.34, 123.28, 123.22, 122.87, 116.56, 107.99, 107.24, 106.86, 106.45, 106.25, 69.92, 69.79, 69.50, 69.24, 68.91, 31.72, 31.66, 31.45, 29.47, 29.45, 29.42, 29.35, 29.18, 25.90, 25.87, 25.84, 25.66, 22.69, 22.66, 22.55, 14.08, 14.06, 13.92. MS (MALDI): m/z calculated for C55H75NO9 (M+, 100%): 894.20 found 893.6.
(4-Methoxybenzoate) ester 8d
Prepared from MHT and 4-methoxybenzoyl chloride following the procedure described for 8a, giving dyad 8d as an off-white solid (0.03 g, 17%). IR (thin film, cm−1) 2928, 2854, 1721, 1605, 1511,1468, 1434, 1392, 1316, 1253. 1H NMR (500 MHz, Chloroform-d) δ 8.25 (d, J = 8.9 Hz, 2 H), 8.20 (s, 1H), 7.91 (s, 1H), 7.88 (s, 1H), 7.84 (s, 1H), 7.83 (s, 1H), 7.80 (s, 1H), 7.02 (d, J = 8.9 Hz, 2 H), 4.33–4.15 (m, 10 H), 3.93 (s, 3 H), 2.04–1.88 (m, 8 H), 1.86–1.69 (m, 2 H), 1.62–1.50 (m, 10 H), 1.47–1.32 (m, 16 H), 1.25–1.18 (m, 4 H), 0.95–0.88 (m, 12 H), 0.82 (t, J = 6.5 Hz, 3 H). 13C NMR (126 MHz, Chloroform-d) δ 164.89, 163.85, 149.68, 149.23, 148.86, 148.79, 140.29, 132.43, 127.92, 124.61, 123.56, 123.28, 123.14, 123.08, 121.97, 116.95, 113.82, 107.96, 107.30, 106.94, 106.41, 77.31, 77.05, 76.80, 69.86, 69.82, 69.52, 69.15, 69.06, 55.52, 31.73, 31.67, 31.53, 29.47, 29.43, 29.35, 29.27, 25.89, 25.83, 25.66, 22.69, 22.66, 22.55, 14.08, 14.05, 13.96. MS (MALDI): m/z calculated for C56H78O8 (M+, 100%): 878.56 found 878.06.
(1-Naphthoate) ester 8e
Prepared from MHT and 1-napthoyl chloride following the procedure described for 8a, giving dyad 8e as an off-white solid (0.116 g, 64%). IR (thin film, cm−1) 2926, 2855, 1727, 1616, 1511, 1467, 1433, 1391, 1285. 1H NMR (500 MHz, Chloroform-d) δ 9.11 (d, J = 8.7 Hz, 1H), 8.59 (dd, J = 8.0, 1.3 Hz, 1H), 8.26 (s, 1H), 8.14 (d, J = 8.0 Hz, 1H), 7.96 (d, J = 8.0 Hz, 1H), 7.94 (s, 1H), 7.90 (s, 1H), 7.84 (s, 1H), 7.83 (s, 1H), 7.82 (s, 1H), 7.69–7.57 (m, 3 H), 4.32–4.16 (m, 10 H), 2.04–1.88 (m, 8 H), 1.84–1.74 (m, 2 H), 1.66–1.50 (m, 10 H), 1.47–1.33 (m, 16 H), 1.25–1.11 (m, 4 H), 1.01–0.83 (m, 12 H), 0.73 (t, J = 7.2 Hz, 3 H). 13C NMR (126 MHz, Chloroform-d) δ 165.92, 149.78, 149.67, 149.29, 148.93, 148.83, 140.12, 134.04, 133.95, 131.72, 131.23, 128.60, 128.09, 128.00, 126.39, 126.35, 126.05, 124.71, 124.59, 123.58, 123.29, 123.19, 123.09, 117.00, 108.08, 107.36, 106.97, 106.51, 106.13, 77.29, 77.03, 76.78, 69.94, 69.84, 69.53, 69.20, 68.95, 31.71, 31.65, 31.50, 29.47, 29.45, 29.42, 29.33, 25.89, 25.86, 25.81, 25.73, 22.69, 22.64, 22.45, 14.08, 14.04, 13.86. MS (MALDI): m/z calculated for C59H78O7 (M+, 100%): 898 found 898.
(2-Naphthoate) ester 8 f
Prepared from MHT and 2-napthoyl chloride following the procedure described for 8a, giving dyad 8 f as an off-white solid (0.05 g, 28%). IR (thin film, cm−1) 2930, 2853, 1731, 1614, 1515, 1434, 1391, 1285. 1H NMR (500 MHz, Chloroform-d) δ 8.90 (s, 1H), 8.29 (dd, J = 8.5, 1.7 Hz, 2 H), 8.26 (s, 1H), 8.04 (d, J = 8.5 Hz, 1H), 7.99 (d, J = 8.5 Hz, 1H), 7.95 (d, J = 8.5, 1H), 7.93 (s, 1H), 7.90 (s, 1H), 7.84 (s, 1H), 7.83 (s, 1H), 7.82 (s, 1H), 7.66 (ddd, J = 8.2, 6.9, 1.3 Hz, 1H), 7.60 (ddd, J = 8.2, 6.9, 1.3 Hz, 1H), 4.29–4.16 (m, 10 H), 2.00–1.86 (m, 8 H), 1.81–1.72 (m, 2 H), 1.64–1.48 (m, 10 H), 1.45–1.30 (m, 16 H), 1.26–1.09 (m, 4 H), 1.01–0.85 (m, 12 H), 0.72 (t, J = 7.1 Hz, 3 H). 13C NMR (126 MHz, Chloroform-d) δ 165.32, 149.76, 149.64, 149.29, 148.93, 148.84, 140.27, 135.87, 132.62, 132.02, 129.52, 128.55, 128.38, 128.07, 127.87, 126.82, 126.80, 125.71, 124.69, 123.57, 123.33, 123.19, 123.08, 116.91, 108.02, 107.36, 106.99, 106.47, 106.43, 77.28, 77.23, 77.03, 76.77, 69.91, 69.84, 69.54, 69.18, 69.08, 31.71, 31.63, 31.45, 29.46, 29.45, 29.42, 29.32, 29.24, 25.88, 25.86, 25.80, 25.66, 22.68, 22.63, 22.48, 14.07, 14.03, 13.84. MS (MALDI): m/z calculated for C59H78O7 (M+, 100%): 898 found 898.
(2-Anthracenoate) ester 8 g
Prepared from MHT and 2-anthracenoyl chloride following the procedure described for 8a, giving dyad 8 g as a green solid (0.03 g, 16%). IR (thin film, cm−1) 2923, 2851, 1728, 1616, 1511, 1433, 1384, 1311, 1258. 1H NMR (500 MHz, Chloroform-d) δ 9.11 (s, 1H), 8.65 (s, 1H), 8.52 (s, 1H), 8.29 (s, 1H), 8.22 (dd, J = 8.5, 1.7 Hz, 1H), 8.13 (d, J = 8.0 Hz, 1H), 8.07 (t, J = 7.7 Hz, 2 H), 7.95 (s, 1H), 7.91 (s, 1H), 7.85 (s, 1H), 7.86 (2 x s, 2 H), 7.60–7.51 (m, 2 H), 4.34–4.17 (m, 10 H), 2.06–1.91 (m, 8 H), 1.87–1.73 (m, 2 H), 1.71–1.55 (m, 10 H), 1.50–1.31 (m, 16 H), 1.25–1.12 (m, 4 H), 1.02–0.85 (m, 12 H), 0.72 (t, J = 7.1 Hz, 3 H). 13C NMR (126 MHz, Chloroform-d) δ 165.34, 149.30, 133.38, 133.31, 132.89, 132.10, 130.46, 128.96, 128.69, 128.55, 128.24, 126.75, 126.34, 125.99, 124.70, 124.39, 116.95, 106.48, 77.27, 77.02, 76.77, 69.91, 69.85, 69.55, 69.18, 69.12, 31.71, 31.63, 31.44, 29.45, 29.42, 29.31, 29.25, 25.88, 25.85, 25.80, 25.67, 22.68, 22.62, 22.48, 14.07, 14.02, 13.83. MS (MALDI): m/z calculated for C63H80O7 (M+, 100%): 948 found 948 (cluster).
(9-Anthracenoate) ester 8 h
MHT (150 mg, 0.0002 mol), anthracene-9-carboxylic acid (0.07 g, 0.0003 mol, 2.4 eq), DMAP (0.004 g, 0.000038 mol, 0.19 eq) and DCC (0.082 g, 0.049 mol, 2.4 eq) were stirred in dry CH2Cl2 (20 ml) under N2 at 0 °C. After 2 days the reaction was stopped and the solvent evaporated. The residue was purified by column chromatography (silica gel, DCM/ pet. ether 2:1) and recrystallised from 2-propanol to give the 8 h as an off-white solid (0.044 g, 23%). IR (thin film, cm−1) 2922, 2855, 1739, 1615, 1513, 1467,1432,1389,1353,1257. 1H NMR (500 MHz, Chloroform-d) δ 8.70 (d, J = 8.7 Hz, 2 H), 8.64 (s, 1H), 8.28 (s, 1H), 8.10 (d, J = 8.7 Hz, 2 H), 8.04 (s, 1H), 7.95 (s, 1H), 7.86 (s, 1H), 7.85 (s, 1H), 7.85 (s, 1H), 7.68–7.53 (m, 4 H), 4.43 (t, J = 6.6 Hz, 2 H), 4.31 − 4.17 (m, 8 H), 2.09–2.04 (m, 2 H), 2.01–1.86 (m, 8 H), 1.67–1.52 (m, 10 H), 1.49–1.35 (m, 16 H), 1.33–1.18 (m, 4 H), 1.02–0.76 (m, 15 H). 13C NMR (126 MHz, Chloroform-d) δ 167.29, 149.94, 149.78, 149.30, 149.15, 148.89, 139.71, 131.08, 130.31, 129.28, 128.67, 128.39, 127.11, 126.71, 125.80, 125.58, 124.84, 123.49, 123.39, 123.32, 123.06, 116.90, 108.28, 107.42, 106.99, 106.04, 77.28, 77.02, 76.77, 70.06, 69.84, 69.52, 69.49, 69.21, 31.74, 31.71, 31.67, 31.65, 29.56, 29.49, 29.44, 29.42, 29.35, 25.90, 25.87, 25.84, 25.82, 22.70, 22.69, 22.65, 22.55, 14.08, 14.04, 13.97. (MALDI): m/z calculated for C63H80O7 (M+, 100%): 948 found 950 (cluster).
Phthalate dyad 12
3,6,7,10,11-Pentakis(hexyloxy)triphenylen-2-ol MHT (0.530 g, 7.11 × 10−4 mol), benzene-1,2-dicarboxylic acid (0.046 g, 2.74 × 10−4 mol), N,N’-dicyclohexylcarbodiimide (DCC) (0.280 g, 1.36 × 10−3 mol) and 4-(dimethylamino)pyridine (DMAP) (0.004 g, 3.36 × 10−5 mol) were stirred in dry DCM (50 ml) at 30°C for 72 h. The mixture was filtered, washed with dilute 2 M HCl (20 ml x 2), washed with dilute sodium hydroxide and finally washed with distilled water and extracted with DCM (3x50 ml), dried with MgSO4 and evaporated to dryness in vacuo. The crude product was loaded onto a silica column and eluted with a 1:20 mixture of ethylacetate/hexane. The solid obtained was recrystallised from DCM/ethanol giving the dyad 12 (0.18 g, 41%). Mp 82°C; IR (thin film, cm−1) 2929, 2858, 1738, 1616, 1509, 1432, 1259; 1H- NMR (500 MHz, Chloroform-d) δ 8.31 (s, 2 H), 8.17–8.15 (m, 2 H), 7.92 (s, 2 H), 7.87 (s, 2 H), 7.80 (s, 2 H),7.76–7.74 (m, 2 H) 7.72 (s, 2 H), 7.50 (s, 2 H), 4.26–4.18 (m, 16 H), 4.09 (t, J = 6.5, 4 H) 2.00–1.77 (m, 20 H), 1.60–0.89 (m, 90 H) ppm. 13C-NMR (126 MHz, Chloroform-d); δ 166.0, 150.0, 149.8,, 139.9, 132.2, 131.7, 129.9, 128.4, 125.0, 123.7, 123.5, 123.0, 117.2, 108.1, 107.8, 69.6, 68.5, 31.9, 29.6, 25.8, 22.7, 14.2 ppm. MS (MALDI): m/z Calculated for C104H146O14 (M+, 100%): 1620.07 found 1619. 45.
Isophthalate twin 9
Prepared following an identical procedure to dyad 12, giving dyad 9 (0.13 g, 29%). IR (thin film, cm−1) 2954, 2858, 2928, 1741, 1616, 1511, 1432, 1260, 1225; 1H- NMR (500 MHz, Chloroform-d) δ 9.16(s, 1H), 8.51 (dd, J = 7.7, 1.5 Hz, 2 H), 8.18 (s, 2 H), 7.87 (s, 2 H), 7.82 (s, 2 H), 7.77 (s, 2 H), 7.76 (s, 2 H), 7.74 (s, 2 H), 7.69 (t, J = 7.7, 1H),, 4.10–4.20 (m, 20 H), 1.80–1.92 (m, 16 H), 1.69–1.76 (m, 4H), 1.25–1.41 (m, 20 H), 1.12–1.24 (m, 10 H) 0.70–0.92 (M, 30 H) ppm. 13C-NMR (126 MHz, Chloroform-d); δ 164.2, 149.8, 148.9, 139.9, 128.2, 124.7, 123.5, 123.3, 123.2, 116.8, 108.0, 107.0, 106.4, 69.9, 69.8, 69.2, 69.0, 31.7, 31.5, 29.6, 29.0, 25.8, 22.7, 22.6, 22.3, 14.1 ppm. MS (MALDI): m/z calculated for C104H146O14 (M+, 100%): 1620.07 found 1620.38
The urea adduct 10 was also isolated from this reaction (0.22 g, 51%). Mp 95°C; IR (thin film, cm−1) 2930, 2857, 1738, 1651, 1514, 1437, 1258. 1H- NMR (500 MHz, Chloroform-d) δ 8.48 (t, J = 1.0, 1H), 8.40–8.35(m, 1H), 8.20 (s, 1H), 7.92 (s, 1H), 7.88 (s, 1H), 7.86–7.82 (m, 3H), 7.79 (s, 1H), 7.60 (t, J = 8.0 Hz, 1H), 4.27–4.05 (m, 11 H), 3.61–3.53 (m, 1H) 2.12–1.19 (m, 60 H), 0.96–0.81 (m, 15 H) ppm. 13C- NMR (126 MHz, Chloroform-d) δ 174.5, 164.3, 154.1, 149.5, 149.1, 149.0, 140.0, 137.7, 132.4, 132.0, 130.2, 129.0, 128.8, 128.3, 123.6, 123.1, 116.9, 108.2, 107.5, 107.1, 106.6, 106.5, 70.1, 69.9, 69.7, 69.7, 58.0, 49.9, 31.8, 29.6, 26.4, 26.0,22.8, 22.7, 14.2, 14.1 ppm. MS (MALDI): m/z C69H99N2O9 (M + H)+: 1099.74 found 1099.55
Terephthalate twin 13
Prepared following an identical procedure to dyad 12, giving dyad 13 (0.15 g, 35%). IR (thin film, cm−1) 2918, 2850, 1731, 1515, 1435, 1261; 1H- NMR (500 MHz, Chloroform-d) δ 8.46 (s, 4H), 8.24 (s, 2 H), 7.92 (s, 2 H), 7.88 (s, 2 H), 7.82 (s, 2 H), 7.79 (s, 4H), 4.19–4.28 (m, 20 H), 1.99–1.89 (m, 16 H), 1.76–1.84 (m, 4H), 1.52–1.65 (m, 20 H), 1.35–1.48 (m, 40 H),0.87–0.98 (m, 30 H) ppm.13C- NMR (126 MHz, Chloroform-d); δ 164.5, 149.8, 149.5, 148.9, 140.0, 133.8, 130.3, 128.4, 124.9, 123.4, 123.1, 116.9, 108.2, 106.5, 69.4, 69.2, 31.9, 29.6, 25.8,, 22.8, 14.1 ppm. MS (MALDI): m/z calculated for C104H146O14 (M+, 100%): 1620.07 found 1619.68.
Thiophene Dyad 14
2,5-Thiophenedicarboxylic acid (19.2 mg, 0.001 mmol, 1eq) was dissolved in 10 ml dry DCM. Then 2 drops of dry DMF and oxalyl chloride (0.3 ml) were added to the solution. The mixture was stirred under nitrogen overnight. After that the solvents were evaporated under reduced pressure the resulting product was used straight away without further analysis.
MHT (0.25 g, 0.33 mmol, 3eq) was dissolved in dry DCM (10 ml). Then 2, 5thiophenedicarbonyl dichloride (0.02 g, 0.0009 mmol, 1eq) was added to the MHT solution after dissolving it in dry DCM (10 ml). Then dry TEA (0.1 ml) was added. The reaction was kept stirring under nitrogen overnight then quenched by adding more DCM and water. The mixture was separated and washed with dilute HCl. The solvent was evaporated, and the product recrystallised from ethanol to yield dyad 14 as a pale yellow sold (0.13 g, 30%). IR (thin film, cm−1) 2981,1725, 1514,1262. 1H- NMR (500 MHz, Chloroform-d) δ 8.24 (s, 2 H), 8.09 (s, 2 H), 7.93 (s, 2 H), 7.89, (s, 2 H), 7.84–7.81 (m, 6H), 4.27–4.21 (m, 20 H), 1.97–1.82 (m, 20 H), 1.58–152 (m, 20), 1.30–1.41 (m, 40), 0.96–0.87 (m, 30 H) ppm. 13C-NMR (126 MHz; Chloroform-d) 158.84, 148.8, 148.29, 148.00, 147,38, 138.44, 137.86, 133.23, 127.32, 123.76, 122.20, 115.75, 107.02, 105.94, 105.40, 68.91, 68.80, 68.51, 68.09, 30.68, 30.63, 30.50, 28.44, 28.41, 28.38, 28.31, 28.21, 24.85, 24.83, 24.80, 24.66, 21.65, 21.63, 21.57, 13.04, 13.02 ppm. MS (MALDI): m/z calculated for C102H144O14S (M+, 100%): 1626.32 found 1626.00.
2,7-Naphthalene dyad 11
2,7 Naphthalenedicarboxylic acid (0.5 g,4.62 mmol. 1eq) was dissolved in DCM (20 ml). Then oxalyl chloride (3.4 g, 27.7 mmol, 6eq) and DMF 2 drops were added to the solution. The reaction was kept stirring under nitrogen overnight. The solvent was evaporated, and the crude product recrystallised from toluene to give the acid chloride as a colourless solid (0.30 g, 30%) that was used directly in the next step.
2,7- Naphthalene dicarbonyl dichloride (0.06 g, 0.268 mmol, 1eq) was dissolved in dry DCM (20 ml) then it was added to a solution of MHT (0.5 g, 0.670 mmol, 2.5eq) in dry DCM (20 ml) followed by dry TEA (0.1 ml). The reaction was kept stirring under nitrogen overnight then quenched by adding more DCM and water. The mixture was separated and washed with dilute HCl. The solvent was evaporated, and the product recrystallised from ethanol to yield dyad 11 as a pale yellow solid (0.2 g, 40%). IR (thin film, cm−1) 2928., 1727, 1513, 1254. 1H-NMR (500 MHz, Chloroform-d) δ 8.98 (s, 2H), 8.41 (d, J = 8.0 Hz, 2H), 8.28 (s, 2H), 8.18 (d, J = 8.0 Hz, 2H), 7.95 (s, 2H), 7.90 (s, 2H), 7.87–7.82 (m, 6H), 4.19–4.28 (m, 20 H), 1.77–1.98 (m, 20 H), 1.17–1.52 (m, 80 H), 0.73–0.98(m, 30 H). 13C-NMR (126 MHz; Chloroform-d) 164.82, 149.75, 149.52, 149.17, 148.82, 148.85, 141.74, 140.14, 130.87, 130.33, 129.00, 126.83, 123.32, 117.04, 116.92, 106.87, 106.39, 69.89, 69.81, 69.56, 69.19, 69.07, 31.73, 31,67, 31.53, 29.46, 29.35, 29.27, 25.90, 25.87, 25.83, 25.69, 22.69, 22.66, 22.56, 14.08, 14.05, 13.97 ppm.
4,4ʹ-Stilbene dyad 15
Prepared from MHT and stilbene-4,4ʹdicarboxylic acid following the procedure described for dyad 11 to give 15 as a colourless solid (0.2 g, 50%). IR (thin film, cm−1)2915, 2850, 1729, 1509, 1432, 1256. 1H-NMR (500 MHz, Chloroform-d) δ 8.33 (d, J = 8.4 Hz, 4H), 8.23 (s,2H), 7.94 (d, J = 8.4 Hz, 4H), 7.89–7.71 (m, 10 H), 7.38 (s, 2H), 4.33–4.15 (m,20 H), 2.02–1.74 (m,20 H), 1.65–1.15 (m, 65 H), 1.00–0.77 (m, 30 H). 13C-NMR (126 MHz; Chloroform-d) 165.79, 161.50, 149.86, 149.41, 149.05, 148.96, 146.52, 140.24, 130.18, 128.08, 127.73, 126.99, 125.98, 124.79, 123.73, 123.31, 123.24, 115.09, 108.17, 107.15, 103.66, 77.41, 77.16, 76.91, 70.05, 69.99, 69.68, 69.34, 69.27, 68.38, 31.84, 31.79, 31.70, 29.58, 29.55, 29.46, 29.42, 29.28, 26.01, 25.99, 25.96, 25.84, 22.81, 22.79, 22.75, 14.20, 14.13 ppm.
Triad 16
1, 3, 5-Benzene tricarbonyl chloride (0.03 g, 0.0001 mmol, 1eq) was dissolved in dry DCM (10 ml) and added to a solution of MHT (0.4 g, 0.536 mmol, 4.5 eq) in dry DCM (10 ml) and TEA (1 ml). The mixture was stirred under nitrogen overnight at room temperature then quenched by adding more DCM and water. The mixture was separated and washed with dilute HCl. The solvent was evaporated then the crude was purified by column chromatography on silica gel eluting with ethyl acetate and petroleum ether (1:15) to give triad 16 as a pale yellow solid (0.09 g, 32%). IR (thin film, cm−1) 2919.83, 1742.70, 1263.73 1H-NMR (500 MHz, CD3COCD3) δ 9.42(s,3H), 8.21(s,3H), 7.69 (s,3H), 7.78(s,3H), 7.51(s,3H), 7.20(s,3H),6.97(s,3H), 3.77–4.36(m,30 H), 1.41–2.09(m,120 H), 0.88–1.04(m, 45 H). 13C-NMR (126 MHz; CD3COCD3) δ = 163.36, 148.33, 139.39, 139.23, 131.21, 123.20, 122.81, 107.15, 106.33, 69.80, 68.84, 31.81, 31.72, 31.54, 30.93, 29.63, 29.45, 29.33, 29.25, 26.00, 25.88, 25.81, 25.63, 22.73, 22.63, 22.50, 14.11, 14.08, 14.01, 13.91 ppm. MS (MALDI): m/z calculated for C153H216O21 (M+, 100%): 2391 found 2392 (cluster).
Conclusions
A short series of monomeric triphenylenes bearing a single aryl ester plus five hexyloxy substituents has been synthesised and investigated. All examples form columnar hexagonal mesophases and the stability of the mesophases is relatively insensitive to the ester employed. Ester linked dyad systems have been prepared from diacids and several examples, which can be considered to have intermediate rigidity between columnar alkyoxy-bridged systems and nematic dialkynes, show narrow-range nematic phases which further implies that a combination of shape and rigidity are necessary to promote nematic behaviour over columnar organisation. The corresponding triad based on benzene-1,3,5-triester linkage is non-mesogenic.
Acknowledgments
We thank Princess Nourah Bint Abdul Rahman university, Taibah university, Technical and Vocational training corporation (Saudi Arabia) and TETFUND Nigeria for financial support.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Chandrasekhar S, Sadashiva BK, Suresh KA. Liquid crystals of disc-like molecules. Pramana – J Phys. 1977;9:471–480.
- Billard J, Dubois JC, Huutinh N, et al. A disk-like mesophase. New J Chem. 1978;2:535–540.
- Destrade C, Mondon MC, Malthete J. Hexasubstituted triphenylenes: a new mesomorphic order. J Phys Supp C3. 1979;40:17–21.
- Cammidge AN, Bushby RJ. Discotic liquid crystals: synthesis and structural features. In: Demus D, Goodby JW, Gray GW, et al., editors. Handbook of Liquid Crystals. Vol. 2. Weinheim: Wiley-VCH; 1998. p. 693–748.
- Kumar S. Recent developments in the chemistry of triphenylene-based discotic liquid crystals. Liq Cryst. 2004;31:1037–1059.
- Kumar S. Chemistry of discotic liquid crystals. Florida: CRC Press; 2010.
- Wöhrle T, Wurzbach I, Kirres J, et al. Discotic liquid crystals. Chem Rev. 2016;116:1139–1241.
- Tinh NH, Gasparoux H, Destrade C. Homologous series of disc-like mesogens with nematic and columnar polymorphism. Mol Cryst Liq Cryst. 1981;68:101–111.
- Hindmarsh P, Watson MJ, Hird M, et al. Investigation of the effect of bulky lateral substituents on the discotic mesophase behaviour of triphenylene benzoates. J Mater Chem. 1995;5:2111–2123.
- Cammidge AN, Gopee H. Design and synthesis of nematic phases formed by disk-like molecules. In: Goodby JW, Collings PJ, Kato T, et al., editors. Handbook of Liquid Crystals. Vol. 3. 2nd ed. Weinheim: Wiley-VCH; 2014. p. 293–334.
- Cammidge AN. The effect of size and shape variation in discotic liquid crystals based on triphenylene cores. Philos Trans R Soc A. 2006;364:2697–2708.
- Cammidge AN, Gopee H. Structural factors controlling the transition between columnar-hexagonal and helical mesophase in triphenylene liquid crystals. J Mater Chem. 2001;11:2773–2783.
- Cammidge AN, Chausson C, Gopee H, et al. Probing the structural factors influencing columnar mesophase formation and stability in triphenylene discotics. Chem Commun. 2009;7375–7377. DOI:https://doi.org/10.1039/b913678a
- Cammidge AN, Gopee H. Mixed alkyl-alkoxy triphenylenes. Mol Cryst Liq Cryst. 2003;397:417–428.
- Cammidge AN, Gopee H. Synthesis and liquid crystal properties of mixed alkynyl-alkoxy-triphenylenes. Liq Cryst. 2009;36:809–819.
- Boden N, Bushby RJ, Cammidge AN, et al. The creation of long-lasting glassy columnar discotic liquid crystals using “dimeric” discogens. J Mater Chem. 1999;9:1391–1402.
- Kumar S, Manickam M, Schonherr H. First examples of functinalized triphenylene discotic dimers: molecular engineering of advance materials. Liq Cryst. 1999;26:1567–1571.
- Kumar S. Triphenylene-based discotic liquid crystal dimers, oligomers and polymers. Liq Cryst. 2005;32:1089–1113.
- Kumar S, Varshney SK. Design and synthesis of discotic nematic liquid crystals. Org Lett. 2002;4:157–159.
- Zhang L, Gopee H, Hughes DL, et al. Antiaromatic twinned triphenylene discotics showing nematic phases and 2-dimensional Π-overlap in the solid state. Chem Commun. 2010;46:4255–4257.
- Zhang L, Hughes DL, Cammidge AN. Discotic triphenylene twins linked through thiophene bridges: controlling nematic behavior in an intriguing class of functional organic materials. J Org Chem. 2012;77:4288–4297.
- Cammidge AN, Turner RJ, Beskeni RD, et al. A modified route to unsymmetrically substituted triphenylenes, new functionalised derivatives and twins, and the smallest reported triphenylene mesogen. Liq Cryst. 2017;44:2018–2028.
- Kumar S, Lakshmi B. A convenient and economic method for the synthesis of monohydroxy-pentaalkoxy- and hexaalkoxytriphenylene discotics. Tetrahedron Lett. 2005;46:2603–2605.
- Ban J, Chen S, Zhang H. Synthesis and phase behaviour of mesogen jacketed liquid crystalline polymer with triphenylene discotic liquid crystal mesogen unit in side chains. RSC Adv. 2014;4:54158–54167.
- Kong X, He Z, Gopee H, et al. Improved synthesis of monohydroxytriphenylenes (MHTs) – important precursors to discotic liquid crystals. Tetrahedron Lett. 2011;52:77–79.