
ABSTRACT
Purpose
To develop a more efficient impression cytology (IC) method for the transfer of ocular surface cells onto glass microscope slides for cytochemical, immunocytochemical, and immunofluorescence studies.
Methods
Cells are lifted off the ocular surface with a mixed cellulose ester membrane and then firmly attached to a glass slide using a novel triblock copolymer comprised of collagen type I, polyethylenimine and poly-L-lysine (CPP), and crosslinking cells and glass slide by heating and cooling. The membrane is removed intact after softening it with a butanol/ethanol solution. Transfer of cells is complete in about 10–15 minutes and is ready for staining. The efficiency of our cell transfer method was compared to current methods based on poly-L-lysine and albumin paste.
Results
Our method ensured almost complete transfer of cells. In contrast, the transfer of rabbit conjunctiva cells onto poly-L-lysine-covered slides was 37.5 ± 6.3% lower, and onto albumin-paste covered slides 62.5 ± 5.6% lower (mean ± SD); the transfer of rabbit goblet cells was even less efficient. The new method was also more efficient for transfer of cells from human oral mucosa obtained by IC. Transferred cells were successfully stained with H&E, chemiluminescence, and immunofluorescence agents. Using our method, we stained ocular surface cells for S100A4 and ATF4, both of which play a role in the pathophysiology of dry eye disease. We obtained similar results with oral mucosal cells, suggesting the generalizability of our approach. We propose an explanation for the strong adhesion of cells to the glass slide, which is based on their interactions with the triblock copolymer.
Conclusions
We developed a novel approach for the efficient and rapid transfer of cells obtained by IC onto glass microscope slides using a novel copolymer. Compared to available methods, our improved approach makes IC robust and simple, and should increase its diagnostic yield and clinical applicability.
Introduction
Impression cytology (IC) is a minimally invasive method for the retrieval of the outermost layer of ocular surface cells using various types of membranes. Introduced in 1977, IC evolved into the primary method of evaluating human conjunctival and corneal epithelial cells.Citation1,Citation2 IC transfers cells from the upper layers of the ocular surface (mainly interpalpebral conjunctiva) by applying on it a membrane either directly or, more recently, using a specialized instrument (Eyeprim, Opia Technologies, France). These cells are then evaluated according to the needs of the study. Thus, IC can be conceptualized as consisting of three distinct steps: cell acquisition from the ocular surface, followed by transfer to a platform, where they are subjected to evaluation by morphological or molecular methods.
What makes IC appealing are its simplicity and versatility. IC, an effective and safe technique, has contributed significantly to both clinical and research ophthalmology. The range of its applications encompasses conventional cytological evaluation of ocular surface cells for squamous metaplasia; examination of molecular and cellular mediators of inflammation; and assessment of the expression of various genes by determining the levels of their transcripts or of the proteins they encode.Citation3–5
IC is still evolving methodologically in order to overcome some persisting limitations. The need for topical anesthesia to minimize patient discomfort during sampling has been viewed as a limitation, as it may affect the molecular analysis of the sample.Citation6 Another limitation is the variability of the number of harvested cells available for evaluation, which depends on the examiner’s skill and the properties of the materials used, especially those of the membrane. Given the clinical and research utility of IC, investigators have attempted to optimize each of the three steps of IC mentioned above.
The properties of the membrane play a major role in IC cell transfer. The ideal membrane should maximize cell retrieval, maintain the structural integrity of the cells, and allow their quantitative release to the final destination, usually a microscope glass slide, or a test tube containing appropriate buffers for the extraction of proteins or nucleic acids. Several membranes have been used including those based on cellulose acetate, polycarbonate, nitrocellulose, or polyethersulfone.Citation2,Citation7,Citation8 The Eyeprim device uses polyethersulfone membranes; despite initial enthusiasm, it appears to perform similarly to the traditional IC method, at least for some applications.Citation9
The inadequate release of cells from membranes led investigators to study their morphology directly on the membrane.Citation10 However, visualization of cells on membranes is hindered by the lack of membrane transparency and/or by the high background when cells are stained, especially when nitrocellulose or cellulose acetate membranes are stained with immunocytochemical stains. To overcome this problem, Krenzer and Freddo harvested cells on pure nitrocellulose membranes, which were then sprayed with a fixative, transferred to poly-L-lysine-coated glass slides and dried.Citation11 This procedure, however, proved cumbersome, as it requires chemical dissolution of the membrane matrix and a long washing step with various solutions or enzymatic digestion until the membrane is totally dissolved. Equally cumbersome and prolonged (includes a 3-day washing step) was a method in which cells were transferred onto a light microscopy slide coated with poly-L-lysine.Citation12 Finally, a method using a double-sided adhesive tape on glass slides had its own limitationsCitation13 .
To improve the transfer of cells from the membrane to the glass microscope slide, we coated the glass slide with a novel triblock copolymer of collagen, polyethyleneimine, and poly-L-lysine, which serves as a glue. This glue provides a pillow-like structure that greatly improves the adhesion of the cells to the glass slide. Here, we report our results, which demonstrate that our approach ensures the quantitative transfer of ocular surface cells onto microscope slides. These cells can then be evaluated with several morphological methods, including conventional staining, immunocytochemistry, and immunofluorescence.
Materials and methods
Reagents
All general solvents and reagents were of HPLC grade or of the highest grade commercially available. Nitrocellulose membranes: mixed cellulose nitrate and acetate esters, 0.22 µm pore size, (cat. no. GSTF14250, Millipore Corp. Bedford, MA). Glutaraldehyde 50% aqueous solution (16320, Electron Microscopy Sciences, Hatfield, PA). Bovine collagen type I 5 mg/mL (A1064401, Thermo Fisher Scientific, CA). Reagents: Sodium metasulfite (161519), Ponceau S (141194), ethanol (T038181000), 1-butanol (B7906), poly-L-lysine 0.1% (w/v) in H2O (P8920), polyethylenimine branched Mn~10,000 (408727), polyethylene glycol 400 (202398), periodic Acid-Schiff PAS kit (395B), hematoxylin (HHS16) and eosin (HT110316), Rhodamine B isothiocyanate 283924 and fluorescein isothiocyanate (46950) were from Sigma (St Louis, MO). VECTASHIELD® Antifade Mounting Medium with DAPI (H-1500, Vector Laboratories, Burlingame, CA). S100A8/9 antibody (48M7C7, Norvus, St Louis, MO). ATF4 (11815, Cell Signalling, Beverly, MA). NFAT5 antibody (GTX23446, GeneTex). Antibodies: CD11b/ITGAM (ab133357, Abcam, Cambridge, MA). HRP-anti-rabbit IgG (ab7090), HRP-anti-mouse (ab47827), FITC-anti-rabbit (ab6717), FITC-anti-mouse (ab6785), Texas Red anti-mouse (ab6787), Texas Red anti-rabbit (ab6719) antibodies, as well as goat IgG (ab37373), rabbit IgG (ab37415) and mouse IgG (ab37355) Isotype controls were from Abcam (Cambridge, MA). Histostain Plus Bulk kit (85–8943, Invitrogen, Frederick, MD). In all instances, we used the antibody dilutions recommended by their respective manufacturers; they ranged between 1:200 and 1:2,000.
Study animals
Nine male Dutch-belted rabbits (DB), each 12-weeks weighing ~2 kg, were from Envigo (Somerset, NJ) and housed singly in a room with a strict 12-h on/off light cycle, temperature of 22 ± 2°C, and humidity of 45 ± 5%. During experiments, animals had free access to standard chow and water without nutritional enrichments that might affect tear production or stability. Rabbits were acclimated to this environment for ≥2 weeks prior to any study. All animal studies were in accord with the Statement for the Use of Animals of the Association for Research in Vision and Ophthalmology and approved by our relevant institutional committees.
Human sample collection
We evaluated conjunctival cells obtained by IC from nine normal male subjects, 45–65 years old. The study was approved by the Institutional Review Board (IRB) of the Stony Brook University Ethics Committee. Written consent was obtained from all human volunteers.
Conventional impression cytology
We employed the conventional IC methodology as follows: Sample acquisition: In humans, cells were obtained from the superior and inferior bulbar conjunctiva (1 o’clock and 7 o’clock position) after topical anesthesia with proparacaine hydrochloride (Sandos). In rabbits, cells were obtained from the superior bulbar conjunctiva after topical anesthesia with 1% lidocaine; sampling of the inferior conjunctiva was difficult. We removed the excess fluid using flat, round-tipped forceps. Then, an 11 mm x 8 mm MF Mixed Cellulose Ester membrane (MCE membrane type 0.22uM GSTF14250, Millipore Corp., Bedford, MA, USA) was applied to the bulbar conjunctiva with gentle pressure for 5 s, then an additional 20-µL drop of the proparacaine solution was added to the center of the membrane to soften it (for rabbits we used lidocaine). Once softened, the membrane was removed with a peeling motion. During conventional IC, at this stage, some practitioners transfer the cells onto a glass microscope slide by applying the membrane on the glass slide and then staining the cells. This approach often leads to unsatisfactory results. Consequently, others fix the cells on the membrane, which is then applied onto a glass microscope slide with double-sided adhesive tape. Following that cells are stained in situ, e.g., with PAS to identify goblet cells. In rabbits, we followed the latter approach.
Proposed method for impression cytology
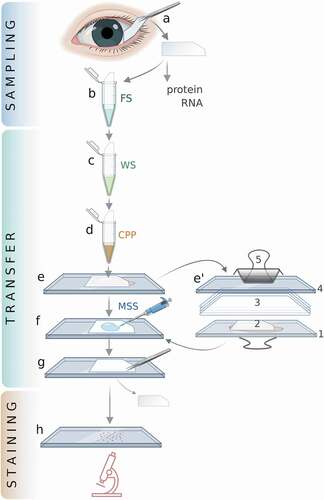
The proposed method is outlined in . Cell acquisition and staining are essentially the same as in the conventional method. The main difference between the two methods lies in the transfer of the cells to the glass microscope slide.
Figure 1. The improved IC method.
Working solutions
Fixing solution (FS)
2% (v/v) glutaraldehyde in PBS. If specific protocols require a lower concentration of glutaraldehyde, e.g., 1%, then adding paraformaldehyde to 4% (w/v) provides adequate fixing of the cells.
Washing solution (WS)
75% (v/v) ethanol in ddH2O.
CPP copolymer (Collagen/Polyethylenimine/Poly-L-lysine
Bovine collagen type I 2.5 mg/mL, branched polyethylenimine 0.05% (w/v), poly-L-lysine 0.05% (w/v), 33% (v/v) ethanol in ddH2O.
To avoid coalescing of its components, this mixture is prepared as follows: Solutions in ddH2O of bovine collagen type I 5 mg/mL, polyethylenimine 0.1% (w/v), and poly-L-lysine 0.1% (w/v), and absolute ethanol are added in a 2:1:1:2 ratio (v/v/v/v) in the sequence as they are presented here and mixed by vortexing.
Membrane softening solution (MSS)
20% 1-butanol in 75% ethanol.
Depending on the properties of a given membrane, the composition of this solution may be modified to achieve the required softening for its removal. For example, the concentration of butanol can be increased to as high as 30% or lowered by adding ddH2O to as low as 10%. Only through trial and error the optimal composition of this solution can be determined. Too high a concentration of butanol may degrade the membrane such that its complete removal is not feasible; the opposite happens if its concentration is too low for a given membrane. Depending on the membrane composition, additional softening agents (10%) can be added including 4-hydroxybutyl nitrate, 4-chloro-1-butanol or γ-undecalactone.
The time of exposure of the membrane to the Membrane Softening Solution can be critical. The higher the butanol concentration, the shorter the duration of its contact with the membrane ought to be to ensure its complete removal. Such exposure may vary between 2 and 30 seconds or even longer.
Immunofluorescence blocking solution
5% normal bovine serum, 0.3% Triton X-100, supplemented with 250 mM L-glycine and 250 mM L-arginine in PBS. This solution blocks the autofluorescence of free cell aldehyde groups.
Freshly prepared working solutions are recommended; however, they are stable for at least 3 months when stored at 4°C in a hermetically sealed container.
In case it is desirable to determine on which side of a membrane the cells have been attached (for example, if there is doubt when membranes are not properly marked), the cells can be readily visualized by dipping the membrane for a few seconds into a solution of 0.05% Ponceau S in PBS. Ponceau S reversibly stains cell proteins and can be washed away in PBS; such washing does not interfere with subsequent steps of this method.
Cell transfer onto glass microscope slides
Each step of our method is outlined in
Cell acquisition and fixing
Upon removal of the IC membrane from the eye, cells are fixed by immersing the membrane in Fixing Solution for 20 minutes. If needed, membranes can be kept in Storage Buffer for up to 2 weeks at 4 C.
Washing the membrane
The Fixing Solution is washed away by dipping the membrane in a tube with 5–10 mL Washing Solution with slow but constant agitation for about one minute.
Application of the copolymer
After washing, the CPP copolymer is applied to the membrane by immersing it in a tube with an adequate amount of CPP solution followed by gentle shaking for a 10 sec. For optimal results, drying of the membrane between this and the next step should be avoided by not delaying the transfer, and 1 drop (20 μL) of the same copolymer can be deposited directly in the middle of a fat-free glass slide. Following that, the membrane is placed on the glass with the cells facing the slide. The membrane is then covered with four layers of soft tissue paper (preferably Kimwipes) and another glass slide is placed on top of the paper. Firm pressure in the middle of the glass is then applied for about 90 sec (or longer if needed) with a binder clip. Fluid extruded during the application of pressure is removed with tissue paper.
After that, the covering glass slide and the tissue paper above the membrane are removed. The glass slide that has the membrane attached to it is dried in a heating block (56°C for 3 min) and cooled on ice for at least 3 min. These two steps allow the polymerization of the triblock components and the binding of cells, copolymer, and glass. If needed, the microscope slide with the membrane can be safely stored overnight at 4°C.
Membrane softening
To soften the membrane glued on the glass slide, a 40-µL drop of Membrane Softening Solution is applied onto the membrane, evenly distributed with a small plastic rod or a pipette tip, and immediately dried with absorbing tissue paper to remove the excess solution. If the membrane is not dried, it will be difficult to remove it intact.
Membrane removal
About 10 seconds after the application of the Membrane Softening Solution, the membrane is grasped at one of its corners with a pair of tweezers (preferably ~20 cm long) and peeled with a rolling movement towards its opposite side. If membrane removal is delayed, the membrane becomes too soft to be removed in toto, usually breaking into small pieces.
Following removal of the membrane, the cells on the glass slide are stained with conventional methods such as H&E or immunocytochemistry. If immunofluorescence staining is desired, the slide should be also washed with the immunofluorescence blocking solution.
Two additional points merit mention. First, in addition to glutaraldehyde, cells can be fixed in 4% paraformaldehyde, 55% ethanol, or 75% isopropanol (2-propanol) applied for 20 minutes. Methanol, ethanol (>60%), butanol, pentanol, and higher alcohols, as well as long chain hydrophobic aldehydes like propionaldehyde are not appropriate fixatives, as they adversely affect the membranes, which in their presence are either excessively softened or disintegrate.
Second, IC membranes can be stored for up to 2 weeks at 4oC in the following Storage Buffer: 0.2% aldehyde (glutaraldehyde or paraformaldehyde) with 2% isopropanol (2-propanol) or at least one of them in PBS. Before transferring cells to a glass slide, stored membranes should be processed from step b () by incubating for in Fixing Solution (4% glutaraldehyde in PBS) that is required for binding by CPP on a glass.
Immunocytochemistry and immunofluorescence staining
The application of these two methods to cells was as previously described.Citation14,Citation15 Briefly, cells transferred onto glass slides were washed with PBST (PBS + 0.05% Tween 20) three times and permeabilized with 0.1% Triton X-100 in PBS for 15 min. Cells are blocked with normal serum or 5% bovine serum albumin for 1 h, and then incubated with an appropriate dilution of primary antibodies at 4°C overnight. After three washes with PBST for 15 min, cells are incubated for 1 h with horseradish peroxidase-conjugated secondary antibody at room temperature. Cells are then developed with chromogen (DAB) for 5–10 min at room temperature after three washes are counterstained with hematoxylin. The slides are dehydrated and mounted with liquid mounting media. For immunofluorescence detection, cells are processed with the same procedure as above but incubated for 1 h with fluorophore-conjugated secondary antibodies (usually diluted 1:400) instead, and covered with mounting medium with 4ʹ-6-diamidino-2-phenylindole (DAPI). All stained slides are examined and photographed using a Nikon fluorescence microscope (Nikon Eclipse Ni, Tokyo, Japan) with a Wide Zoom Camera.
Results and discussion
Our novel method, outlined in , was applied successfully to the IC study of rabbit and human eyes. We consistently achieved essentially complete transfer of the ocular surface cells from membranes onto microscope slides.
(top row) illustrates the application of this method to the rabbit eye. As expected, the nitrocellulose membrane (NCM) was covered with ocular surface cells. With our transfer method, these cells were quantitatively deposited onto the glass slide. The extent of this transfer is demonstrated by the continuous carpet of cells on the glass slide visualized by PAS staining, and by the practically complete absence of cells on the NCM following the transfer step. The transfer of cells can be accomplished in 10–15 minutes.
Figure 2. Application of the IC method to rabbits.
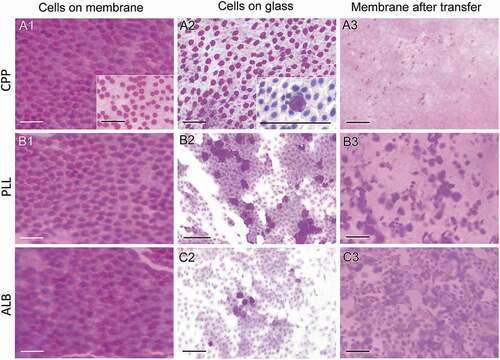
We compared our method to two conventional approaches, which use glass microscope slides covered with either poly-L-lysine or albumin paste. As can be seen in (middle and lower rows), representative cytological pictures indicate that the transfer of cells from the NCM to these slides is suboptimal. Consistent with this observation is that a large number of cells remain on the corresponding NCMs after the transfer to the glass slides, which contrast with the absence of cells on the NCM when using the new method.
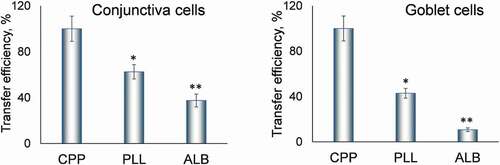
We quantified the efficiency of cell transfer by each of these three methods (). Compared to our method, the transfer of conjunctiva cells onto poly-L-lysine-covered slides was 37.5 ± 6.3% lower (mean ± SD for this and all subsequent values), and onto albumin-paste covered slides 62.5 ± 5.6% lower. The transfer of goblet cells was even less efficient. Compared to our method, it was 57.1 ± 4.3% lower onto poly-L-lysine-covered slides, and 89.3 ± 1.6% lower onto albumin-paste covered slides (p < .01 for both). Of note, no transfer of cells was possible onto fat-free microscope slides without coating them with agents such as poly-L-lysine.
Figure 3. Quantitative assessment of the transfer efficiency.
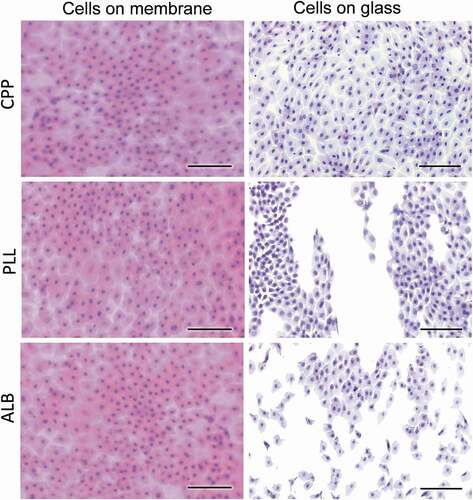
The method was applied to human subjects as well. As indicated in , the transfer of human cells was also complete. Similar to the rabbit cells, the transfer of conjunctiva cells to glass slides covered with poly-L-lysine or albumin paste was incomplete. This figure also shows an interesting variation of our approach. Cells can be stained on the membrane immediately after they are lifted off the ocular surface and then deposited onto the glass slide, requiring no further staining (Suppl. Figure 1). Viewing cells on the membrane hinders their morphological evaluation because the optical properties of the membrane differ from those of glass. All cells shown in this figure were stained with H&E.
Figure 4. Application of the IC method to humans.
In addition to PAS and H&E staining, cells transferred by our method can be evaluated using a variety of methods. We have successfully stained conjunctival cells with additional stains, including crystal violet, Coomassie blue, and Ponceau S (Suppl. Figure 2) and in a preliminary manner with Masson trichrome, Prussian blue, toluidine blue O and Sudan black B. In addition, we stained these using immunocytochemistry and immunofluorescence to identify cellular proteins. For example, ocular cells were stained by immunocytochemistry for the expression of the S100A8/9 protein, which plays a role in the pathophysiology of dry eye disease,Citation16,Citation17 a common ocular diseaseCitation18 (). An example of immunofluorescence staining of conjunctiva cells was the detection of the transcription factor ATF4, of importance to ocular diseases,Citation17,Citation18 shown in . More cellular proteins have been detected in transferred cells by immunofluorescence, such as S100A8/9, p65, NFAT5, and ITGAM (Suppl. –5). Furthermore, we also studied by confocal microscopy cells obtained with this method (data not shown). It is likely that the glutaraldehyde-fixed cells could be studied by electron-microscopy as well. Finally, fluorescent labeled small molecules that enter conjunctiva cells can be detected in cells obtained by IC and transferred to glass slides (Suppl. Figure 6).
Figure 5. Immunohistochemical detection of S100A8/9.
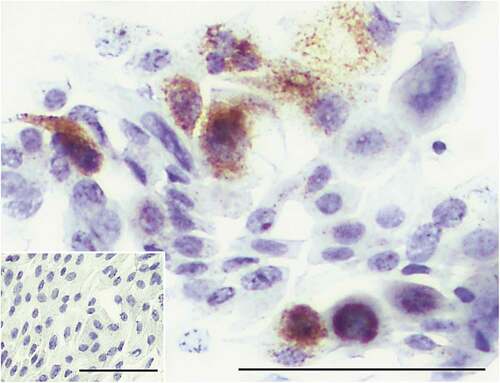
Figure 6. Immunofluorescence detection of ATF4.
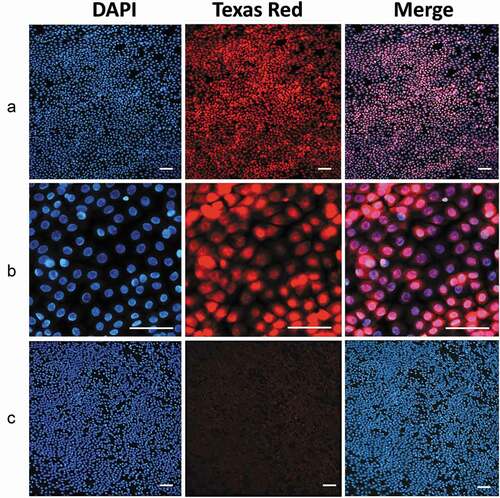
Of interest, goblet cells among those transferred with our method can be detected by using fluorescent stains such as fluorescein isothiocyanate or rhodamine isothiocyanate. Both of them fail to stain the mucins of the goblet cells and generate a “negative image” of the goblet cells (Suppl. Figure 7).
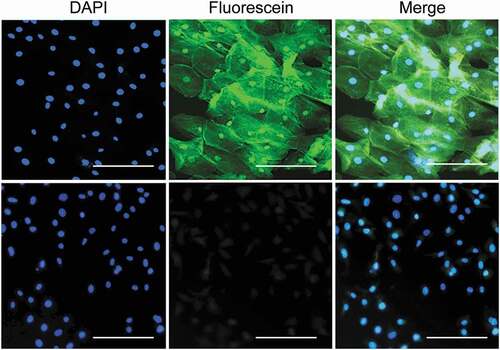
Our improved IC methodology can be applied to extra-ocular tissues. For example, we obtained cells from the oral cavity by IC using our transfer step and stained them with fluorescein isothiocyanate used as a conjugate with a novel salirasib derivative () or control conjugate of fluorescein isothiocyanate and ethanolamine.Citation19,Citation20 Staining methods mentioned above can be successfully applied to these cells, e.g., Suppl. Figure 2.
Figure 7. Application of the IC method to human oral cavity cells.
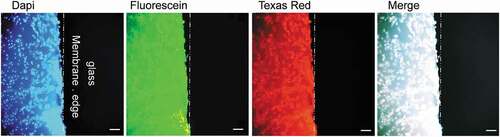
We compared the immunofluorescence staining of conjunctival cells directly on NCMs to that after their transfer onto class slides. As seen in , staining cells directly on NCMs was accompanied by a high background that made the evaluation of cellular features difficult or impossible. Such background was not encountered when cells were deposited on glass slides.
Figure 8. The high fluorescence background of membranes used in IC.
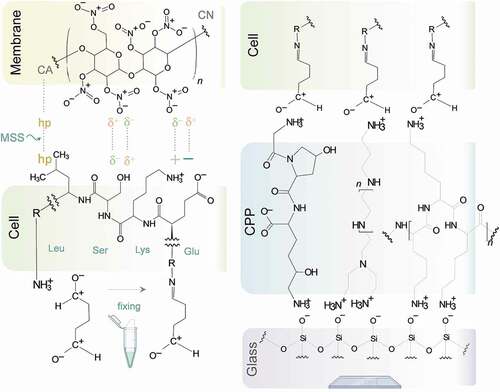
indicates the chemical interactions that likely underlie critical steps of the method. The attachment of cells to the membrane is facilitated by an array of hydrophobic interactions and electrostatic attractions between the cell surface and the nitrocellulose membrane, which have been reported in detail.Citation21,Citation22 Prominent among them are hydrophobic (hp) and van der Waals interactions of lipophilic molecules, e.g., extracellular lipoproteins. The electrostatic interactions include dipole–dipole (δ+δ−: δ+δ−) forces; attractions between the permanent dipoles of the nitrate groups (at neutral pH they are negatively charged) and the dissociated polar and polarizable groups of the cellular proteins; dipole-zwitterions; and dipole-ion attractions between the nitrate groups of the membrane and cations of the cell surface molecules, like arginine, lysine or histidine residues. Of note, the adsorption of proteins to membranes of different chemical composition has been the subject of significant research.Citation23,Citation24
Figure 9. Proposed chemical interactions underlying the improved IC method.
Glutaraldehyde fixes the cells by binding to -NH2 groups of amino acid residues of proteins exposed on cell surface.Citation25
The strong adhesion of the cells to the glass slide is achieved through electrostatic attractions (and perhaps other forces) between the one side of the triblock copolymer and the negatively charged surface and between the other side of the copolymer and the negatively charged surface of the glass slide (the copolymer is sandwiched between them).
The nature of the interactions between cells, membrane, copolymer, and glass perhaps explains how our Membrane Softening Solution can help remove the membrane off the attached cells while not disrupting their attachment to the glass through the copolymer. We hypothesize that the electronegative, polar Softening Solution lowers the hydrophobic attractions and neutralizes bond dipole moments of cellulose nitrate, thus facilitating the disruption of dipole–dipole, dipole-zwitterion and dipole-ion attractions. The result is the dissociation of the membrane from the cells. But since the Membrane Dissociation Solution has no such effect on the interactions of the copolymer with the cells and the glass microscope slide, cells remain on the slide.
Our findings convincingly support the notion that our novel triblock copolymer greatly improves the critical step of cell transfer from the membrane to the glass slide where they can be evaluated. Indeed, with our method such transfer is essentially complete. This improved transfer step will likely minimize the number of membrane applications onto the ocular surface to obtain an adequate sample, making IC faster and less burdensome to both patient and examiner.
The improved method is simple to perform and robust, only requiring the quick optimization of the Membrane Softening Solution for each type of membrane and of the duration of its application. The wide applicability of this method is demonstrated by the examples shown here that represent ocular and extraocular cells stained with conventional as well as immunocytochemical and immunofluorescence staining.
In conclusion, we present a novel improvement of IC that makes it robust, simpler and with increased diagnostic yield compared to its current variations.
Acknowledgments
Basil Rigas is the guarantor of this work and, as such, had access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. We thank Dr. Ernest Natke for his assistance with the manuscript. This work was supported by the National Institutes of Health [Grant R44EY031193].
Disclosure statement
The authors declare no competing interests except for BR who has an equity position in Medicon Pharmaceuticals, Inc. and Apis Therapeutics, LLC; and LH, who has served as an employee of Medicon Pharmaceuticals, Inc. and is currently an employee with an equity position in Apis Therapeutics, LLC.
Additional information
Funding
References
- Egbert PR, Lauber S, Maurice DM. A simple conjunctival biopsy. Am J Ophthalmol. 1977;84:798–801. doi:https://doi.org/10.1016/0002-9394(77)90499-8.
- Hagan S. Biomarkers of ocular surface disease using impression cytology. Biomark Med. 2017;11:1135–47. doi:https://doi.org/10.2217/bmm-2017-0124.
- Singh R, Joseph A, Umapathy T, Tint NL, Dua HS. Impression cytology of the ocular surface. Br J Ophthalmol. 2005;89:1655–59. doi:https://doi.org/10.1136/bjo.2005.073916.
- Lopin E, Deveney T, Asbell PA. Impression cytology: recent advances and applications in dry eye disease. Ocul Surf. 2009;7:93–110. doi:https://doi.org/10.1016/S1542-0124(12)70301-4.
- Thia -Z-Z, Tong L. Update on the role of impression cytology in ocular surface disease. Taiwan J Ophthalmol. 2019;9:141–49. doi:https://doi.org/10.4103/tjo.tjo_57_19.
- Hyun C, Filippich L, Hughes I. The inhibitory effect of pentobarbitone on reverse transcription-PCR. J Biochem Biophys Methods. 2005;62:63–68. doi:https://doi.org/10.1016/j.jbbm.2004.09.005.
- Roy P, Cimbolini N, Feraille L, Elena -P-P. Evaluation of membranes material for impression cytology. Invest Ophthalmol Vis Sci ARVO. 2011;52:1937–1937.
- Nelson JD, Havener VR, Cameron JD. Cellulose acetate impressions of the ocular surface. Dry eye states. Arch Ophthalmol. 1983;101:1869–72.
- López-Miguel A, Gutiérrez-Gutiérrez S, García-Vázquez C, Enríquez-de-salamanca A. RNA collection from human conjunctival epithelial cells obtained with a new device for impression cytology. Cornea. 2017;36:59–63. doi:https://doi.org/10.1097/ICO.0000000000000977.
- Calonge M, Diebold Y, Sáez V, Enríquez de Salamanca A, García-Vázquez C, Corrales RM, Herreras JM. Impression cytology of the ocular surface: a review. Exp Eye Res. 2004;78:457–72. doi:https://doi.org/10.1016/j.exer.2003.09.009.
- Krenzer KL, Freddo TF. Cytokeratin expression in normal human bulbar conjunctiva obtained by impression cytology. Invest Ophthalmol Vis Sci. 1997;38:142–52.
- Luzeau R, Carlier C, Ellrodt A, Amédée-Manesme O. Impression cytology with transfer: an easy method for detection of vitamin A deficiency. Int J Vitam Nutr Res. 1988;58:166–70.
- Haller-Schober E-M, Schwantzer G, Berghold A, Fischl M, Theisl A, Horwath-Winter J. Evaluating an impression cytology grading system (IC score) in patients with dry eye syndrome. Eye. 2006;20:927–33. doi:https://doi.org/10.1038/sj.eye.6702058.
- Mattheolabakis G, Papayannis I, Yang J, Vaeth BM, Wang R, Bandovic J, Ouyang N, Rigas B, Mackenzie GG. Phospho-aspirin (MDC-22) prevents pancreatic carcinogenesis in mice. Cancer Prev Res (Phila). 2016;9:624–34. doi:https://doi.org/10.1158/1940-6207.CAPR-15-0344.
- Mattheolabakis G, Wang R, Rigas B, Mackenzie GG. Phospho-valproic acid inhibits pancreatic cancer growth in mice: enhanced efficacy by its formulation in poly-(L)-lactic acid-poly(ethylene glycol) nanoparticles. Int J Oncol. 2017;51:1035–44. doi:https://doi.org/10.3892/ijo.2017.4103.
- Tong L, Lan W, Lim RR, Chaurasia SS. S100A proteins as molecular targets in the ocular surface inflammatory diseases. Ocul Surf. 2014;12:23–31. doi:https://doi.org/10.1016/j.jtos.2013.10.001.
- Wang S, Song R, Wang Z, Jing Z, Wang S, Ma J. S100A8/A9 in inflammation. Front Immunol. 2018;9:1298. doi:https://doi.org/10.3389/fimmu.2018.01298.
- Clayton JA. Dry eye. N Engl J Med. 2018;378:2212–23. doi:https://doi.org/10.1056/NEJMra1407936.
- Swoboda G, Hasselbach W. Reaction of fluorescein isothiocyanate with thiol and amino groups of sarcoplasmic atpase. Z Naturforsch C Biosci. 1985;40:863–75. doi:https://doi.org/10.1515/znc-1985-11-1220.
- Heilkenbrinker A, Reinemann C, Stoltenburg R, Walter JG, Jochums A, Stahl F, Zimmermann S, Strehlitz B, Scheper T. Identification of the target binding site of ethanolamine-binding aptamers and its exploitation for ethanolamine detection. Anal Chem. 2015;87:677–85. doi:https://doi.org/10.1021/ac5034819.
- Low SC, Shaimi R, Thandaithabany Y, Lim JK, Ahmad AL, Ismail A. Electrophoretic interactions between nitrocellulose membranes and proteins: biointerface analysis and protein adhesion properties. Colloids Surf B Biointerfaces. 2013;110:248–53. doi:https://doi.org/10.1016/j.colsurfb.2013.05.001.
- Přistoupil TI, Kramlová M, Štěrbíková J. On the mechanism of adsorption of proteins to nitrocellulose in membrane chromatography. J Chromatogr. 1969;42:367–75. doi:https://doi.org/10.1016/S0021-9673(01)80636-1.
- Holstein CA, Chevalier A, Bennett S, Anderson CE, Keniston K, Olsen C, Li B, Bales B, Moore DR, Fu E, et al. Immobilizing affinity proteins to nitrocellulose: a toolbox for paper-based assay developers. Anal Bioanal Chem. 2016;408:1335–46. doi:https://doi.org/10.1007/s00216-015-9052-0.
- Kurien BT, Scofield RH. Other notable methods of membrane protein detection: a brief review. Methods Mol Biol. 2015;1314:357–70.
- Hopwood D. Theoretical and practical aspects of glutaraldehyde fixation. Histochem J. 1972;4:267–303. doi:https://doi.org/10.1007/BF01005005.