
ABSTRACT
Objective: We aimed to demonstrate non-inferiority of once-daily fluticasone furoate/vilanterol 100/25 µg (FF/VI) to twice-daily fluticasone propionate/salmeterol 250/50 µg (FP/SAL) in adults/adolescents with asthma well controlled on inhaled corticosteroid/long-acting β2 agonist (ICS/LABA). Methods: This was a randomized, double-blind, double-dummy, parallel-group, 24-week study (NCT02301975/GSK study 201378). Patients whose asthma met study-defined criteria for control were randomized 1:1:1 to receive FF/VI, FP/SAL or twice-daily FP 250 µg for 24 weeks. Primary endpoint was change from baseline in evening trough forced expiratory volume in 1 second (FEV1). Secondary endpoints included rescue-/symptom-free 24-hour periods. Safety was also assessed. Results: The intent-to-treat (ITT) population included 1504 randomized and treated patients (504 FF/VI; 501 FP/SAL; 499 FP); mean age 43.5 years, 64% female. FF/VI demonstrated non-inferiority (using a margin of −100 mL) to FP/SAL for evening trough FEV1 at Week 24 (ITT: 19 mL [95% confidence interval (CI) −11 to 49]; per protocol population [N = 1336]: 6 mL [95% CI −27 to 40]). Improvement in evening trough FEV1 at Week 24 for both FF/VI (123 mL; p < 0.001) and FP/SAL (104 mL; p < 0.001) was greater than FP. FF/VI increased rescue-/symptom-free 24-hour periods by 1.2%/1.2% compared with FP/SAL. All treatments were well tolerated. On-treatment adverse event (AE) rates were 43% to 45% across arms; there were no drug-related serious AEs. Conclusions: FF/VI was non-inferior to FP/SAL for evening trough FEV1 at 24 weeks. These data suggest that patients well controlled on FP/SAL could step across to FF/VI without loss of control.
Introduction
International treatment guidelines recommend the use of an inhaled corticosteroid (ICS)/long-acting β2 agonist (LABA) combination as maintenance therapy for adult and adolescent patients with persistent asthma uncontrolled on ICS alone (Citation1–3). Previous studies have demonstrated that ICS/LABA combination therapy can improve asthma control compared with ICS alone (Citation2, 4). In adults and children with persistent asthma, the addition of a LABA to ICS monotherapy has been shown to improve lung function and to reduce symptoms, rescue medication use and exacerbations (Citation2).
ICS/LABA combinations currently approved for the treatment of persistent asthma in adult and adolescent patients include fluticasone propionate/salmeterol (FP/SAL), fluticasone furoate/vilanterol (FF/VI), FP/formoterol, beclometasone/formoterol, budesonide/for-moterol and mometasone/formoterol (Citation1). The majority of approved combinations are indicated for twice-daily (BID) administration (Citation5–11); however, FF/VI is indicated for once-daily (OD) administration (Citation12, 13). Reducing the frequency of inhaler use from BID to OD administration could simplify treatment, improve convenience for patients and improve treatment adherence (Citation14, 15).
Data from a recent systematic review/meta-analysis suggest that the efficacy of OD FF/VI is comparable with alternative BID ICS/LABA combinations in improving lung function and health status in patients with persistent asthma (Citation16). A previous head-to-head study found FF/VI and FP/SAL to be similarly efficacious in improving lung function in patients with persistent asthma uncontrolled on mid-dose ICS (Citation17).
Few studies have evaluated whether asthma control can be maintained when patients are stepped across from one ICS/LABA to another (Citation18, 19); therefore, this Phase IIIa study was conducted to evaluate whether FF/VI 100/25 µg OD was non-inferior to FP/SAL 250/50 µg BID in adult and adolescent patients with persistent asthma already well controlled on BID ICS/LABA. The study examined lung function and tolerability, as well as patient-reported symptoms and quality of life.
Methods
Patients
Male or non-pregnant female patients aged ≥12 years with an asthma diagnosis according to National Institutes of Health 2007 criteria (Citation3) no less than 12 weeks prior to Visit 1 were eligible. Patients were required to have a forced expiratory volume in 1 second (FEV1) of ≥80% of the predicted normal value according to the Global Lung Function Initiative equations (Citation20) for spirometry reference values at Visit 1, and to have received treatment with ICS/LABA (equivalent to FP/SAL 250/50 BID), either as a fixed-dose combination or through separate inhalers, for at least 12 weeks prior to Visit 1. The ability for patients to replace their current short-acting β2-agonist with albuterol/salbutamol was required from Visit 1 for the duration of the study. All female patients of childbearing potential had to agree to use a proven highly effective contraception method throughout the study.
Key exclusion criteria were: history of life-threatening asthma in the previous 5 years; evidence of concurrent respiratory disease or other clinically significant medical condition; ongoing respiratory infection within 4 weeks prior to Visit 1; use of tobacco products within 3 months prior to Visit 1 or a historical use of ≥10 pack years; severe milk protein allergy or specific drug allergy; or asthma exacerbation that required oral corticosteroids within 12 weeks prior to Visit 1, or that resulted in overnight hospitalization requiring additional asthma treatment within 6 months prior to Visit 1.
The study was approved by relevant national, regional, or investigational center ethics committees/institutional review boards in accordance with International and applicable country-specific requirements. In addition, the study was conducted in accordance with the principles founded in the Declaration of Helsinki and Good Clinical Practice Guidelines. All patients provided written informed consent.
Study design and treatment
This was a 24-week, multicenter, randomized, Phase IIIa, double-blind, double-dummy, parallel-group study evaluating non-inferiority of FF/VI 100/25 µg OD to FP/SAL 250/50 µg BID in patients aged ≥12 years with persistent asthma already well controlled on BID ICS/LABA equivalent to FP/SAL 250/50 µg BID (NCT02301975; GSK study 201378).
From Visit 1, patients received the ICS component of their current ICS/LABA treatment during a 5 (±2)-day LABA washout period. Following washout (Visit 2), patients who demonstrated reversibility in FEV1 of ≥150 mL within 10−40 minutes after four inhalations of albuterol/salbutamol aerosol received open-label FP/SAL during a 4-week run-in period. Following run-in (Visit 3), eligible patients were randomized 1:1:1 using an Interactive Voice Response System to receive one of three blinded study treatments over 24 weeks: FF/VI 100/25 µg OD via the ELLIPTA inhaler (evening) plus placebo BID via the DISKUS inhaler; FP/SAL 250/50 µg BID via DISKUS plus placebo OD via ELLIPTA (evening); or FP 250 µg BID via DISKUS plus placebo OD via ELLIPTA (evening). Patients were provided with albuterol/salbutamol rescue medication for use as necessary.
To be eligible for randomization at Visit 3, patients had to be symptom free during the daytime and not have used rescue/reliever medication on >2 days each week for the last 14 consecutive days of the run-in period, or have had any asthma-related night-time awakenings during the last 14 consecutive days of run-in. Patients had to show compliance with completion of morning/evening e-Diary data on ≥4 of the last 7 consecutive days of the run-in period. Patients could not have changed asthma medication except for the planned change from ICS/LABA to the same ICS alone at Visit 1 and from ICS alone to open-label FP/SAL at Visit 2, or have experienced a respiratory infection or severe asthma exacerbation between Visits 1 and 3.
Patients attended four on-treatment visits at Weeks 4, 8, 16 and 24 (Visits 4 to 7) and a follow-up telephone safety assessment was conducted approximately 1 week after treatment completion.
Endpoints
The primary efficacy endpoint was change from baseline in FEV1 at the evening clinic visit (pre-bronchodilator and pre-dose) at the end of the 24-week treatment period.
Secondary efficacy endpoints included change from baseline to 24 weeks in the percentage of rescue-free and symptom-free 24-hour periods, and morning and evening peak expiratory flow (PEF), as well as the percentage of patients with controlled asthma (defined as an Asthma Control Test [ACT] score ≥20) after 24 weeks.
Other efficacy endpoints were change from baseline to 24 weeks in ACT score and in total Asthma Quality of Life Questionnaire for 12 years and older (AQLQ [12+]) score, and percentage of patients with a change from baseline in total AQLQ (12+) score of ≥0.5.
Safety (incidence of adverse events [AEs], serious AEs [SAEs], AEs of special interest and severe asthma exacerbations) was also assessed.
Assessments
FEV1 was measured in the evening at Visits 1−7 using standardized electronic spirometry equipment. Patients were required to withhold albuterol/salbutamol for ≥6 hours before lung function measurements.
Morning and evening PEF, daytime and night-time asthma symptom scores and the number of inhalations of rescue albuterol/salbutamol used during the day and night were recorded daily by patients in an e-Diary. Daytime asthma symptoms were self-rated according to a 6-point scale ((Citation1) indicating no symptoms during the day through to (Citation6) indicating symptoms severely impacting patients' ability to go to work or perform normal daily activities). Night-time symptoms were rated according to a 5-point scale ((Citation1) indicating no symptoms during the night through to (Citation5) indicating symptoms severely impacting patients' ability to sleep at all). Patients measured their PEF each evening and morning prior to medication dosing and/or albuterol/salbutamol use.
Asthma control was self-reported using the 5-item ACT questionnaire (Citation21). Patients' quality of life was assessed using the disease-specific, self-administered AQLQ (12+) questionnaire (Citation22) and assumed a minimal clinically important difference of 0.5 points (Citation23). Patients completed the ACT and AQLQ (12+) questionnaires at Visit 3 (randomization) and either Visit 7 (end of treatment), or at the early treatment discontinuation/study withdrawal visit. AEs/asthma exacerbations were monitored by investigators/site staff and were coded according to the Medical Dictionary for Regulatory Activities.
Patient compliance with medication was assessed at each visit from Visit 4 to the end of study/early withdrawal by reviewing the inhaler dose counters and calculating the % compliance based on the number of doses used/number of doses expected to be used.
Statistical analyses
Sample size calculations were based on the primary efficacy endpoint of evening FEV1. The primary comparison of FF/VI versus FP/SAL used a non-inferiority limit of −100 mL and a 0.025 one-sided significance level, with equal importance for the intent-to-treat (ITT) and per protocol (PP) populations. The inequality comparison of FF/VI versus FP assumed a true population effect of 100 mL and used a two-sided 5% significance level. The stated non-inferiority margin and true population effect were both agreed with regulatory agencies prior to finalization of the protocol. Approximately 1461 patients (487 per arm) were planned to provide 90% power for both comparisons.
The ITT population comprised all randomized patients who received at least one dose of study medication. The PP population comprised patients in the ITT population without full protocol deviations. Additional pre-defined analysis populations included subsets of patients in the ITT and PP populations aged ≥15 years at screening (defined for consistency with the licensed population for FF/VI in Japan).
To demonstrate assay sensitivity, FF/VI was compared with FP for the primary and secondary/other efficacy endpoints. Additionally, FP/SAL was compared with FP for all efficacy endpoints. To account for multiplicity across key endpoints, a step-down closed testing procedure was applied to the inequality comparison of FF/VI versus FP.
The primary efficacy analysis was conducted using a mixed model repeated measures model, with an additional sensitivity analysis performed on the ITT population, whereby missing FEV1 data at Week 24 were imputed using a last observation carried forward (LOCF) approach. Analyses of 24-week changes from baseline in secondary and other endpoints were performed using analysis of covariance. Analyses of the percentage of patients controlled (ACT score ≥20) and those with a change from baseline in total AQLQ (12+) score of ≥0.5 at Week 24 were conducted using logistic regression. Data were analyzed using SAS Version 9.4 or later.
More detailed information on the statistical analyses is included in the Online Supplement.
Results
Study conduct
This study was conducted at 157 sites across 12 countries between 16 March 2015 and 25 November 2016.
Patients
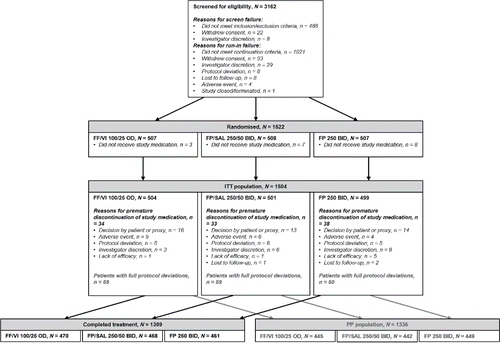
summarizes patient flow through the study. Overall, 3162 patients were screened for eligibility, with 1522 patients randomized. Of these, 1504 (99%) received ≥1 dose of study medication and were included in the ITT population (n = 504 FF/VI; n = 501 FP/SAL; n = 499 FP). The majority of patients (n = 1399; 93%) completed treatment. Treatment discontinuation rates were consistent across arms; the most common reason for treatment discontinuation was patient or proxy decision (n = 43, 3%). Twenty-seven patients (2%) discontinued treatment, but completed the study. The PP population comprised 1336 patients.
Figure 1. Study disposition. Abbreviations: BID, twice daily; FF, fluticasone furoate; FP, fluticasone propionate; ITT, intent-to-treat; OD, once daily; PP, per protocol; SAL, salmeterol; VI, vilanterol.
The majority (82%) of patients were white, 64% were female, and most (96%) were aged ≥15 years (mean 43.5 years) (). Fifty-eight percent had an asthma history of ≥10 years. At randomization (Visit 3; baseline), patients had a mean pre-dose FEV1 of 2.827 L and mean percent predicted FEV1 of 90.24%. Across the treatment arms, the mean percent/absolute reversibility in FEV1 was 15.8%/376.2 mL, and 96% of patients in each treatment group reported an ACT score ≥20. At baseline, 409 patients (27%) had comorbidities, the most common being hypertension (22%) and hypercholesterolemia (7%). The most frequently used asthma medications pre-treatment were salbutamol (75−76%), FP (66−68%) and FP/SAL (47−51%); budesonide/formoterol was used by 28−30% of patients.
Table 1. Patient demographics and baseline characteristics (ITT population).
Demographics/baseline characteristics of the PP population and the subset of patients aged ≥15 years in the ITT and PP populations were similar to the overall ITT population (data not shown).
Exposure and treatment compliance
The mean duration of exposure was 161.7 days (range 3 to 284) in the FF/VI arm (ELLIPTA inhaler), and 161.4 days (4 to 190) in the FP/SAL arm and 161.6 days (11 to 231) in the FP arm (DISKUS inhaler). Mean overall compliance rates were approximately 98% and 96% for ELLIPTA and DISKUS inhalers, respectively. The majority of patients were between 95% to 105% compliant for both ELLIPTA (79−83% across arms) and DISKUS (72−74% across arms) inhalers.
Efficacy
Primary endpoint – evening trough FEV1
At Week 24, the treatment difference between FF/VI and FP/SAL for evening trough FEV1 was 19 mL (95% confidence interval [CI] −11 to 49) in the ITT population and 6 mL (95% CI −27 to 40) in the PP population (). In both populations, the lower bound of the evening trough FEV1 95% CI was greater than the pre-defined non-inferiority margin of −100 mL, thus demonstrating non-inferiority of FF/VI to FP/SAL for the primary endpoint.
Table 2. Statistical analysis of change from baseline in evening trough FEV1 (L) at Week 24a.
In the ITT population, the least squares (LS) mean improvement in evening trough FEV1 at Week 24 was significantly greater for FF/VI than with FP (123 mL, p < 0.001) and for FP/SAL than with FP (104 mL, p < 0.001) ( and ).
Figure 2. Change from baseline in evening trough FEV1 in (a) the ITT population and (b) the PP population (repeated measures analysis). (a) ITT population. (b) PP population. Abbreviations: BID, twice daily; CI, confidence interval; FEV1, forced expiratory volume in 1 second; FF, fluticasone furoate; FP, fluticasone propionate; ITT, intent-to-treat; LS, least squares; OD, once daily; PP, per protocol; SAL, salmeterol; VI, vilanterol.
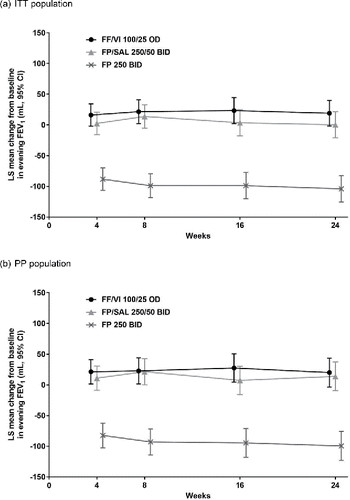
A sensitivity analysis using a LOCF approach in the ITT population produced similar results to the repeated measures analysis. FF/VI was non-inferior to FP/SAL with an evening trough FEV1 treatment difference of 16 mL (95% CI −13 to 46). The LS mean improvement in evening trough FEV1 was significantly greater for FF/VI than with FP (124 mL [95% CI 95 to 153]; p < 0.001) and for FP/SAL than with FP (107 mL [95% CI 78 to 137]; p < 0.001).
Data for the primary endpoint in the subset of patients aged ≥15 years were consistent with findings in the overall ITT and PP populations, demonstrating non-inferiority of FF/VI to FP/SAL for evening trough FEV1 at Week 24 (treatment difference, ITT: 23 mL [95% CI −7 to 53]; PP: 12 mL [95% CI −21 to 46]) (Supplementary ).
Secondary/other efficacy endpoints
Baseline percentages of rescue-free 24-hour periods were comparable across the arms (range 97.7% to 98.4%). The change from baseline in rescue-free 24-hour periods over 24 weeks was similar for FF/VI versus FP/SAL (1.2% [95% CI −0.5 to 3.0]), while a statistically significant 2.7% difference was observed for FF/VI versus FP (95% CI 0.9 to 4.4; p = 0.002). The difference between FP/SAL and FP was 1.4% (). Findings were consistent in the subset of ITT patients aged ≥15 years (Supplementary ).
Figure 3. Treatment effect for primary/secondary/other efficacy endpoints (ITT analysis). (a) FF/VI 100/25 OD versus FP/SAL 250/50 BID. (b) FF/VI 100/25 OD versus FP 250 BID. (c) FP/SAL 250/50 BID versus FP 250 BID. Abbreviations: ACT, Asthma Control Test; AQLQ (12+), Asthma Quality of Life Questionnaire for 12 years and older; AM, morning; BID, twice daily; CI, confidence interval; FEV1, forced expiratory volume in 1 second; FF, fluticasone furoate; FP, fluticasone propionate; OD, once daily; PEF, peak expiratory flow; PM, evening; SAL, salmeterol; VI, vilanterol.
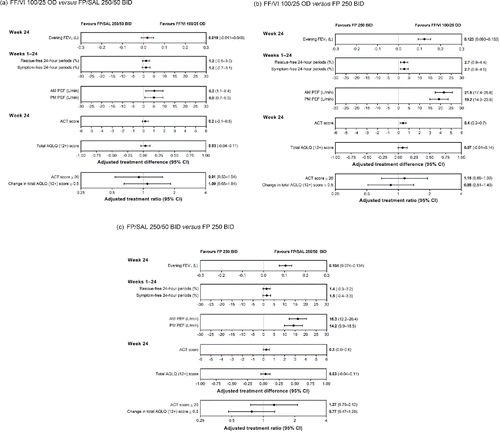
Baseline percentages of symptom-free 24-hour periods were comparable across the treatment arms (range 97.1−98.4%). The change from baseline in symptom-free 24-hour periods over 24 weeks was similar for FF/VI versus FP/SAL (1.2% [95% CI −0.7 to 3.1]), while a statistically significant 2.7% difference was observed for FF/VI versus FP (95% CI 0.8 to 4.5; p = 0.004). The difference between FP/SAL and FP was 1.5% (). Findings were similar in the subset of ITT patients aged ≥ 15 years (Supplementary ).
Baseline mean morning PEF values were similar across the treatment arms (range 407.4−414.4 L/min). The LS mean change from baseline to 24 weeks in morning PEF was similar for FF/VI and FP/SAL, but improved significantly with FF/VI versus FP (21.5 L/min; 95% CI 17.4 to 25.6; p < 0.001) and with FP/SAL versus FP (16.3 L/min; 95% CI 12.2 to 20.4; p < 0.001) ().
Baseline mean ACT scores were comparable across the treatment arms (range 23.4−23.6). At Week 24, the proportion of patients with an ACT score ≥ 20 was maintained in all treatment groups (92% FF/VI; 93% FP/SAL; 91% FP). LS mean change from baseline to 24 weeks in ACT score for FF/VI was similar to FP/SAL, whereas FF/VI and FP/SAL demonstrated differences in ACT scores versus FP of 0.4 (95% CI 0.2 to 0.7) and 0.3 (95% CI 0.0 to 0.6), respectively (). Findings were consistent in the subset of ITT patients aged ≥15 years (Supplementary ).
Table 3. Summary of AEs, SAEs and AEs of special interest (ITT population; N = 1504).
Baseline mean total AQLQ (12+) scores were similar across treatment groups (range 6.40−6.46). LS mean change from baseline to Week 24 in total AQLQ (12+) score was similar for FF/VI and FP/SAL, and for FF/VI and FP/SAL compared with FP ().
Safety
AEs
AEs, SAEs and AEs of special interest are summarised in . Rates of on-treatment AEs were comparable across treatment arms (); the most common on-treatment AEs were nasopharyngitis and headache. Post-treatment AEs were rare (n = 17; 1%); events occurring in >1 patient were nasopharyngitis (n = 4), spontaneous abortion (n = 3), bronchitis (n = 2), influenza (n = 2) and urinary tract infection (n = 2). Among the three patients who experienced spontaneous abortion, two were receiving FP/SAL and one was receiving FP; two were first trimester pregnancies. Rates of drug-related AEs were 3% for FF/VI, 3% for FP/SAL and 2% for FP. The most common drug-related AEs were dysphonia (n = 7, <1% each arm) and oral candidiasis (n = 7, <1% each arm). Nineteen patients (1%) experienced AEs leading to treatment discontinuation/study withdrawal (n = 9 FF/VI; n = 6 FP/SAL; n = 4 FP); events occurring in >1 patient were oral candidiasis (n = 2) and insomnia (n = 2). On-treatment SAEs were reported in 15 patients (1%) and were comparable between treatment arms (). No individual SAE occurred in >1 patient and none of the events were deemed to be drug related. There were no on-treatment deaths.
The most frequently reported on-treatment/post-treatment AEs of special interest were lower respiratory tract infection excluding pneumonia, local steroid effects, and hypersensitivity (). There were two reports of pneumonia, both in the FF/VI arm.
Asthma exacerbations
In total, 66 patients (4%) experienced on-treatment severe asthma exacerbations (requiring systemic corticosteroids for ≥3 days or inpatient hospitalization or an emergency department visit); rates were similar across the treatment arms (4% FF/VI; 4% FP/SAL; 5% FP). None of these events required hospitalization; however, seven patients were treated in the emergency department (n = 2 each FF/VI and FP/SAL; n = 3 FP). One patient in the FP arm experienced a severe asthma exacerbation post-treatment.
Discussion
The overall aim of this study was to determine whether patients with asthma already well controlled on BID ICS/LABA could be switched to OD ICS/LABA without loss of control (assessed by lung function, symptoms, rescue medication use and ACT questionnaire). To address this hypothesis, the present Phase III trial tested non-inferiority of FF/VI to FP/SAL in patients with asthma well controlled on mid-dose ICS/LABA. More than 1500 adults and adolescents with persistent asthma were enrolled across 12 countries globally. The patient population had well-controlled asthma at baseline with a mean percent predicted FEV1 of 90.24%, and 96% of patients had ACT scores ≥20.
The study met its primary endpoint, demonstrating non-inferiority of FF/VI to FP/SAL for evening trough FEV1 at Week 24 (treatment differences of 19 mL [95% CI −11 to 49] in the ITT population and 6 mL [95% CI −27 to 40] in the PP population, with the lower bounds of the 95% CI surpassing the pre-defined non-inferiority margin of −100 mL). This was supported by the secondary/other efficacy endpoints, whereby findings for rescue-free and symptom-free 24-hour periods, morning/evening PEF, ACT scores and AQLQ (12+) scores were all similar for FF/VI compared with FP/SAL. With the exception of ACT score ≥20, the point estimates for the secondary and other efficacy endpoints favored FF/VI over FP/SAL.
Assay sensitivity was demonstrated with superiority of both FF/VI and FP/SAL over FP. Statistically significant improvements from baseline in evening trough FEV1 at Week 24 were shown for both FF/VI versus FP (123 mL, p < 0.001) and FP/SAL versus FP (104 mL, p < 0.001). This was supported by statistically significant improvements for FF/VI over FP of 2.7% (p = 0.002) for rescue-free 24-hour periods, 2.7% (p = 0.004) for symptom-free 24-hour periods and 21.5 L/min (p < 0.001) for morning PEF.
The efficacy of FF/VI has been previously compared with FF, FP and FP/SAL in patients with uncontrolled asthma; however, there is a paucity of direct comparative data for FF/VI versus alternative ICS/LABA combination therapies. At the time of writing, FP/SAL is the only ICS/LABA to have been directly compared with FF/VI in a randomized controlled trial, despite other ICS/LABA combinations being commonly prescribed in general practice (Citation16, 17). The prior head-to-head study in patients with persistent asthma uncontrolled on mid-dose ICS did not show a difference in efficacy between FF/VI and FP/SAL with respect to lung function improvement, asthma control and health-related quality of life (Citation17). Taken together, the results of the current study and the previous study suggest that FF/VI is as effective as FP/SAL for both gaining and maintaining asthma control in a double-blind, randomized clinical trial setting. These results are further supported by the first period of a study conducted in Japanese patients, which demonstrated that patients maintained control when they stepped across from FP/SAL to FF/VI (Citation19). In a recent study set in everyday clinical practice, FF/VI was shown to provide significant improvements in asthma control compared with usual care (ICS [± LABA]) (Citation24).
As expected in a double-blind randomized study where compliance is monitored and actively encouraged, treatment exposure was similar across the arms and compliance was high. All three treatments were well tolerated in this patient population, as evidenced by ≤2% of patients discontinuing treatment due to AEs. The incidence and pattern of on-treatment AEs was similar across the arms (AE rates 43% to 45%; most common were nasopharyngitis and headache). The safety profile was consistent with previous studies of FF/VI 100/25 OD (Citation17) and with the known AE profile of FF/VI in asthma (Citation12, 13). Rates of drug-related AEs and SAEs were low and comparable across the treatment groups. Pneumonia events were reported in only two patients (both FF/VI 100/25 OD) and severe asthma exacerbations in only 4% to 5% of patients across arms.
The design of this study is one of its strengths. FP/SAL was selected as the non-inferiority comparator as it is a recognized ICS/LABA with well-documented efficacy in the treatment of persistent asthma. The trial was planned to carefully select a patient population who, although well controlled at study entry, were not limited by a ‘ceiling’ effect, and in whom a treatment difference between FF/VI and FP/SAL could be demonstrated should one exist. The 5-day LABA washout period, followed by the minimum FEV1 reversibility barrier of 150 mL ensured that patients who retained a high level of lung function on ICS alone (and therefore did not require a LABA component to maintain control over their asthma) were excluded. Inclusion of an ICS-only arm demonstrated assay sensitivity with respect to the ICS/LABA arms and ensured that any demonstrated non-inferiority of FF/VI to FP/SAL was not due to a ceiling effect. The treatment duration of 24 weeks (approximately 6 months) in the present study was deemed sufficient within which to demonstrate any loss of asthma control (Global Initiative for Asthma guidelines for asthma recommend review approximately every 3 months (Citation1)). A placebo arm was not necessary to prove the hypothesis of the study and, for that reason, was not ethically justified.
There are two limitations to the current study; firstly, the patient population was highly selective, with a 48% screen failure rate, reducing the generalizability of the findings. Secondly, the double-blind nature of the study meant that any compliance advantage associated with once-daily versus twice-daily medication could not be evaluated.
Conclusions
Our findings, demonstrating the non-inferiority of FF/VI to FP/SAL, suggest that patients with asthma already well controlled on FP/SAL could step across to FF/VI without loss of efficacy, giving physicians and/or patients the option of a once-daily ICS/LABA if preferred.
Declaration of interest
DB has received payment from GSK for serving as Principal Investigator in GSK-sponsored clinical trials and as advisory board consultant for GSK. RF, LY, LA, and LJ are employed by, and hold stock in, GSK.
Supplementary_material_clean.docx
Download MS Word (21.1 KB)Acknowledgements
The authors would like to thank all patients for their participation in this study. This study was funded by GSK (GSK study 201378; NCT02301975). Editorial support (in the form of writing assistance, collating author comments, assembling tables/figures, grammatical editing, fact checking and referencing) was provided by Emma Landers, PhD, and David Mayes, MChem, of Gardiner-Caldwell Communications (Macclesfield, UK) and was funded by GSK.
Additional information
Funding
Related Research Data
References
- Global strategy for asthma management and prevention [Internet]. Global Initiative for Asthma; updated 2017 [cited 2017 Jun 5]. Available from: http://ginasthma.org/2017-gina-report-global-strategy-for-asthma-management-and-prevention/
- Ducharme FM, Ni Chroinin M, Greenstone I, Lasserson TJ. Addition of long-acting beta2-agonists to inhaled steroids versus higher dose inhaled steroids in adults and children with persistent asthma. Cochrane Database Syst Rev. 2010 Apr 14;(4):CD005533.
- Guidelines for the diagnosis and management of asthma – expert panel report 3. Bethesda (MD): National Institutes of Health (NIH), US Department of Health and Human Services; 2007. ( NIH publication; no. 07–4051.)
- Bateman ED, Boushey HA, Bousquet J, et al. Can guideline-defined asthma control be achieved? Am J Respir Crit Care Med. 2004;170;836–844. doi:10.1164/rccm.200401-033OC. PMID: 15256389
- Seretide 100, 250, 500 Accuhaler. Summary of product characteristics [Internet]. GlaxoSmithKline [cited 2017 Jun 5]. Available from: https://www.medicines.org.uk/emc/medicine/2317
- ADVAIR DISKUS (fluticasone propionate and salmeterol inhalation powder), for oral inhalation. Prescribing information [Internet]. GlaxoSmithKline; updated 2017 Feb [cited 2017 Jun 5]. Available from: https://www.gsksource.com/pharma/content/dam/GlaxoSmithKline/US/en/Prescribing_Information/Advair_Diskus/pdf/ADVAIR-DISKUS-PI-MG-IFU.PDF
- Flutiform 50 mcg/5 mcg, 125 mcg/5 mcg and 250 mcg/10 mcg per actuation pressurised inhalation, suspension. Summary of product characteristics [Internet]. Napp Pharmaceuticals Ltd [cited 2017 Jun 5]. Available from: https://www.medicines.org.uk/emc/medicine/26954
- Fostair 100/6 micrograms per actuation pressurised inhalation solution. Summary of product characteristics [Internet]. Chiesi Ltd [cited 2017 Jun 5]. Available from: https://www.medicines.org.uk/emc/medicine/21006
- Symbicort® Turbohaler® 200 micrograms/6 micrograms/inhalation, inhalation powder. Summary of product characteristics [Internet]. AstraZeneca [cited 2017 Jun 5]. Available from: https://www.medicines.org.uk/emc/medicine/4821
- SYMBICORT 80/4.5 (budesonide 80 mcg and formoterol fumarate dihydrate 4.5 mcg); SYMBICORT 160/4.5 (budesonide 160 mcg and formoterol fumarate dihydrate 4.5 mcg) Inhalation Aerosol FOR ORAL INHALATION. Prescribing information [Internet]. AstraZeneca; updated 2010 Jun [cited 2017 Jun 5]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/021929s021lbl.pdf
- DULERA (mometasone furoate 100 mcg and formoterol fumarate dihydrate 5 mcg; 200 mcg/5 mcg (mometasone furoate 200 mcg and formoterol fumarate dihydrate 5 mcg) Inhalation Aerosol. Prescribing information [Internet]. Merck; updated 2011 Jul [cited 2017 Jun 5]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/022518s002s004lbl.pdf
- Relvar Ellipta. Summary of product characteristics [Internet]. GlaxoSmithKline [cited 2017 Jun 5]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002673/WC500157633.pdf
- BREO ELLIPTA 100/25 (fluticasone furoate 100 mcg and vilanterol 25 mcg inhalation powder), for oral inhalation; fluticasone furoate 200 mcg and vilanterol 25 mcg inhalation powder), for oral inhalation. Prescribing information [Internet]. GlaxoSmithKline; updated 2016 Jul [cited 2017 Jun 5]. Available from: https://www.gsksource.com/pharma/content/dam/GlaxoSmithKline/US/en/Prescribing_Information/Breo_Ellipta/pdf/BREO-ELLIPTA-PI-MG.PDF
- Price D, Robertson A, Bullen K, et al. Improved adherence with once-daily versus twice-daily dosing of mometasone furoate administered via a dry powder inhaler: a randomized open-label study. BMC Pulm Med. 2010;10:1. doi:10.1186/1471-2466-10-1. PMID: 20051135
- Campbell LM. Once-daily inhaled corticosteroids in mild to moderate asthma: improving acceptance of treatment [discussion 52]. Drugs. 1999;58(Suppl 4):25–33. doi:10.2165/00003495-199958004-00004. PMID: 10711856
- Svedsater H, Stynes G, Wex J, Frith L, Leather D, Castelnuovo E, Detry M, Berry S. Once-daily fluticasone furoate/vilanterol versus twice daily combination therapies in asthma-mixed treatment comparisons of clinical efficacy. Asthma Res Pract. 2016;2:4. doi:10.1186/s40733-015-0016-0. PMID: 27965772
- Woodcock A, Bleecker ER, Lötvall J, O'Byrne PM, Bateman ED, Medley H, Ellsworth A, Jacques L, Busse WW. Efficacy and safety of fluticasone furoate/vilanterol compared with fluticasone propionate/salmeterol combination in adult and adolescent patients with persistent asthma: a randomized trial. Chest. 2013;144:1222–1229. doi:10.1378/chest.13-0178. PMID: 23846316
- Barnes N, van Noord JA, Brindicci C, Lindemann L, Varoli G, Perpiña M, Guastalla D, Casula D, Patel S, Chanez P. Stepping-across controlled asthmatic patients to extrafine beclometasone/formoterol combination. Pulm Pharmacol Ther. 2013;26:555–561. doi:10.1016/j.pupt.2013.01.011. PMID: 23524015
- Adachi M, Goldfrad C, Jacques L, Nishimura Y. Efficacy and safety comparison: Fluticasone furoate and fluticasone propionate, after step down from fluticasone furoate/vilanterol in Japanese patients with well-controlled asthma, a randomized trial. Respir Med. 2016;120:78–86. doi:10.1016/j.rmed.2016.09.018. PMID: 27817819
- Quanjer PH, Stanojevic S, Cole TJ, Baur X, Hall GL, Culver BH, Enright PL, Hankinson JL, Ip MS, Zheng J, Stocks J. Multi-ethnic reference values for spirometry for the 3–95-yr age range: the global lung function 2012 equations. Eur Respir J. 2012;40:1324–1343. doi:10.1183/09031936.00080312. PMID: 22743675
- Nathan RA, Sorkness CA, Kosinski M, Schatz M, Li JT, Marcus P, Murray JJ, Pendergraft TB. Development of the asthma control test: a survey for assessing asthma control. J Allergy Clin Immunol. 2004;113:59–65. doi:10.1016/j.jaci.2003.09.008. PMID: 14713908
- Juniper EF, Svensson K, Mörk AC, Ståhl E. Modification of the asthma quality of life questionnaire (standardised) for patients 12 years and older. Health Qual Life Outcomes. 2005;3:58. doi:10.1186/1477-7525-3-58. PMID: 16168050
- Juniper EF, Guyatt GH, Willan A, Griffith LE. Determining a minimal important change in a disease-specific Quality of Life Questionnaire. J Clin Epidemiol. 1994;47:81–87. doi:10.1016/0895-4356(94)90036-1. PMID: 8283197
- Relvar Ellipta significantly improved asthma control in Salford Lung Study patients compared with their usual care [Internet]. GlaxoSmithKline; updated 2017 May 5 [cited 2017 Jun 5]. Available from: http://www.gsk.com/en-gb/media/press-releases/relvar-ellipta-significantly-improved-asthma-control-in-salford-lung-study-patients-compared-with-their-usual-care/