
ABSTRACT
Objective: To assess the efficacy and safety of fluticasone propionate (Fp) and Fp/salmeterol (FS) administered via a novel multidose dry powder inhaler (MDPI) that is easy to use correctly in asthma patients. Methods: This phase-3, multicenter, double-blind, parallel-group study evaluated asthmatic patients (≥12 years of age) previously treated with either low- or mid-dose inhaled corticosteroids (ICSs) or ICS/long-acting beta agonists. After a 14- to 21-day run-in, patients were randomized to Fp MDPI 50 mcg, Fp MDPI 100 mcg, FS MDPI 50/12.5 mcg, FS MDPI 100/12.5 mcg, or placebo twice daily for 12 weeks. Change from baseline in forced expiratory volume in 1 second (FEV1; primary endpoint) was evaluated at week 12, and serial spirometry was collected at day 1 and week 12 (subset of patients). Safety was assessed by adverse events (AEs). Results: The full analysis and serial spirometry subset included 640 and 312 patients, respectively. At week 12, FS MDPI significantly improved FEV1 from baseline at each dose vs corresponding Fp MDPI doses (p < 0.05). Change from baseline in FEV1 for active treatment groups was significantly greater vs placebo (p < 0.05). After 12 weeks, serial spirometry was significantly greater at all time points in the FS MDPI groups vs corresponding Fp MDPI groups (p < 0.05). Improvements in serial spirometry on day 1 were maintained through week 12. AEs were similar across groups. Conclusions: Pulmonary function was significantly improved with Fp MDPI and FS MDPI vs placebo and FS MDPI vs Fp MDPI. Active treatments had a safety profile comparable to placebo.
CLINICALTRIALS.GOV REGISTRATION IDENTIFIER:
Introduction
Daily long-term medications, particularly those that reduce inflammation and hyperresponsiveness of the airways, are recommended for patients with persistent asthma for adequate control of symptoms Citation[1]. Based on current asthma treatment guidelines, inhaled corticosteroids (ICSs) are the preferred single agents for daily use for symptom control Citation[1]. Fluticasone propionate (Fp), an ICS, was approved by the US Food and Drug Administration in 1994 for twice-daily dosing for maintenance treatment as prophylactic therapy in patients with asthma aged 4 years and older Citation[2,3]. It is currently available as a pressurized metered dose inhaler (MDI) utilizing a hydrofluoroalkane (HFA) propellant and as a dry powder inhaler (DPI) (Flovent® HFA and Flovent® Diskus, respectively, GlaxoSmithKline, Research Triangle Park, NC) Citation[2,3]. For patients whose asthma symptoms are not well controlled with low- (fluticasone DPI 100–300 mcg or equivalent) to mid-dose (fluticasone DPI > 300–500 mcg or equivalent) ICS monotherapy Citation[1], the use of long-acting beta agonists (LABAs) in combination with ICS is a recommended next step in treatment Citation[1]. The combination of Fp with the LABA salmeterol (FS) was approved for asthma maintenance in the same patient population in 2000 Citation[4]. The addition of salmeterol to moderate doses of ICS has consistently demonstrated a significantly greater clinical benefit than increased doses of ICS monotherapy in patients symptomatic on their current asthma medication Citation[5–7].
The improper use of inhaler devices, such as MDIs and conventional DPIs, is a common problem Citation[8,9] that may significantly limit the effectiveness of these devices Citation[8,10–12]. A novel inhalation-driven device (multidose dry powder inhaler [MDPI]; Teva Pharmaceuticals, Inc., Frazer, PA) has been developed that uses a cyclone design that facilitates efficient de-agglomeration and aerosolization of the drug particles from the lactose carrier. It is easy to use correctly, is intuitive, does not require patients to coordinate device actuation with inhalation, and operates in three steps for the administration of a dose: open the cap, inhale the dose, and close the cap. The MDPI may reduce the issues of improper administration of asthma medications that have been associated with the use of different inhaler devices for commonly co-prescribed medications, and thus help overcome a key barrier to effective treatment. Due to the optimization of the formulation and device features of the MDPI products, the delivery of Fp and salmeterol has been improved such that lower doses can be administered with similar efficacy and similar or less systemic exposure. A previous dose-ranging study with Fp MDPI demonstrated that a dose of 50 mcg of Fp MDPI provides comparable efficacy to that of 100 mcg of Fp DPI with similar to slightly less systemic exposure Citation[13]. A second study evaluating the salmeterol dose for the development of FS MDPI demonstrated that a salmeterol dose of 12.5 mcg in FS MDPI provided similar bronchodilation to 50 mcg of salmeterol in FS DPI with less systemic exposure Citation[14]. The ability to use lower doses of these drugs may offer advantages for patients who might not tolerate the higher doses in the Fp and FS DPI products.
The objectives of this study were to assess the efficacy, safety, and tolerability of twice-daily Fp MDPI and FS MDPI over 12 weeks in patients with persistent asthma aged 12 years and older.
Methods
Study description
This 12-week, multicenter, randomized, double-blind, parallel-group, placebo-controlled phase-3 study was conducted at 129 centers in the United States, Canada, Czech Republic, Hungary, Poland, Russian Federation, South Africa, and Ukraine between July 2014 and September 2015. The study assessed the safety and efficacy of Fp MDPI (50 and 100 mcg doses twice daily) and the corresponding FS MDPI (50/12.5 and 100/12.5 mcg doses twice daily) in patients 12 years of age and older with persistent asthma who were previously treated with either low- or mid-dose ICS or ICS/LABA. The study was conducted in accordance with International Council for Harmonisation Good Clinical Practice Consolidated Guideline (E6) and with all applicable national and local laws and regulations. The study protocol was approved by the applicable Independent Ethics Committee or Institutional Review Board prior to study initiation. Written informed consent was obtained from all patients (or from parents/legal guardians of minor patients) in accordance with local regulations. The study was registered on ClinicalTrials.gov (identifier number NCT02139644).
Patients
Key inclusion criteria
Male and female patients 12 years of age or older with a diagnosis of persistent asthma (minimally, symptoms or rescue medication usage >2 days/week, or nighttime awakenings >3 times/month without the use of daily medication) Citation[1] were eligible for inclusion in the study. At the screening visit, patients were required to have a forced expiratory volume in 1 second (FEV1) ≥40% and ≤85% of the predicted value for age, height, sex, and race as per the National Health and Nutrition Examination Survey III reference values Citation[15]. Previous treatment had to include either low- or mid-dose ICS or ICS/LABA for at least one month prior to providing consent (those taking ICS/LABA were required to have a prescreening visit to change to a comparable dose of ICS monotherapy). All patients were required to be able to replace their current short-acting beta agonist (SABA) with albuterol/salbutamol HFA MDI inhalation aerosol at the screening visit for use as required for the duration of the study. Patients were to withhold all inhaled SABA bronchodilators for at least 6 hours prior to all study visits. In addition, patients had to demonstrate reversibility of disease (≥15% reversibility [all patients] and ≥200-mL increase [patients ≥18 years] from baseline FEV1) within 30 minutes following SABA administration at the screening visit. Female patients were eligible if they were not currently pregnant, breastfeeding, or attempting to become pregnant, and were willing to use birth control or of non-childbearing potential.
Key exclusion criteria
Patients were excluded from the study if they had a his-tory of life-threatening asthma exacerbation. Additional criteria for exclusion included asthma exacerbation which required systemic corticosteroids 30 days prior to the screening visit, or any hospitalization for asthma two months prior to the screening visit, the use of immunosuppressive medications four weeks prior to the screening visit, initiation or dose escalation of immunotherapy planned during the study period, documented or suspected bacterial or viral infection of the upper or lower respiratory tract, sinus, or middle ear unresolved within two weeks of the screening visit, history of a positive human immunodeficiency virus test or active hepatitis B virus or hepatitis C infection, the presence of untreated oral candidiasis at the screening visit, or the presence of any disease or condition judged by the study investigator as putting the patient's safety at risk during the study. In addition, patients, who were current smokers, had a smoking history of 10 pack years or more, or had used any tobacco products within the past year, patients with a history of alcohol or drug abuse within two years preceding the screening visit, and patients who had previously participated in a study of Fp MDPI or FS MDPI were excluded.
Key randomization criteria
Patients were required to meet the following criteria for randomization to treatment: FEV1 within 40–85% of predicted normal, no clinically significant abnormal electrocardiograms (ECGs) or clinical visual evidence of oral candidiasis, and no changes in asthma medications, asthma exacerbations, upper respiratory infection or lower respiratory infection during the run-in period. In addition, patients were required to have asthma scores ≥1 (daytime or nighttime) and/or albuterol/salbutamol use on ≥1 occasion on at least four of the seven days immediately preceding the randomization visit, and demonstrate compliance (on at least four of the final seven days of the run-in period) with completion of their daily asthma diaries.
Study design
The study design is schematically presented in . Between the screening visit and the first treatment visit (day 1), patients completed a 14- to 21-day run-in period during which they replaced their existing SABA inhalers with albuterol/salbutamol HFA MDIs for as-needed immediate relief of asthma symptoms during the run-in and treatment periods. During the run-in period, patients discontinued their current ICS or ICS/LABA and received one inhalation twice daily of placebo MDPI and one puff twice daily of open-label beclomethasone dipropionate 40 mcg HFA MDI or equivalent. At the randomization visit, patients were randomized to one of the five treatment groups in equal proportions: Fp MDPI 50 mcg, Fp MDPI 100 mcg, FS MDPI 50/12.5 mcg, FS MDPI 100/12.5 mcg, or placebo, all administered twice daily. Predose spirometry was performed in the morning at the randomization visit and at each treatment visit within 1 hour of the baseline measurement time using standard spirometry; patients were permitted eight efforts, with the highest FEV1 value from three technically acceptable and two repeatable efforts used. All morning doses of the study drug were administered at the investigational center. Serial spirometry was conducted at investigational centers that were preselected based on their capabilities and prior experience with this testing at randomization and week 12, in a subset of patients who performed serial spirometry at predefined postdose intervals for 12 hours following the morning dose. During the double-blind treatment period (from day 1 [baseline] to week 12), patients used paper diaries to record relevant events (e.g., daytime and nighttime asthma symptoms, morning and evening peak expiratory flows [PEFs], rescue medication use, treatment compliance, health-related events, and changes in medication). Patients returned weekly during weeks 1–4 and then every two weeks during weeks 6–12.
Figure 1. Study design. aRequired for patients whose prestudy asthma therapy included a LABA in addition to an ICS. BID, twice daily; ET, early termination; FP MDPI, fluticasone propionate multidose dry powder inhaler; FS MDPI, fluticasone propionate/salmeterol multidose dry powder inhaler; ICS, inhaled corticosteroid; LABA, long-acting beta agonist; RV, randomization visit; SV, screening visit; TV, treatment visit.

Efficacy
Primary efficacy endpoints
The co-primary efficacy endpoints were the change from baseline in trough (morning predose and prerescue bronchodilator) FEV1 at week 12, and the standardized baseline-adjusted area under the effect curve for FEV1 from time 0 to 12 hours after dosing (FEV1 AUEC0–12h) at week 12 in a subset of patients performing serial spirometry after dosing.
Secondary efficacy endpoints
Secondary efficacy endpoints measured over the 12-week treatment period were the change from baseline in weekly averages of the daily trough morning PEF, total daily asthma symptom score, total daily rescue medication use (albuterol/salbutamol HFA MDI), the time to patient withdrawal due to worsening asthma, the change from baseline to endpoint in the Asthma Quality of Life Questionnaire with Standardized Activities [AQLQ(S)] (collected in patients ≥18 years of age), and the time to 15% and 12% improvement from baseline in FEV1 after dosing on day 1 (collected in the serial spirometry subset).
Safety
Safety was assessed by monitoring of vital signs, physical and oropharyngeal examinations, ECGs, concomitant medication usage, and adverse events (AEs). All AEs were coded using the Medical Dictionary for Research Activities (MedDRA; International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, McLean, VA), version 17.10, preferred terms. Patients who demonstrated oropharyngeal signs consistent with oral candidiasis were to have a culture to confirm the diagnosis. Serious AEs (SAEs) were defined as an AE occurring at any dose that resulted in death, a life-threatening AE, inpatient hospitalization or prolongation of existing hospitalization, persistent or significant disability or incapacity, or a congenital anomaly or birth defect. Asthma exacerbations, defined as any worsening of asthma requiring an emergency department visit or hospitalization, were documented.
Statistics
The primary population for the efficacy endpoint analysis was the full analysis set (FAS), which included patients in the intent-to-treat (ITT) population (i.e., all randomized patients) who had received at least one dose of study drug and had at least one postbaseline trough FEV1 assessment. The safety population included all ITT patients who received at least one dose of study drug. Finally, the subset of approximately 300 patients who performed postdose serial spirometry (i.e., serial spirometry subset) was used for the analysis of standardized baseline-adjusted FEV1 AUEC0–12h at week 12 and other postdose spirometry endpoints. All data were processed and summarized using SAS® version 9.1.3 (SAS Institute, Cary, NC).
Analysis for the primary efficacy endpoint (change from baseline in trough FEV1 at week 12) was performed using an analysis of covariance (ANCOVA) model with effects due to baseline trough morning FEV1, sex, age, center (pooled), previous therapy (ICS or ICS/LABA), and treatment. The comparisons of the four treatment arms vs placebo were part of the prespecified multiplicity control procedure.
Standardized baseline-adjusted FEV1 AUEC0–12h was calculated using the trapezoidal rule, and the analysis was performed using an ANCOVA model with fixed effects of treatment, sex, center (pooled), previous therapy (ICS or ICS/LABA), and with covariates of age and baseline FEV1. The four comparisons of interest that were part of the preplanned multiplicity plan were the comparisons of the two FS MDPI arms vs their corresponding Fp MDPI arms and the comparisons of the two FS MDPI arms vs placebo. The order of the eight inferential comparisons of interest in the co-primary analysis was prespecified, and if all comparisons were significant, then inferential testing was extended to the secondary analysis. The order and comparisons of interest for secondary endpoints were prespecified. Change from baseline in morning PEF, total daily asthma symptom scores, and rescue medication use were analyzed using a mixed model for repeated measures method with an unstructured covariance matrix and with effects due to baseline value, sex, age, center (pooled), previous therapy (ICS or ICS/LABA), week, treatment, and week-by-treatment interaction.
The number of patients who withdrew due to worsening asthma and the proportion of patients who achieved 15% and 12% improvements from baseline in FEV1 after dosing on day 1 were calculated. The analysis of time to patient withdrawal for worsening asthma and time to 15% and 12% improvements from baseline in FEV1 after dosing on day 1 was performed using a log-rank test. Time to withdrawal was displayed graphically with the Kaplan–Meier figure. Median times to withdrawal for worsening asthma and associated 95% confidence intervals were estimated.
The change from baseline in the AQLQ(S) score at the endpoint (i.e., last postbaseline observation) was analyzed using an ANCOVA model with effects due to baseline AQLQ(S) score, sex, age, center (pooled), previous therapy (ICS or ICS/LABA), and treatment.
The incidence of AEs was summarized using descriptive statistics (counts and percentages) by MedDRA preferred term.
The planned study enrollment was 125 patients per treatment group (i.e., 625 patients overall, with a subset of approximately 300 patients who performed serial spirometry at day 1 and week 12). The planned number of patients was based on an assumed dropout rate of 15% to yield a statistical power of at least 85%, at a significance level of 0.05, for demonstrating the superiority of Fp MDPI 50 mcg twice daily over placebo for change from baseline in trough FEV1 at week 12 and a statistical power of >99%, at a significance level of 0.05, for demonstrating superiority of FS MDPI 50/12.5 mcg twice daily over Fp MDPI 50 mcg twice daily for the standardized baseline-adjusted FEV1 AUEC0–12h at week 12.
Results
Patients
Of the 1363 patients that were screened, 787 patients were enrolled in the study, and 647 patients were randomized to the five treatment groups (). The demographic and baseline characteristics of the randomized patients (ITT population) are summarized in . The FAS, safety population, and serial spirometry subset comprised 640, 641, and 312 patients, respectively. Overall, 45 (7%) patients discontinued study treatment (17 [13%], 8 [6%], 9 [7%], 8 [6%], and 3 [2%] in the placebo, Fp MDPI 50 mcg, Fp MDPI 100 mcg, FS MDPI 50/12.5 mcg, and FS MDPI 100/12.5 mcg groups, respectively). Of note, AEs were the most frequent reason for study withdrawal, occurring in 12 patients overall, including six patients in the placebo group. Another 10 patients withdrew due to disease progression or lack of efficacy, including six in the placebo group.
Figure 2. Patient disposition. FAS, full analysis set; Fp MDPI, fluticasone propionate multidose dry powder inhaler; FS MDPI, fluticasone/salmeterol multidose dry powder inhaler; ICS, inhaled corticosteroid; ITT, intent to treat; LABA, long-acting beta agonist.
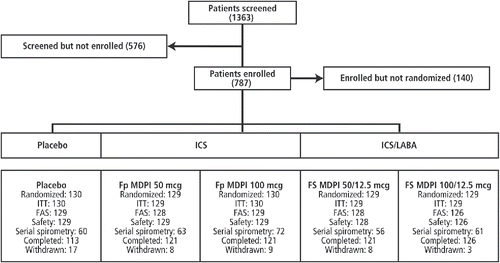
Table 1. Patient demographics and baseline characteristics (ITT population).
Efficacy
Primary efficacy endpoints
At week 12, significant improvements from baseline in trough FEV1 were observed in the Fp MDPI 50 and 100 mcg and FS MDPI 50/12.5 and 100/12.5 mcg treatment groups compared with the placebo group (p < 0.05). At week 12, the FS MDPI 50/12.5 mcg group demonstrated significant improvements from baseline in FEV1 compared with the Fp MDPI 50 mcg group (p < 0.05), and both FS MDPI 50/12.5 mcg and 100/12.5 mcg treatment groups demonstrated significant improvements compared with the Fp MDPI 100 mcg treatment group (p ≤ 0.05) ().
Table 2. Primary efficacy endpoints by the treatment group (full analysis set).
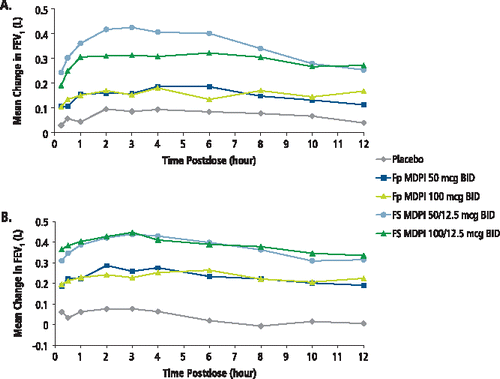
In the serial spirometry subset of patients, standardized baseline-adjusted FEV1 AUEC0–12h (L) was significantly improved at week 12 compared with placebo for the Fp MDPI 50 and 100 mcg and FS MDPI 50/12.5 and 100/12.5 mcg groups (p ≤ 0.05) (; ). In addition, the baseline-adjusted FEV1 AUEC0–12h (L) was significantly improved at week 12 in the FS MDPI 50/12.5 mcg group compared with the Fp MDPI 50 mcg group (p < 0.05), and in the FS MDPI 50/12.5 and 100/12.5 mcg treatment groups compared with the Fp MDPI 100 mcg treatment group (p ≤ 0.05). Of note, substantial improvements in FEV1 AUEC0–12h were evident at day 1 () and were maintained over the 12-week study period ().
Figure 3. Serial spirometry: mean change from baseline in FEV1 (L) at (A) day 1 and (B) week 12 (serial spirometry subset). BID, twice daily; FEV1, forced expiratory volume in 1 second; Fp MDPI, fluticasone propionate multidose dry powder inhaler; FS MDPI, fluticasone propionate/salmeterol multidose dry powder inhaler.
Secondary efficacy endpoints
Analyses of secondary efficacy endpoints are summarized in . Patients treated with FS MDPI 50/12.5 mcg and FS 100/12.5 mcg experienced greater improvements in weekly average of daily trough morning PEF compared with patients treated with Fp MDPI 50 mcg and both Fp MDPI 50 and 100 mcg, respectively (p ≤ 0.05). In addition, improvements in morning PEF for patients in the Fp MDPI 100 mcg and both FS MDPI treatment groups were significantly greater compared with the placebo group (p ≤ 0.05). Changes from baseline in weekly averages of the total daily asthma symptom scores, in total daily use of albuterol/salbutamol, and in the AQLQ(S) score were similar between the Fp MDPI and FS MDPI groups and significantly greater than placebo (p ≤ 0.05) (). The times to patient withdrawal for worsening asthma symptoms were similar between Fp MDPI and FS MDPI groups, and were also similar between Fp MDPI groups and FS MDPI 50/12.5 mcg group and the placebo group due to few withdrawals. There was a trend toward a decrease in the times to patient withdrawal due to worsening asthma in the active treatment groups compared with the placebo group ().
Table 3. Secondary efficacy endpoints by treatment group (full analysis set).
In the serial spirometry subset, the comparison between active treatment groups demonstrated that the proportions of patients who met the predefined thresholds of 15% and 12% improvements from baseline in FEV1 postdose at day 1 were greater in the FS treatment groups vs the corresponding Fp treatment groups (). In addition, the proportions of patients with time to 15% and 12% improvements from baseline in FEV1 postdose were greater for both FS MDPI doses compared with placebo. The time to improvement in the FS MDPI groups was significantly greater than that in the placebo group for 15% improvement (p < 0.05) and for 12% improvement (p < 0.0001).
Safety
Of the 641 patients in the safety population, 214 (33%) experienced at least one treatment-emergent AE (TEAE) during the study (). The most frequent TEAEs were nasopharyngitis, cough, and upper respiratory tract infections, reported in 5.9%, 2.7%, and 3.7% of patients across the four active treatment groups, respectively. Six patients treated with placebo experienced at least one TEAE that required withdrawal from the study; six other patients across the four active treatment groups also withdrew due to TEAEs. AEs reported by at least 2% of the patients in any treatment group during the study are summarized in . There were no clinically meaningful trends in change from baseline values for any vital sign or ECG findings. Any shifts in physical examination findings from normal to abnormal were sporadic across treatment groups. No deaths occurred during the study.
Table 4. Summary of treatment-emergent adverse events (safety population).
Table 5. Incidence of treatment-emergent adverse events reported by at least 2% of the patients (safety population).
AEs of special interest
Positive swab test results for oral candidiasis were reported by one patient in the placebo group and 10 patients in the active treatment groups (Fp MDPI 50 mcg, n = 4; Fp MDPI 100 mcg, n = 2; FS MDPI 50/12.5 mcg, n = 1; and FS MDPI 100/12.5 mcg, n = 3); while all cases of oral candidiasis events were resolved, some patients had more than one event. Of note, four patients had AEs of oral candidiasis without a reported positive swab test (Fp MDPI 50 mcg, n = 1 [event occurred before the treatment period], and one patient each in the Fp MDPI 100 mcg, FS MDPI 50/12.5 mcg, and FS MDPI 100/12.5 mcg groups).
Cardiac disorders were reported in two patients in the placebo group (atrial fibrillation [n = 1], supraventricular extrasystoles and T wave amplitude decrease [n = 1]—all moderate in severity), two patients in the Fp MDPI 100 mcg group (supraventricular extrasystoles [n = 1], ECG QRS complex prolonged [n = 1]—all mild), and three patients in the FS MDPI 50/12.5 mcg group (palpitations [n = 1], tachycardia [n = 1], and ventricular extrasystoles [n = 1]—all mild). These cardiac findings were resolved/resolving in all but two patients (one each in the Fp MDPI 100 mcg and FS MDPI 50/12.5 groups) and were not considered to be SAEs.
SAEs
Four patients experienced at least one SAE as follows: in the placebo group, one patient experienced a mild spontaneous abortion and one patient experienced moderate asthma; in the Fp MDPI 100 mcg group, one patient had plasma cell myeloma; and in the FS MDPI 100/12.5 mcg group, one patient experienced severe pancreatitis, severe cholecystitis, and severe cholelithiasis. None of these SAEs was considered to be related to the study drugs.
Asthma exacerbations
A total of 13 patients reported at least one asthma exacerbation: seven (5%) in the placebo group and six (1.2%) in the active treatment groups (Fp MDPI 50 mcg, n = 1; Fp MDPI 100 mcg, n = 1; FS MDPI 50/12.5 mcg, n = 3; FS MDPI 100/12.5 mcg, n = 1). Four patients in the placebo group experienced at least one asthma exacerbation resulting in a hospitalization or study discontinuation compared with only two patients in the active treatment groups (one in the Fp MDPI 100 mcg group and one in the FS MDPI 50/12.5 mcg group).
Discussion
In the present study of patients with persistent asthma, 12 weeks of treatment with twice-daily Fp MDPI 50 and 100 mcg and FS MDPI 50/12.5 and 100/12.5 mcg demonstrated significantly greater improvement in the change from baseline in trough FEV1 at week 12 compared with placebo, demonstrating efficacy of both doses of Fp MDPI and FS MDPI in this patient population. The addition of salmeterol to Fp significantly improved the primary efficacy measures, with patients treated with FS MDPI 50/12.5 and 100/12.5 mcg experiencing greater improvements in both primary efficacy measures compared with their respective Fp MDPI comparators. This confirms the results obtained from other studies that support improved efficacy with ICS/LABA combination therapy compared with ICS monotherapy Citation[5–7,16–18]. The improvements in pulmonary function were sustained for the length of the study and were consistent with the twice-daily dosing regimen. Significant benefits of ICS/LABA treatment compared with ICS monotherapy were also observed for improvements in morning PEF and the proportion of patients who achieved 15% and 12% improvements from baseline FEV1 postdose Citation[5–7]. The availability of ICS monotherapy and ICS/LABA combination asthma medications in multiple dosages allows for an evidence-based, stepwise approach to asthma management, with current recommendations considering the addition of a LABA to a low-dose ICS equally acceptable to an increase in the ICS dose from low to medium, and better tailoring of treatment regimens to provide optimal asthma control Citation[1].
Significant improvements vs placebo were demonstrated in all Fp and FS treatment arms for the primary efficacy endpoints, and significant improvements vs placebo were generally observed in morning PEF, asthma symptom scores, rescue medication use, and quality of life. Improvements in pulmonary function and asthma symptoms appeared to be generally dose dependent and occurred at a lower dose than the recommended doses of commercially available Fp DPI (100–500 mcg twice daily in patients ≥12 years of age) or FS DPI (100/50–500/50 mcg twice daily in patients ≥12 years of age) for this patient population Citation[2,4].
Overall, the safety profiles for Fp MDPI and FS MDPI observed in this study were consistent with those reported in previous Fp and FS clinical studies and in the current product information for Fp HFA, Fp DPI, and FS DPI Citation[2–4,19]. In the present study, all doses of Fp MDPI and FS MDPI were demonstrated to be safe and well tolerated when administered twice daily for 12 weeks. Incidence rates of AEs were similar across the treatment and placebo groups. Asthma exacerbations, discontinuations due to AEs, and treatment discontinuations occurred more frequently in the placebo group than in the active treatment groups. In addition, no serious asthma exacerbations were reported and no SAE was considered to be related to study drug.
The results of this study are encouraging, although they may be limited by the duration of treatment and the lack of an active comparator. However, the results are consistent with previous studies conducted to evaluate the efficacy and safety of Fp and the combination FS administered via a DPI Citation[16–18].
Conclusions
In summary, this randomized, double-blind, placebo-controlled study demonstrated that 12 weeks of treatment with FS MDPI significantly improved pulmonary function compared with corresponding doses of Fp MDPI in patients with persistent asthma aged 12 years or older. Both FS MDPI and Fp MDPI were associated with greater pulmonary function, better symptom control, and improved quality of life compared with placebo, and both were safe and well tolerated with safety profiles consistent with those of other inhaled asthma medications in the same classes.
Declaration of interest
Dr. Raphael has conducted contracted research for Circassia, Lupin, and Teva.
Ms. Yiu and Dr. Liu are employees of Teva Pharmaceuticals, Frazer, PA.
Dr. Sakov is an employee of Teva Pharmaceuticals, Netanya, Israel.
Dr. Caracta was an employee of Teva Pharmaceuticals, Frazer, PA, at the time of study conduct and manuscript preparation.
Acknowledgements
This study was sponsored by Teva Branded Pharmaceutical Products R&D, Inc. Medical writing assistance was provided by Lisa Feder, PhD, of Peloton Advantage and was funded by Teva Branded Pharmaceutical Products R&D, Inc. Teva provided a full review of the article.
References
- Expert Panel Report 3 (EPR3). Guidelines for the diagnosis and management of asthma. Bethesda, MD: National Heart, Lung, and Blood Institute, US Department of Health and Human Services; 2007 7/2007.
- Flovent Diskus [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014 5/2014.
- Flovent HFA [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014 5/2014.
- Advair Diskus [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014 July 2014.
- Bateman ED, Boushey HA, Bousquet J, Busse WW, Clark TJ, Pauwels RA, et al. Can guideline-defined asthma control be achieved? The Gaining Optimal Asthma ControL study. Am J Respir Crit Care Med 2004;170(8):836–844.
- Masoli M, Weatherall M, Holt S, Beasley R. Moderate dose inhaled corticosteroids plus salmeterol versus higher doses of inhaled corticosteroids in symptomatic asthma. Thorax 2005;60(9):730–734.
- Ducharme FM, Ni CM, Greenstone I, Lasserson TJ. Addition of long-acting beta2-agonists to inhaled corticosteroids versus same dose inhaled corticosteroids for chronic asthma in adults and children. Cochrane Database Syst Rev 2010(5):CD005535.
- Price DB, Pearce L, Powell SR, Shirley J, Sayers MK. Handling and acceptability of the Easi-Breathe device compared with a conventional metered dose inhaler by patients and practice nurses. Int J Clin Pract 1999;53(1):31–36.
- Brocklebank D, Ram F, Wright J, Barry P, Cates C, Davies L, et al. Comparison of the effectiveness of inhaler devices in asthma and chronic obstructive airways disease: a systematic review of the literature. Health Technol Assess 2001;5(26):1–149.
- Virchow JC, Crompton GK, Dal Negro R, Pedersen S, Magnan A, Seidenberg J, et al. Importance of inhaler devices in the management of airway disease. Respir Med 2008;102(1):10–19.
- O'Conor R, Wolf MS, Smith SG, Martynenko M, Vicencio DP, Sano M, et al. Health literacy, cognitive function, proper use and adherence to inhaled asthma controller medications among older adults with asthma. Chest 2015;147(5):1307–1315.
- Giraud V, Roche N. Misuse of corticosteroid metered-dose inhaler is associated with decreased asthma stability. Eur Respir J 2002;19(2):246–251.
- Kerwin EM, Gillespie M, Song S, Steinfeld J. Randomized, dose-ranging study of a fluticasone propionate multidose dry powder inhaler in adolescents and adults with uncontrolled asthma not previously treated with inhaled corticosteroids. J Asthma 2017;54(1):89–98.
- Miller DS, Yiu G, Hellriegel ET, Steinfeld J. Dose-ranging study of salmeterol using a novel fluticasone propionate/salmeterol multidose dry powder inhaler in patients with persistent asthma. Allergy Asthma Proc 2016;37(4):291–301.
- The Third National Health and Nutrition Examination Survey (NHANES III), 1988–1994 Series 11, No. 9A Data Release. Atlanta, GA: Centers for Disease Control and Prevention; 2001. Available from: https://wwwn.cdc.gov/nchs/data/nhanes3/9a/Readme.txt.
- Pearlman DS, Stricker W, Weinstein S, Gross G, Chervinsky P, Woodring A, et al. Inhaled salmeterol and fluticasone: a study comparing monotherapy and combination therapy in asthma. Ann Allergy Asthma Immunol 1999;82(3):257–265.
- Kavuru M, Melamed J, Gross G, Laforce C, House K, Prillaman B, et al. Salmeterol and fluticasone propionate combined in a new powder inhalation device for the treatment of asthma: a randomized, double-blind, placebo-controlled trial. J Allergy Clin Immunol 2000;105(6Pt 1):1108–1116.
- Nathan RA, Rooklin A, Schoaf L, Scott C, Ellsworth A, House K, et al. Efficacy and tolerability of fluticasone propionate/salmeterol administered twice daily via hydrofluoroalkane 134a metered-dose inhaler in adolescent and adult patients with persistent asthma: a randomized, double-blind, placebo-controlled, 12-week study. Clin Ther 2006;28(1):73–85.
- Lasserson TJ, Ferrara G, Casali L. Combination fluticasone and salmeterol versus fixed dose combination budesonide and formoterol for chronic asthma in adults and children. Cochrane Database Syst Rev 2011(12):CD004106.