
Abstract
Purpose
The purpose of the study is to explore the mRNA and protein expression of FUNDC1 in cataract cells and tissues, and clarify the function and mechanism of FUNDC1 in cataract cells under oxidative stress.
Methods
We used bioinformatic analysis to screen differentially expressed genes in cataract cells from GSE153933. The expression of FUNDC1 in cataract specimens and cells was measured by reverse transcription quantitative polymerase chain reaction and western blotting. MethPrimer was used to predict CpG island of FUNDC1 promoter. The methylation of FUNDC1 in cataract specimens and cells was determined by methylation-specific polymerase chain reaction assay. Flow cytometry assay was used to measure cell apoptosis in FUNDC1-knockdown and -overexpression SRA01/04 cells. The expression of LC3 was analyzed by immunofluorescence assay. The expression of apoptosis-related proteins, autophagy, and PI3K/Akt/mTOR-related proteins was determined by western blotting.
Results
The results of bioinformatic analysis revealed that FUNDC1 was upregulated in cataract. FUNDC1 was high expression in SRA01/04 cells with H2O2 treatment, whereas hypomethylation of FUNDC1 in cataract lens cells under oxidative stress. The knockdown of FUNDC1 decreased cell apoptosis and autophagy in comparison with the negative control of SRA01/04 cells. While the overexpression of FUNDC1 elevated cell apoptosis and autophagy compared to the empty vector group in SRA01/04 cells. Mechanically, FUNDC1 reduced the phosphorylation of PI3K/Akt/mTOR pathway under oxidative stress in SRA01/04 cells.
Conclusion
Our study suggested that FUNDC1 deficiency restrains cell apoptosis and autophagy by inhibiting PI3K/Akt/mTOR signal pathway.
Introduction
Cataract is a priori reason for blindness and aging is the main cause for cataracts.Citation1 Ultraviolet raysCitation2 and oxidative stressCitation3 have been identified as pivotal factors in leading to cataract formation. Previous study demonstrated that there is a meaningful relationship between aging elicited related to cataract and oxidative stress.Citation4 Endogenous reactive oxygen species (ROS) damage exceeds the antioxidant capacity of cells, thus affecting signal transduction system, DNA damage, lipids, and other macromolecules.Citation5 Oxidative stress plays important roles in regulating normal physiological functions, including cell cycle,Citation6 cell migration,Citation7 and cell death.Citation8
Previous studies indicated that oxidative stress facilitated cell apoptosisCitation9 and autophagyCitation10 of lens epithelial cells (LECs). There are many studies uncovering the potential mechanisms of autophagy and apoptosis as new targets for treatment of age-related cataracts.Citation11 Ma et al.Citation12 showed that HO-1 protects human LECs from hydrogen peroxide (H2O2) damage by regulating antioxidant enzyme activity and reducing the production of ROS, thereby inhibiting caspase family-dependent cell apoptosis and inducing oxidative stress. Morishita et al.Citation13 demonstrated that the loss of Atg5 and Pik3c3 genes in the lens cells lead to the degeneration of cataracts without the degradation of programmed organelles dependence.Citation13 However, the molecular mechanism of age-related cataract formation remains unclear.
In this study, we demonstrated that the expression of FUNDC1 in an animal model of cataract was significantly increased as revealed by bioinformatic analysis. Furthermore, many studies have shown that FUNDC1 expression is low under hypoxic conditions, which is closely related to autophagy. Under oxidative stress, it can be phosphorylated for better binding to light chain 3 (LC3),Citation14,Citation15 thereby promoting the development of the prolonged phase of mitochondrial autophagy. In addition, numberous researches has shown that lens mitochondria become disordered and deteriorate with age, and ROS levels also increase with age ROS is closely related to autophagy and apoptosis.Citation16 We speculated that the aging of mitochondria is malfunctional, the microenvironment homeostasis of lens is broken with the increase in age,Citation17 oxidative stress induces autophagy and apoptosis of lens cells by mediating the expression of FUNDC1, thus resulting in the occurrence of cataracts.
Materials and methods
Cells, tissues, and reagents
SRA01/04(HLE) were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and cultured in Dulbecco’s modified Eagle’s medium containing 10% FBS and 100 U/ml penicillin and streptomycin, which were maintained at 37 °C in 5% CO2 incubator. Si-FUNDC1 and H2O2 were provided by Sigma (Darmstadt, Germany). Cataract and noncataract tissue specimens were obtained from The Second Affiliated Hospital of Qiqihar Medical College (). Ethical approval was in accordance with the standards upheld by the Ethics Committee of Qiqihar Medical College and with those of the 1964 Helsinki Declaration and its later amendments for ethical research involving human subjects. Written informed consent was obtained from a legally authorized representative(s) for anonymized patient information to be published in this article. The experiments were manipulated according to the previously reported work.Citation18
Table 1. Demographic and clinical characteristics of cataract patients and normal controls.
Bioinformatic analysis, prediction of FUNDC1 promoter CpG Island, and MSP
The expression of FUNDC1 in cataract and noncataract specimens was analyzed by bioinformatics from GSE153933Citation19 as described previously. The prediction of FUNDC1 promoter CpG island was derived from MethPrimer online (http://www.urogene.org/cgi-bin/methprimer2/MethPrimer.cgi). For methylation-specific polymerase chain reaction (MSP), the DNA was denatured to generate single-stranded DNA, subsequently modified with sodium bisulfite, and then PCR amplification was performed with two pairs of primers, one of which is for methylated DNA and the other is for unmethylated DNA.
RNA extraction and real-time quantitative PCR
Total RNA was isolated using TRIzol reagent (Ambion, CA, USA). About 1 µg of RNA was reverse-transcribed with the ImProm-IITM Reverse Transcription System (Promega, WI, USA). Real-time quantitative PCR (RT-qPCR) was performed using SYBR GREEN qPCR Super Mix (Invitrogen, CA, USA). Primers are listed as below.
FUNDC1 forward: 5′-CCTCCCCAAGACTATGAAAGTGA-3′;
FUNDC1 reverse: 5′-AAACACTCGATTCCACCACTG-3′;
GAPDH forward: 5′-AGACAGCCGCATCTTCTTGT-3′;
GAPDH reverse: 5′-CTTGCCGTGGGTAGAGTCAT-3′.
Cell apoptosis analysis
Cell apoptosis was conducted as described previously.Citation20 SRA01/04 cells with indicated plasmids treatment were seeded, cultured, and treated with H2O2 for 24 h. Next, cells were collected, fixed, and incubated. Later, they were stained with Annexin V-FITC/PI apoptosis detection kit to perform apoptosis assay, and analyzed by a flow cytometry (Becton Dickinson, CA).
Immunofluorescence assay
The immunofluorescence assay was performed according to the published work.Citation20 SRA01/04 cells with indicated treatment were treated with H2O2 for 24 h. Next, cells were washed, fixed, permeabilized, blocked, and incubated with anti-FUNDC1 antibody and Alexa Fluor goat anti-mouse IgG(H + L) antibody and images were visualized under confocal microscopy. Staining results were measured by light microscopy.
Western blotting
Western blotting was performed as described as previously depicted.Citation21 Cells were washed with phosphate-buffered saline and the proteins were isolated with radioimmunoprecipitation buffer with PMSF. The total proteins were separated with sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred with polyvinylidene fluoride, and incubated with the corresponding primary antibodies ().
Table 2. Antibody.
Statistical analysis
The comparison between different groups was performed using one-way analysis of variance. Data from at least three independent experiments were presented as mean ± SEM. A significant difference was presented as p < 0.05.
Results
Oxidative stress promotes the expression of FUNDC1
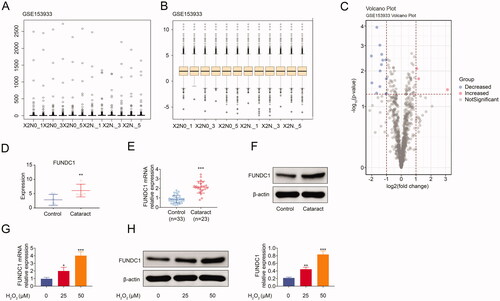
Previous studies demonstrated that ultraviolet raysCitation2 and oxidative stressCitation3 acted as key factors in cataract formation. However, the underlying regulatory mechanism was poorly understood. It was important to screen out the key genes determining cataract progression. Firstly, we screened different expression genes, including decreased (blue) and increased (red), in cataract and the control group (). Moreover, FUNDC1 expression was upregulated in cataract compared to the control group from GSE153933 (). In order to further confirm the expression of FUNDC1 in cataract patient specimens, we collected 23 cases of cataract specimens and 33 cases of noncataract specimens. The results showed that both the mRNA and protein expression of FUNDC1 were upregulated in cataract specimens. Besides, we also measured the expression of FUNDC1 at cell level. As shown in and (), RT-qPCR () and western blotting () showed that FUNDC1 was upregulated in SRA01/04 cells treated with H2O2 for 24 h. Collectively, our data demonstrated that H2O2 treatment facilitated the expression of FUNDC1 in SRA01/04 cells.
Figure 1. FUNDC1 was high expression in cataract specimens. (A–C) Screening of DEGs associated with contract from GSE153933. (D) The mRNA expression of FUNDC1 in cataract was analyzed by bioinformatics from GSE153633. (E and F) The expression of FUNDC1 in cataract was measured by RT-qPCR (E) and western blotting (F). (G and H) SRA01/04(HLE) cells were treated with or without H2O2 treatment for 24 h, and then the expression of FUNDC1 was determined by RT-qPCR (G) and western blotting (H). Error bars represent data from three independent experiments (mean ± SD). *p<.05, **p<.01, ***p<.001.
Hypomethylation of FUNDC1 in SRA01/04 cells under oxidative stress
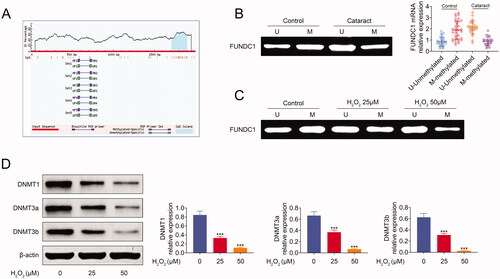
Methylation as a modification of DNA exerted key role in the regulation of gene expression. We speculated that the expression of FUNDC1 may be regulated by its methylation. In order to confirm this hypothesis, we used the MethPrimer online software to predict the CpG island of FUNDC1 promoter. There was a CpG island (blue) as shown in . Next, we used MSP assay to measure the methylation of FUNDC1 in cataract specimens. As shown in , FUNDC1 was low methylated in cataract specimens. We also used the same method to measure methylation in SRA01/04 cells. FUNDC1 was also hypomethylated in SRA01/04 cells after treatment with H2O2 for 24 h (). Previous studies demonstrated that DNA methyltansferase was involved in the regulation of gene methylation. Here, we analyzed the expression of three important methyltransferases at cell level, where all of the three methyltransferases (DNMT1, DNMT3a, and DNMT3b) were downregulated expressed in SRA01/04 cells treated with H2O2 for 24 h (). In summary, our data demonstrated that H2O2 treatment suppressed the methylation of FUNDC1.
Figure 2. Hypomethylation of FUNDC1 in SRA01/04 cells under oxidative stress. (A) FUNDC1 promotor CpG island was predicted by MethPrimer online software. (B) MSP assay analyzed the methylation expression of FUNDC1 between cataract groups and control group. (C) SRA01/04(HLE) cells were treated with or without H2O2 treatment for 24 h, and then the methylation expression of FUNDC1 was determined by MSP. (D) The expression of DNA methyltransferases was measured by western blotting. Error bars represent data from three independent experiments (mean ± SD). ***p<.001.
Fundc1 facilitated SRA01/04 cell apoptosis and autophagy under oxidative stress
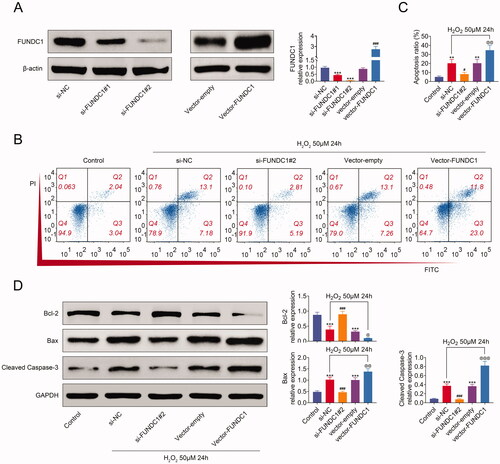
In order to clarify the function of FUNDC1 in SRA01/04, we first constructed FUNDC1-knockdown cell lines by transfection with si-FUNDC1, and FUNDC1 overexpression cell lines by transfection with vector-FUNDC1 (). Then, cell apoptosis was analyzed by flow cytometry. As shown in and (), H2O2 treatment increased cell apoptosis of SRA01/04 cell lines. The knockdown of FUNDC1 antagonized increased cell apoptosis in SRA01/04 cells treated with H2O2, while the overexpression of FUNDC1 further aggravated cell apoptosis. Additionally, H2O2 treatment reduced Bcl-2 expression, whereas it increased Bax and c-caspase3 expression. However, FUNDC1-knockdown promoted the expression of Bcl-2, and inhibited Bax and c-caspase3 expression in SRA01/04 cells. However, FUNDC1-overexpression got an opposite effect from the knockdown of FUNDC1 (). Collectively, our data demonstrated that FUNDC1 facilitated cell apoptosis in SRA01/04 cells treated with H2O2.
Figure 3. FUNDC1 promoted SRA01/04 cell apoptosis under oxidative stress. (A) SRA01/04(HLE) cells were transfected with the indicated plasmids, treating with 50 uM H2O2 for 24 h, and then the expression of FUNDC1 was measured by western blotting. (B and C) SRA01/04(HLE) cells were treated as in (A), and cell apoptosis was determined by flow cytometry (B) and quantified (C). (D) Collecting the same cells as in (B), western blotting analyzed the expression of apoptosis-related proteins. Error bars represent data from three independent experiments (mean ± SD). *Compared to a control group, #compared with the si-NC group, @compared to the vector empty group. *p<.01, ***p<.001, #p<.05, ###p<.001, @p<.05, @@p<.01, @@@p<.001.
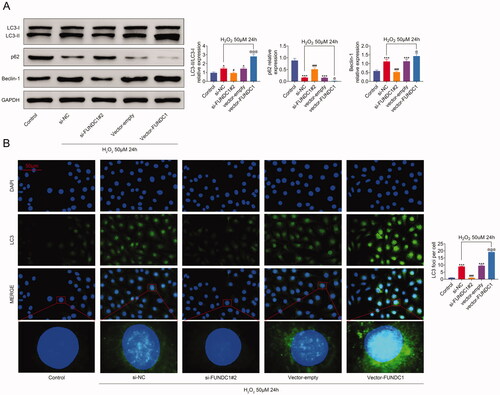
Previous studies indicated that oxidative stress not only induced apoptosisCitation9 but also autophagyCitation10 in LECs. Similarly, the expression of autophagy-related proteins was also measured in SRA01/04 cell lines. As shown in H2O2 treatment reduced p62 expression, whereas it induced the expression of LC3-II and Beclin-1. However, FUNDC1-knockdown upregulated the expression of p62 and inhibited LC3-II and Beclin-1 expression in SRA01/04 cells. In contrast, FUNDC1-overexpressed SRA01/04 cells showed an inverse expression pattern of p62, LC3-II, and Beclin-1. Moreover, similar result of LC3 expression in SRA01/04 cells treated with H2O2 was acquired by immunofluorescence staining (). Taken together, our data demonstrated that FUNDC1 increased SRA01/04 cells autophagy upon H2O2 treatment.
Figure 4. FUNDC1 increased autophagy in SRA01/04 cells under oxidative stress. (A) SRA01/04(HLE) cells were transfected with the indicated plasmids, treating with 50 uM H2O2 for 24 h, and then the expression of autophagy-related proteins was measured by western blotting. Error bars represent data from three independent experiments (mean ± SD). *p<.01, ***p<.001, #p<.05, ###p<.001, @p<.05. (B) Immunofluorescence assay analyzed the LC3 expression in different treatment SRA01/04(HLE) cell lines.
Fundc1 inhibited PI3K/Akt/mTOR pathway activity under oxidative stress
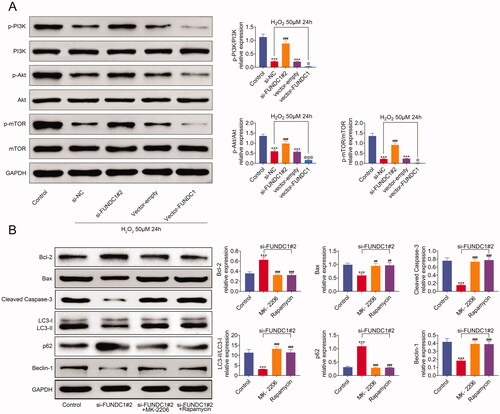
Earlier studies demonstrated that H2O2) induced the phosphorylation of PI3K and Akt to increase cell proliferation. Similarly, western blotting was used to analyze the phosphorylation of PI3K/Akt/mTOR signal pathway. As shown in , H2O2 treatment suppressed the phosphorylation of PI3K, Akt, and mTOR in SRA01/04 cell lines. The knockdown of FUNDC1 antagonized the inhibition of p-PI3K, p-Akt, and p-mTOR in SRA01/04 cells with H2O2 treatment, while the overexpression of FUNDC1 further aggravated the reduction of p-PI3K, p-Akt, and p-mTOR in H2O2 treatment cell lines. In addition, FUNDC1 deficiency induced the expression of Bcl-2 and p62, and increased the expression of Bax, c-caspase3, LC3II, and Beclin1. However Bax, c-caspase3, LC3II, and Beclin1 expression were inhibited by MK‐2206 or Rapamycin treatment, while Bcl-2 and p62 were increasing expression in si-FUNDC1 cells treated with MK‐2206 or Rapamycin (). Collectively, our data demonstrated that FUNDC1 inhibits the PI3K/Akt/mTOR pathway activity in SRA01/04 cells with H2O2 treatment.
Figure 5. FUNDC1 inhibited PI3K/Akt/mTOR pathway activity under oxidative stress. (A and B) Western blotting assay analyzed the expression of PI3K/Akt/mTOR signaling pathway, apoptosis, and autophagy-related proteins. Results presented represent the means of triplicate experiments ± SEM. ***p<.001, ###p<.001, @p<.05.
Discussion
Cataract is one of the leading causes of visual impairment affecting millions of people worldwide, especially in diabetes mellitus patients.Citation22 Cataracts are divided into pediatric cataracts, senile cataracts, and cataracts secondary to other causes, including radiation (X-rays, ultraviolet rays), aging, diabetes, drugs, and genetic factors.Citation1 Furthermore, free radical-induced oxidative stress is the main cause of cataracts recently. In this study, we demonstrated that FUNDC1 expression was upregulated in cataract tissues compared to the control. Cataract cells treated with H2O2 further increased the expression of FUNDC1 through negatively regulating methylation of FUNDC1. Functionally, FUNDC1 positively mediated cell apoptosis by increasing Bax and c-caspase3 expression and autophagy via augmenting the ratio of LC3II/LC3I. Mechanically, FUNDC1 negatively regulated the activation of PI3K/Akt/mTOR pathway in cataract cells.
Living cells, including cataract cells, are always affected by dangerous exogenous or endogenous reactive oxidative molecules. It is well known that oxidative stress acts as one of the most important risk factors in cataractogenesis. The loss of the balance between ROS and endogenous antioxidants contributes to the formation of cataracts. As a source of ROS, H2O2 also played an important role in cataract development. Previous study reported that BDS-II, a Kv3 channel blocker, plays vital roles in oxidative stress-related cataract, which is beneficial for protecting LECs from H2O2-induced cell death.Citation23 H2O2 is a molecule produced immediately after starvation, which is essential for the execution of autophagy. Cataract patients have early oxidative stress, and the level of atrial water H2O2 is significantly increased in cataract patients, which is 30 times that of normal people.Citation24 In vitro studies have revealed that in the lens of cataract patients, the same concentration of H2O2 can cause cell apoptosis and opacity and induce similar pathological manifestations in the eyes, but the mechanism of elevated H2O2 eliciting the pathological changes of the lens is still unclear.
Previous study demonstrated that oxidative stress responses were the primary pathological reason for the development of cataract. Sustained high intensity of oxidative stress response can further activate apoptosis-related pathways, leading to the abnormal expression of downstream effectors such as Bax and Bcl-2. Here, bioinformatics analysis was used to screen the differentially expressed genes (DEGs) of cataract cells, and the results showed that FUNDC1 increased DEGs. As one of the mammalian autophagy receptors, FUNDC1 interacts with mitochondria and recruits LC3 to mitochondria to participate in autophagy.Citation15 FUNDC1 plays an important role in inducing autophagy and apoptosis of hypoxic cells. FUNDC1 deficiency reduces CSE‐stimulated mitochondrial autophagy and apoptosis through suppressing DRP1, thus inhibiting the development of chronic obstructive pulmonary disease.Citation25 Additionally, FUNDC1 also decreased apoptosis and increased mitochondrial function to protect neurons against cerebral ischemia-reperfusion-induced injury.Citation26
As a vital cellular signal transduction pathway, PI3K/Akt/mTOR axis acts as an important regulator in diverse progresses, including cell apoptosis, migration, proliferation, necrosis, and oxidative stress responses.Citation27 PI3K/Akt signaling pathway is a canonical anti-oxidative stress and anti-apoptosis pathway.Citation28 A recent study demonstrated that miR-182 activated the PI3K/Akt signaling pathway by promoting the phosphorylation of Akt to restrain oxidative stress and apoptosis of epithelial cell in the lens of cataract rats.Citation29 Similarly, the phosphorylation of PI3K/Akt/mTOR signaling pathway was also analyzed, which revealed that FUNDC1 inhibited PI3K/Akt/mTOR signaling activity by reducing their phosphorylation under oxidative stress, which indicated that FUNDC1 may induce cell apoptosis and autophagy in cataract cells by inactivating PI3K/Akt/mTOR pathway under oxidative stress.
In conclusion, we demonstrated that FUNDC1 is upregulated in cataract cells and tissues. FUNDC1 is further increased in cataract cells treated with H2O2, which is associated with methylation of FUNDC1. Additionally, FUNDC1 positively regulates cell apoptosis and autophagy by inactivating PI3K/Akt/mTOR signaling. However, the molecular regulatory mechanism of FUNDC1 in regulating oxidative stress of cataract cells needs to be further clarified. Currently, our data indicated that FUNDC1 may act as a target for treating oxidative stress-induced cataract in the future.
Ethics approval
Ethical approval was in accordance with the standards upheld by the Ethics Committee of Qiqihar Medical College and with those of the 1964 Helsinki Declaration and its later amendments for ethical research involving human subjects.
Informed consent
Written informed consent was obtained from a legally authorized representative(s) for anonymized patient information to be published in this article.
Author contributions
Duo Dong and Jing Wu designed the study and supervised the data collection; Lijie Sheng, Xuewu Gong, and Zhichang Zhang analyzed the data, interpreted the data; Caihan Yu prepare the manuscript for publication and reviewed the draft of the manuscript. All authors have read and approved the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
References
- Liu YC, Wilkins M, Kim T, Malyugin B, Mehta JS. Cataracts. Lancet. 2017;390(10094):600–612. doi:https://doi.org/10.1016/S0140-6736(17)30544-5.
- Li G, Song H, Chen L, Yang W, Nan K, Lu P. Tug1 promotes lens epithelial cell apoptosis by regulating mir-421/caspase-3 axis in age-related cataract. Exp Cell Res. 2017;356(1):20–27. doi:https://doi.org/10.1016/j.yexcr.2017.04.002.
- Pinazo-Duran MD, Gallego-Pinazo R, Garcia-Medina JJ, Zanon-Moreno V, Nucci C, Dolz-Marco R, Martinez-Castillo S, Galbis-Estrada C, Marco-Ramirez C, Lopez-Galvez MI, et al. Oxidative stress and its downstream signaling in aging eyes. Clin Interv Aging. 2014;9:637–652. doi:https://doi.org/10.2147/CIA.S52662.
- Babizhayev MA, Yegorov YE. Reactive oxygen species and the aging eye: specific role of metabolically active mitochondria in maintaining lens function and in the initiation of the oxidation-induced maturity onset cataract—a novel platform of mitochondria-targeted antioxidants with broad therapeutic potential for redox regulation and detoxification of oxidants in eye diseases . Am J Ther. 2016;23(1):e98–e117. doi:https://doi.org/10.1097/MJT.0b013e3181ea31ff.
- Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol. 2003;15(2):247–254. doi:https://doi.org/10.1016/s0955-0674(03)00002-4.
- Liang WZ, Jan CR, Hsu SS. Cytotoxic effects of gastrodin extracted from the rhizome of Gastrodia elata Blume in glioblastoma cells, but not in normal astrocytes, via the induction of oxidative stress-associated apoptosis that involved cell cycle arrest and p53 activation. Food Chem Toxicol. 2017;107(Pt A):280–292. doi:https://doi.org/10.1016/j.fct.2017.07.013.
- Wang WT, Ye H, Wei PP, Han BW, He B, Chen ZH, Chen YQ. LncRNAs H19 and HULC, activated by oxidative stress, promote cell migration and invasion in cholangiocarcinoma through a ceRNA manner. J Hematol Oncol. 2016;9(1):117. doi:https://doi.org/10.1186/s13045-016-0348-0.
- Zhang W, Song J, Zhang Y, Ma Y, Yang J, He G, Chen S. Intermittent high glucose-induced oxidative stress modulates retinal pigmented epithelial cell autophagy and promotes cell survival via increased HMGB1. BMC Ophthalmol. 2018;18(1):192. doi:https://doi.org/10.1186/s12886-018-0864-5.
- Yao H, Tang X, Shao X, Feng L, Wu N, Yao K. Parthenolide protects human lens epithelial cells from oxidative stress-induced apoptosis via inhibition of activation of caspase-3 and caspase-9. Cell Res. 2007;17(6):565–571. doi:https://doi.org/10.1038/cr.2007.6.
- Zhou J, Yao K, Zhang Y, Chen G, Lai K, Yin H, Yu Y. Thioredoxin binding protein-2 regulates autophagy of human lens epithelial cells under oxidative stress via inhibition of Akt phosphorylation. Oxid Med Cell Longevity. 2016;2016:1–17. doi:https://doi.org/10.1155/2016/4856431.
- Zhang F, Meng W, Tong B. Down-regulation of microrna-133b suppresses apoptosis of lens epithelial cell by up-regulating bcl2l2 in age-related cataracts. Med Sci Monit. 2016;22:4139–4145. doi:https://doi.org/10.12659/msm.896975.
- Ma T, Chen T, Li P, Ye Z, Zhai W, Jia L, Chen W, Sun A, Huang Y, Wei S, et al. Heme oxygenase-1 (HO-1) protects human lens epithelial cells (SRA01/04) against hydrogen peroxide (H2O2)-induced oxidative stress and apoptosis. Exp Eye Res. 2016;146:318–329. doi:https://doi.org/10.1016/j.exer.2016.02.013.
- Morishita H, Eguchi S, Kimura H, Sasaki J, Sakamaki Y, Robinson ML, Sasaki T, Mizushima N. Deletion of autophagy-related 5 (atg5) and pik3c3 genes in the lens causes cataract independent of programmed organelle degradation. J Biol Chem. 2013;288(16):11436–11447. doi:https://doi.org/10.1074/jbc.M112.437103.
- Liu L, Sakakibara K, Chen Q, Okamoto K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 2014;24(7):787–795. doi:https://doi.org/10.1038/cr.2014.75.
- Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, et al. . Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14(2):177–185. doi:https://doi.org/10.1038/ncb2422.
- Li L, Tan J, Miao Y, Lei P, Zhang Q. ROS and autophagy: interactions and molecular regulatory mechanisms. Cell Mol Neurobiol. 2015;35(5):615–621. doi:https://doi.org/10.1007/s10571-015-0166-x.
- Brennan LA, Kantorow M. Mitochondrial function and redox control in the aging eye: role of MSRA and other repair systems in cataract and macular degenerations. Exp Eye Res. 2009;88(2):195–203. doi:https://doi.org/10.1016/j.exer.2008.05.018.
- Yu L, Dong L, Wang Y, Liu L, Long H, Li H, Li J, Yang X, Liu Z, Duan G, et al. Reversible regulation of SATB1 ubiquitination by USP47 and SMURF2 mediates colon cancer cell proliferation and tumor progression. Cancer Lett. 2019;448:40–51.
- Olsvik PA, Finn RN, Remo SC, Fjelldal PG, Chauvigne F, Glover KA, Hansen T, Waagbo R. A transcriptomic analysis of diploid and triploid Atlantic salmon lenses with and without cataracts. Exp Eye Res. 2020;199:108150. doi:https://doi.org/10.1016/j.exer.2020.108150.
- Dong L, Yu L, Bai C, Liu L, Long H, Shi L, Lin Z. USP27-mediated cyclin E stabilization drives cell cycle progression and hepatocellular tumorigenesis. Oncogene. 2018;37(20):2702–2713. doi:https://doi.org/10.1038/s41388-018-0137-z.
- Yu L, Dong L, Li H, Liu Z, Luo Z, Duan G, Dai X, Lin Z. Ubiquitination-mediated degradation of SIRT1 by SMURF2 suppresses CRC cell proliferation and tumorigenesis. Oncogene. 2020;39(22):4450–4464. doi:https://doi.org/10.1038/s41388-020-1298-0.
- Peterson SR, Silva PA, Murtha TJ, Sun JK. Cataract surgery in patients with diabetes: management strategies. Semin Ophthalmol. 2018;33(1):75–82. doi:https://doi.org/10.1080/08820538.2017.1353817.
- Song MS, Sim HJ, Kang S, Park S, Seo K, Lee SY. Pharmacological inhibition of Kv3 on oxidative stress-induced cataract progression. Biochem Biophys Res Commun. 2020;533(4):1255–1261. doi:https://doi.org/10.1016/j.bbrc.2020.09.138.
- Kaliaperumal R, Venkatachalam R, Nagarajan P, Sabapathy SK. Association of serum magnesium with oxidative stress in the pathogenesis of diabetic cataract. Biol Trace Elem Res. 2021;199(8):2869–2873. doi:https://doi.org/10.1007/s12011-020-02429-9.
- Wen W, Yu G, Liu W, Gu L, Chu J, Zhou X, Liu Y, Lai G. Silencing fundc1 alleviates chronic obstructive pulmonary disease by inhibiting mitochondrial autophagy and bronchial epithelium cell apoptosis under hypoxic environment. J Cell Biochem. 2019;120(10):17602–17615. doi:https://doi.org/10.1002/jcb.29028.
- Cai Y, Yang E, Yao X, Zhang X, Wang Q, Wang Y, Liu J, Fan W, Yi K, Kang C, et al. Fundc1-dependent mitophagy induced by tPA protects neurons against cerebral ischemia-reperfusion injury. Redox Biol. 2021;38:101792. doi:https://doi.org/10.1016/j.redox.2020.101792.
- LoPiccolo J, Blumenthal GM, Bernstein WB, Dennis PA. Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resist Updat. 2008;11(1-2):32–50. doi:https://doi.org/10.1016/j.drup.2007.11.003.
- Mao S, Luo X, Li Y, He C, Huang F, Su C. Role of PI3K/Akt/mTOR pathway associated oxidative stress and cardiac dysfunction in Takotsubo syndrome. Curr Neurovasc Res. 2020;17(1):35–43. doi:https://doi.org/10.2174/1567202617666191223144715.
- Yao L, Yan H. Mir-182 inhibits oxidative stress and epithelial cell apoptosis in lens of cataract rats through PI3K/Akt signaling pathway. Eur Rev Med Pharmacol Sci. 2020;2 4(23):12001–12008.