
Abstract
Background
Schwannomatosis is a relatively rare disorder and is related to neurofibromatosis type 2. Although there is clinical overlap between schwannomatosis and neurofibromatosis type 2, these diseases have to be regarded as separate entities due to the genetic origin and course of the disease.
Methods
A comprehensive review of the literature was conducted for relevant studies using Pubmed and Cochrane databases to discuss the epidemiology, clinical presentation, diagnostic criteria, pathological and imaging features, treatment and genetics of schwannomatosis.
Results
Germline mutations SMARCB1 and LZTRI together with the NF2 gene play a role in the pathophysiology of schwannomatosis. The most common symptom is pain with affection of the spine and peripheral nerves in the majority of patients. High quality contrast enhanced MRI scan is the imaging modality of choice. Treatment is conservative if asymptomatic and surgical if symptomatic. The goal is symptom control with preservation of neurological function.
Conclusion
Schwannomatosis is a relatively rare disorder in which the main goal is to preserve neurological function.
Introduction
The neurocutaneous disorders neurofibromatosis type 1 and 2 are very well known by neurosurgeons for decades. More recently related forms of these two disorders have been reported. Legius syndrome or NF1-like syndrome is caused by mutations in the SPRED1 gene and is associated with similar multiple café-au-lait spots as neurofibromatosis type 1 but the tumoral phenotype is absent.Citation1 Schwannomatosis is a disease related to neurofibromatosis type 2 and is characterized by peripheral and spinal schwannomas in the absence of bilateral vestibular schwannomas.Citation2–6
Familial schwannomatosis is an autosomal dominant disorder characterized by heterozygous inactivating mutations in the tumor suppressor genes, SMARCB1 and LZTR1 in the majority of familial cases and in some sporadic cases of schwannomatosis.Citation2–6
It is clear that schwannomatosis must be distinguished from type 1 and especially from type 2 neurofibromatosis because the genetic origin, course of the disease and its repercussions are different. Although there is, in part, clinical overlap between schwannomatosis and especially the mosaic form of NF2, these diseases have to be regarded as separate entities.
In this review, we describe the characteristics of schwannomatosis relevant for the neurosurgeon.
Epidemiology
Schwannomatosis is a very rare disorder with an estimated birth incidence of 1/40000 to 1/70000 and an incidence of 0.58 cases per 1000000 persons/year.Citation2,Citation5,Citation7,Citation8
It is characterized by a predisposition for multiple schwannomas and less commonly meningiomas. In schwannomatosis patients, the diagnostic criteria for NF2 are occasionally met. There are no bilateral vestibular schwannomas in schwannomatosis but unilateral vestibular schwannomas can be seen in patients with an LZTR1 mutation.Citation1,Citation2,Citation5–9
The majority of schwannomatosis patients occur sporadically whereas familial cases occur in 13%–25% of cases.Citation3,Citation7,Citation8
Clinical presentation
In patients with schwannomatosis, schwannomas commonly affect the spine in 74% of cases and the peripheral nerves in 89% of cases. Cranial nerve schwannomas are uncommon and upon appearance they mostly affect the trigeminal nerve (present in 8% of cases).
In 33% of cases with schwannomatosis, the lesions are anatomically limited to a specific segment of the body: a single limb, several continguous spinal segments or one half of the body.
Schwannomatosis patients mostly develop symptoms in the second or third decade of life with a diagnostic delay of more or less 10 years. There is no predilection for race or sex. Patients most commonly present with chronic pain in 46% of cases or a mass in 27% of cases. In 11% of patients the clinical presentation is a combination of both.
In schwannomatosis patients, depression and anxiety are common comorbidities resulting from chronic pain and affecting 17% to 39% of patients. Additional tumors such as lipomas and angiolipomas occur in 11% and 3% of patients respectively. Meningeomas occur in 5% of patients with schwannomatosis, most often as a solitary lesion.
The risk of malignancy is low and patients with schwannomatosis in general do not have a decreased life expectancy. But there seems to be an increased risk for malignant peripheral nerve sheath tumors in patients with a SMARCB1 mutation.Citation10
In contrast to neurofibromatosis type 2, vestibular schwannomas are rare and the occurrence of bilateral vestibular lesions must prompt the search for an alternative diagnosis. Unilateral vestibular schwannomas however may occur in schwannomatosis patients. On the other hand, intradermal schwannomas, ependymomas, gliomas, neurofibromas, eye abnormalities, skinfold freckling, greater than 6 café-au-lait spots and Lisch noduli which all typically seen in type 1 and/or type 2 neurofibromatosis do not occur in schwannomatosis.
Diagnostic criteria
Since research criteria were published in 1997 and MacCollin et. al. postulated the first consensus diagnostic criteria for schwannomatosis in 2005, revisions of these criteria were published onto a background of an evolving genetic knowledge of this disease.
Currently, the diagnosis of schwannomatosis is based on molecular and/or clinical diagnostic criteria according to Plotkin et al. and Ostrow et al.Citation11
These criteria, together with exclusion criteria, are summarized in .
Table 1. Diagnosis of schwannomatosis based on molecular and/or clinical diagnostic criteria according to Plotkin et al.Citation6 and Pstrow et al.Citation11
Pathological features
A schwannoma arises from Schwann cells. It is a benign tumor that is encapsulated by epineurium and grows eccentrically to the underlying nerve, thereby displacing the nerve fibers. In schwannomas, Schwann cells form fascicles with a wavy aspect of the cytoplasmic processes. Vessels with prominent hyaline walls are often seen (). Antoni A and Antoni B regions can be found. Antoni A regions are characterized by high cellularity and spindle-shaped nuclei arranged in palisades separated by nuclear-free zones. Antoni B regions are less cellular than Antoni A regions and have a more disorganized matrix. Degenerative features like myxoid/cystic degeneration, inflammation and fibrosis are not rare in longstanding tumors (‘ancient’ Schwannoma). Whatever it looks like, Schwannomas are always diffusely positive for S-100 protein ().
Figure 1. (A) Low power view of a schwannoma, showing an encapsulated spindle cell lesion and vessels with hyaline walls. (B) Presence of diffuse S-100 staining.
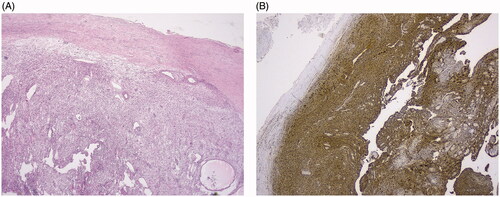
Schwannomas are as such clearly different from neurofibromas which are located centrally in the nerve with a diffuse infiltration between the axonal structures.
Imaging features
High quality MRI-scan with 3 mm thick slices through the internal auditory canal is the cornerstone of radiologic evaluation for vestibular schwannomas and as such the imaging modality of choice.
Schwannomas present as discrete well-defined round-to-oval lesions situated along the course of the nerve.
On MRI the lesions are low-to-intermediately intense on T1-weighted imaging () and hyperintense on T2- and STIR-weighted sequences (). They show an intense contrast enhancement and have a typical dumbbell-shape.
Figure 2. (A) T1-weighted MRI scan with contrast demonstrating the contrast enhanced schwannomas. (B) T1-weighted MRI scan without contrast.
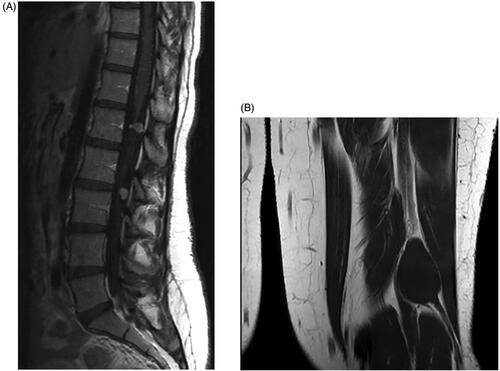
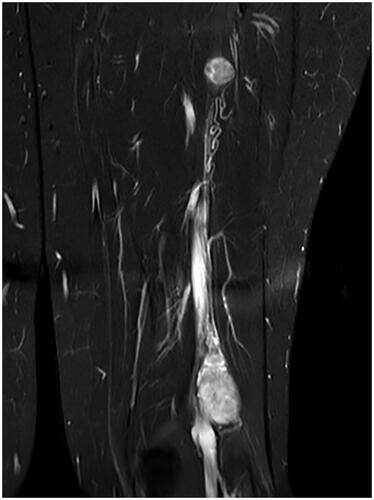
Figure 3. Hyperintense lesions on T2-weighted MRI scan without contrast.
On CT, schwannomas are hypo- to isodense with a variable degree of contrast enhancement.
Treatment
Management of patients with schwannomatosis is symptom-oriented.
The rule is observation for the asymptomatic schwannoma patient. Surgical excision is only indicated for spinal cord compression and for symptoms clearly related to the schwannoma such as pain or a cumbersome mass.
The goal of treatment should be symptom control with preservation of neurological function.
In schwannomatosis patients there is no defined role for radiotherapy or chemotherapy. Radiotherapy should be reserved for life-threatening enlarging schwannomas that cannot be addressed surgically or for the very rare patient with a malignant schwannoma. Individual reports debate on the efficacy of bevacizumab. This however has not been tested in clinical trials, but there are a number of anecdotical reports suggesting a beneficial effect of bevacizumab in schwannomatosis patients.
Genetics
The genetics of schwannomatosis is not yet entirely unraveled.
To date, germline mutations in 2 genes have been identified in schwannomatosis patients: SMARCB1 and LZTR1. These are tumor suppressor genes located on chromosome 22, centromeric to NF2. Germline SMARCB1 mutations account for 48% of familial and 10% of sporadic schwannomatosis patients. Germline LZTR1 mutations on the other hand explain 38% of familial and 30% of sporadic schwannomatosis cases. The genetic cause is unknown in 14% of familial cases and in 60% of sporadic cases. Recent data show that there are still undiscovered genes for familial schwannomatosis and this is the topic of current research. Sporadic cases are often clinically indistinguishable from mosaic neurofibromatosis type 2.Citation9
The schwannomas of patients with a SMARCB1 or LZTR1 germline mutation frequently show loss of the chromosome 22 in the tumor harboring the wild type SMARCB1 or LZTR1 gene. In addition independent tumor specific somatic mutations inactivating the NF2 allele on the remaining chromosome 22 are frequently found in these schwannomas, meaning that these schwannomas are hemizygous for chromosome 22 genes and nullizygous for the NF2 gene AND the SMARCB1 or LZTR1 gene. This led to the three-step, four-hit model of oncogenesis ().Citation6
Figure 4. The four-hit, three-step model for tumorigenesis in schwannomatosis (here shown for SMARCB1): germline mutation in SMARCB1 (hit 1), loss of a region of chromosome 22 (hit 2 and 3) and pathogenic mutation in remaining WT NF2 allele (m = pathogenic mutation; + = WT). The same mechanism has also been demonstrated for germline LZTR1 mutations.
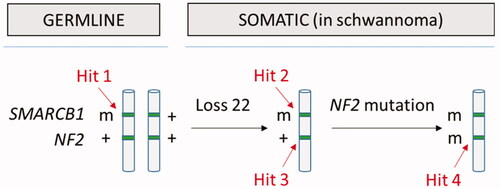
The fact that somatic NF2 mutations often coexist with constitutional SMARCB1 or LZTR1 mutations in schwannomatosis patients demonstrate that the classical 2-hit model of tumorigenesis is insufficient to account for the development of schwannomas in schwannomatosis . Genetic testing is possible on blood- and tumor-derived DNA and a comprehensive mutation analysis of LZTR1, SMARCB1 NF2 should be performed in patients with schwannomatosis.
It is important to point out here that mutations in the SMARCB1 gene can be responsible for schwannomatosis but also for pediatric atypical teratoid/rhomboid tumors and rare cases of Coffin-Siris syndrome, a syndromic form of intellectual disability. The three different syndromes are usually associated with a specific set of mutations in SMARCB1 but some overlap exists and a case of Coffin-Siris syndrome with schwannomatosis has been reportedCitation12 and we are also aware of one unreported case (unpublished data: EL).
On the other hand mutations in LZTR1 have been reported in schwannomatosis but also in patients with Noonan syndrome, a syndromic form of short stature and learning disabilities.Citation13 Schwannomatosis is genetically heterogeneous because mutations in more than one gene can cause the disease.
Conclusion
Schwannomatosis is a very rare disorder which has to be distinguished from the other, better known, neurofibromatosis. To date, germline mutations in SMARCB1 and LZTR1 have been identified in schwannomatosis however there most probably is also a role for the NF2 gene.
The most common symptom in schwannomatosis patients is pain and the spine and peripheral nerves are affected in the majority of patients. In schwannomatosis there can be no diagnostic criteria for NF2 in general and no (bilateral) vestibular schwannomas as well as no first-degree relatives with NF2 and no known constitutional NF2 mutation in particular.
High quality contrast enhanced MRI-scan is the imaging modality of choice.
Treatment of schwannomatosis patients is symptom-oriented with observation for the asymptomatic patient and with surgical excision for spinal cord compression and for symptoms clearly related to the schwannoma such as pain or a cumbersome mass.
The goal of treatment is symptom control with preservation of neurological function. Radiotherapy or systemic therapy are reserved for lesions non-accessible for surgery or for malignancy.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Beert E, Brems H, Renard M, et al. Biallelic inactivation of NF1 in a sporadic plexiform neurofibroma. Genes Chromosomes Cancer 2012;51:852–7.
- Gonzalvo A, Fowler A, Cook RJ, et al. Schwannomatosis, sporadic schwannomatosis, and familial schwannomatosis: A surgical series with long-term follow-up. Clinical article. J Neurosurg 2011;114:756–62.
- Jacoby LB, MacCollin M, Parry DM, et al. Allelic expression of the NF2 gene in neurofibromatosis 2 and schwannomatosis. Neurogenetics 1999;2:101–8.
- Koontz NA, Wiens AL, Agarwal A, Hingtgen CM, Emerson RE, Mosier KM. Schwannomatosis: The overlooked neurofibromatosis? AJR Am J Roentgenol 2013;200:W646–53.
- Merker VL, Esparza S, Smith MJ, Stemmer‐Rachamimov A, Plotkin SR. Clinical Features of Schwannomatosis: A Retrospective Analysis of 87 Patients. Oncologist 2012;17:1317–22.
- Plotkin SR, Blakeley JO, Evans DG, et al. Update from the 2011 International Schwannomatosis Workshop: From genetics to diagnostic criteria, in. Am J Med Genet 2013;161:405–16.
- Antinheimo J, Sankila R, Carpén O, Pukkala E, Sainio M, Jääskeläinen J. Population-based analysis of sporadic and type 2 neurofibromatosis-associated meningiomas and schwannomas. Neurology 2000; 54:71–6.
- MacCollin M, Chiocca EA, Evans DG, et al. Diagnostic criteria for schwannomatosis. Neurology 2005;64:1838–45.
- Smith MJ, Bowers NL, Bulman M, et al. Revisiting neurofibromatosis type 2 diagnostic criteria to exclude LZTR1-related schwannomatosis. Neurology 2017;88:87–92.
- Evans DGR, Huson SM, Birch JM. Malignant peripheral nerve sheath tumours in inherited disease. Clin Sarcoma Res 2012; 2:17
- Ostrow KL, Donaldson K, Blakeley J, Belzberg A, Hoke A. Immortalized human schwann cell lines derived from tumors of schwannomatosis patients. PLoS ONE 2015;10:e0144620.
- Gossai N, Biegel JA, Messiaen L, Berry SA, Moertel CL. Report of a patient with a constitutional missense mutation in SMARCB1, Coffin-Siris phenotype, and schwannomatosis. Am J Med Genet 2015;167:3186–91.
- Yamamoto GL, Aguena M, Gos M, et al. Rare variants in SOS2 and LZTR1 are associated with Noonan syndrome. J Med Genet 2015;52:413–21.